









-
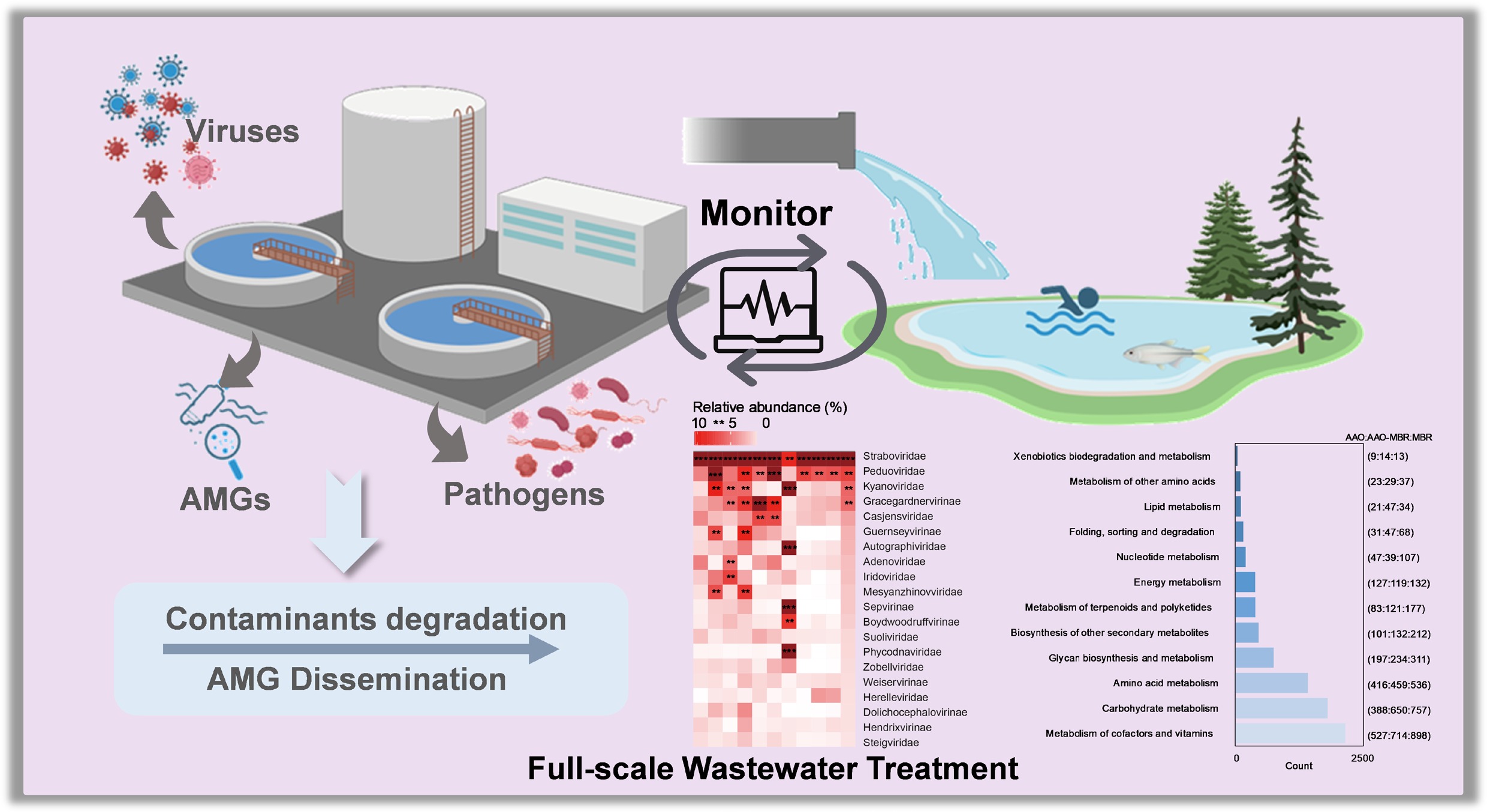
Viruses pose significant threats to global public health and are frequently detected in engineered aquatic environments[1,2]. Wastewater treatment plants (WWTPs) play a pivotal role at the nexus of natural and social water cycles[3], and they are crucial for pollutant removal, water quality improvement, and the protection of human and environmental health[4]. Amid growing pressure on freshwater resources, wastewater reclamation has become an indispensable alternative water source in China[5]. Furthermore, viruses constitute essential members of the microbial consortia within WWTPs, exhibiting broad host ranges and diverse ecological roles[6]. Their transmission dynamics and ecological functions are intricately linked to the treatment process[7], influencing microbial community structure and function, and contributing to biogeochemical cycling through virus-encoded auxiliary metabolic genes (AMGs)[8]. Notably, viruses may also act as vectors for antibiotic resistance genes (ARGs), thereby serving as reservoirs and facilitators of resistance dissemination[9]. Since the outbreak of the COVID-19 pandemic, human society has gradually realized the importance of monitoring pathogenic factors in wastewater for early risk warning[10−12]. When co-occurring with bacterial pathogens, viruses engage in multifaceted interactions that extend beyond classical infection cycles[13]. For instance, a host-pathogen metabolic synchrony that facilitates disease tolerance[14], leading to pathogen persistence and posing unresolved risks to wastewater reclamation safety and human health.
With the rapid development of molecular systems biology and sequencing technologies, metagenomics has emerged as a powerful approach for virus identification and host prediction[15]. In recent years, studies have shown that the treatment process exerts a profound influence on viral diversity, abundance, and virus-host associations within WWTPs[16,17]. The presence of viruses can also affect the function of microorganisms in the treatment system, thereby influencing pollutant removal and biochemical cycling[7]. For instance, viruses may influence biogeochemical processes by modulating host metabolism through the expression of AMGs, thereby affecting the cycling of carbon, sulfur, and phosphorus[18]. However, exploration of the distribution of viral communities across various treatment units throughout the full-scale wastewater treatment process remains insufficient, with much of the focus narrowed on activated sludge[19]. In particular, the dynamic changes in virus–host interactions, especially those involving pathogenic hosts, remain poorly systematically characterized. Previous research has focused mainly on targeted detection of individual or a few viral taxa, lacking broader exploration of the full-spectrum viral diversity and its associated risks[20,21]. A systematic analysis of the distribution characteristics of viruses and hosts across WWTPs can help reveal the ecological functions of viral communities and their interactions with hosts during wastewater treatment[22]. Identifying key viruses and pathogen hosts in wastewater treatment processes, providing a scientific basis for risk assessment and the safety of wastewater reclamation.
Building on our previous work, in which we established a metagenomic-based platform for pathogen identification[2] and compiled a foundational catalog of pathogens distributed across WWTPs[3], metagenomic analysis of 28 real samples collected from three full-scale WWTPs was performed, arranging all treatment units from influent to effluent. In this study, the core composition and dynamic structural shifts of viral communities (phages) within full-scale wastewater treatment systems were systematically characterized. A subset of highly abundant viral taxa was consistently observed throughout the entire treatment process, indicating their potential ecological significance. Notably, the abundance trajectories of Pseudomonas aeruginosa and Aeromonas caviae exhibited strong correlations with those of these persistent viral populations, suggesting their potential as biological indicators of viral dynamics across treatment stages. Furthermore, AMGs were analyzed in WWTPs and their association with wastewater treatment efficiency examined. The results demonstrate that viral AMGs contribute not only to the removal of conventional pollutants but also to the horizontal transmission of ARGs among pathogens. Additionally, by integrating machine learning algorithms for virus-host prediction, a virus-host-specific network can be constructed based on MAGs recovery and annotation. This approach explores the distribution characteristics of viruses and their hosts throughout the full-scale treatment process, clarifying the interactions between viruses and their hosts, and their potential impacts on wastewater treatment. This research provides robust data to support optimizing WWTPs and controlling the risks posed by viruses and pathogens. It provides a scientific basis for broadening the risk-factor surveillance list, optimizing wastewater treatment processes, and improving the safety of wastewater reclamation.
-
A total of 28 samples (17 wastewater samples and 11 sludge samples; 22 samples were self-sampled and six were collected from published work) were collected from three municipal WWTPs. Two self-sampling WWTPs located in Shenzhen, Guangdong, and Zhuozhou, Hebei (China) with different secondary treatment strategies as the mainstay. Another was from published literature[23], and the WWTP was located in Singapore with an MBR process as the mainstay. The WWTP located in Shenzhen (WWTP CHNSZ) was designed to treat 560,000 t/d and to use UV disinfection, achieving a comprehensive water quality compliance rate of 100% (Class I–A) throughout the year. The WWTPs secondary treatment unit utilizes the MUCT (Modified University of Cape Town) process, a modification of the AAO process, which adds an anoxic tank in series with the AAO process. WWTP CHNSZ has a total of nine sampling points, which are influent water samples (AAO-IFW), anoxic pond 1 sludge-wastewater mixture (AAO-AP1W, AAO-AP1S), anoxic pond 2 sludge-wastewater mixture (AAO-AP2W, AAO-AP2S), aerobic pond sludge-wastewater mixture (AAO-AW, AAO-AS), secondary sedimentation tank effluent water samples (AAO-SSTW), concentrated activated sludge (AAO-CCS), tertiary treatment effluent and sludge (AAO-TTW, AAO-TTS), and disinfection effluent water (AAO-DEW). The sampling volumes at the sampling point were as follows: 1 L, 1 L, 1 L, 1 L, 1 L, 50 mL, 3 L, 10 L, and 20 L, respectively.
The WWTP (WWTP AAO-MBR) located in Zhuozhou, Hebei, China, is designed to treat 40,000 t/d, with sodium hypochlorite disinfection, and has a comprehensive compliance rate of 100% (Class I–A) for water quality throughout the year. The secondary treatment unit utilizes a multi-stage A/O-MBR process, with seven sampling points: influent water sample (AAO-MBR-IFW), multi-stage A/O pond sludge-wastewater mixture (AAO-MBR-APW, AAO-MBR-APS, AAO-MBR-AW, AAO-MBR-AS), MBR pond sludge-wastewater mixture (AAO-MBR-MBRW, AAO-MBR-MBRS), secondary sedimentation tank effluent water sample (AAO-MBR-SSTW), disinfection effluent water (AAO-MBR-DEW), and receiving water (AAO-MBR-RW). The sampling points were sampled in the following order: 200 mL, 200 mL, 200 mL, 200 mL, 1 L, 1 L, 1 L, and 50 mL of activated sludge from each section. The specific arrangement of sampling points is schematically illustrated in Supplementary Fig. S1. Samples were transported back to the laboratory for pre-treatment as soon as possible after collection to ensure microbial activity and avoid degradation of DNA.
Samples from the secondary treatment unit of the Singapore WWTP (WWTP MBR), which primarily utilizes the MBR process, were collected at six sampling points. They are influent water sample (MBR-IFW), primary sedimentation tank effluent (MBR-PSW), MBR effluent (MBR-MBRW), secondary sedimentation tank effluent (MBR-SSTW), final effluent (MBR-EFW), and reflux activated sludge (MBR-AS), where the secondary sedimentation tanks are discharged in parallel with the MBR treatment ponds, and the effluents of both converge into the wet wells for discharge. Data were obtained from the publicly available database National Center for Biotechnology Information (NCBI), Project No. PRJNA438174. Containing a total of six samples from this project[23]: SRR6837553, SRR6837554, SRR6837557, SRR6837558, SRR6837571, SRR6837572. Details of pre-treatment and DNA extraction procedures are described in Supplementary Text S1.
DNA extraction and metagenomic sequencing
-
Sequencing libraries of extracted DNA were generated using the ALFA-SEQ DNA Library Prep Kit according to the manufacturer's recommendations, and index codes were added. Library quality was assessed using the Qubit 4.0 Fluorometer (Life Technologies, Grand Island, NY, USA), and the Qsep400 High-Throughput Nucleic Acid Protein Analysis system (Houze Biological Technology Co, Hangzhou, China). Finally, the library was sequenced on an Illumina NovaSeq 6000 platform, generating 150 bp paired-end reads with a target raw data sequencing depth of at least 20 Gbp per sample.
Viral contig assembly and prediction
-
Data pre-treatment includes quality control for raw data via Trimmomatic (v0.39)[24], Flexbar (v3.5.0)[25], Skewer (v0.2.2)[26], and SortMeRNA (v2.1b)[27]. Metagenomic contigs were assembled using MEGAHIT (v1.2.9)[28] with parameter meta-sensitive, -m 0.9. Only contigs ≥ 5 Kb, high-quality assessed by CheckV (v0.6.0)[29], were retained using seqmagick (v0.8.0) (
https://github.com/fhcrc/seqmagick/ ). Candidate viral contigs were identified from assembled contigs by three tools, including DeepVirFinder (v1.0)[30], Virsorter2 (v2.2.4)[31], and VIBRANT (v1.2.1)[32] in parallel. A combined set of viral contig candidates from these tools was collected for further analysis. AMGs on viral contigs were identified via VIBRANT (v1.2.1) and DRAM (v1.5.0)[33] in parallel. ARGs were identified using AMRFinderPlus (v3.11.4)[34]. Viral hosts of contigs prediction were performed using iPHoP (v1.3), developed with machine learning, based on the ICTV database[35].MAGs recovery and annotation
-
After data pre-treatment, MAGs were retrieved from the assembled 2 Kb contigs via a binning approach using metaWRAP (v1.2) (completeness > 50% and contamination < 10%)[36]. GTDB-Tk (v2.1.1)[37] was applied to assign the taxonomy of MAGs based on the Genome Taxonomy Database (GTDB,
http://gtdb.ecogenomic.org ) taxonomy release 214 (Fig. 1). Figures in this study were visualized with R (v4.1.1).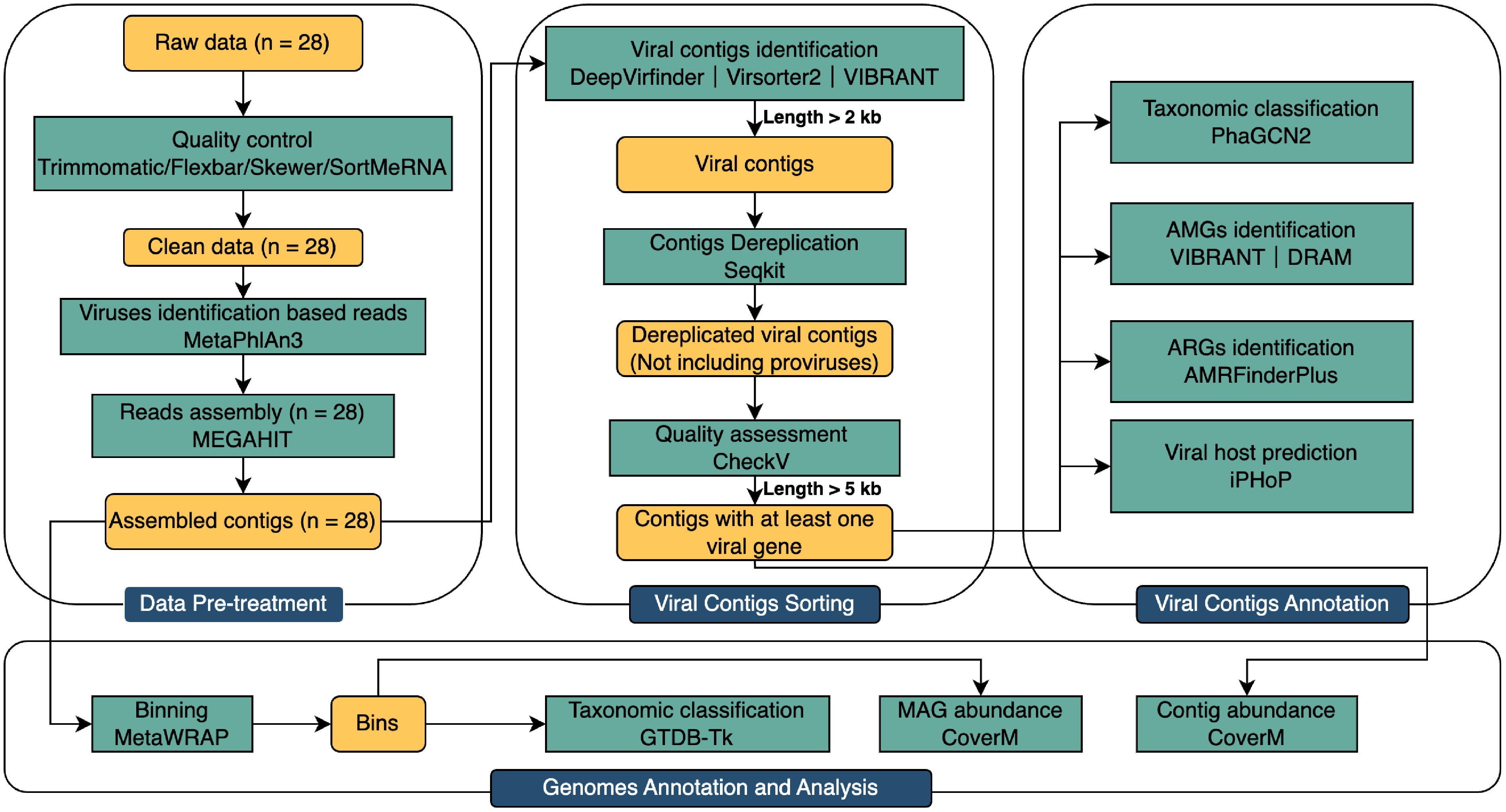
Figure 1.
Analytical diagram of the metagenomic virome data from raw data to binning.
Analysis of α-diversity and correlation test
-
Shannon's diversity index of viruses was calculated using the vegan R package. Bray-Curtis similarity and MDS were calculated using the R package, and the results were plotted using ggplot2 and pheatmap. Wilcox and PERMANOVA tests were used to evaluate the inter-sample significance using the ggsignif and ggplot2 R packages.
Relative abundance heatmap and linear regression analyses
-
Heatmaps of viruses and AMGs were generated using the ComplexHeatmap R package. The regression analyses were performed in R using the Spearman method.
-
Across 28 metagenomes, 29,708, 14,138, and 13,961 viral contigs were recovered with lengths of at least 5 Kb from the AAO, AAO-MBR, and MBR WWTPs, respectively, following data preprocessing. The number of viral contigs in each sample is presented in Supplementary Table S1. Contigs exceeding 5 Kb in length were selected for further analysis since the quality assessment classified them as high quality and completeness (Supplementary Fig. S2a−S2c). A total of 99 family-level viruses were identified throughout the full-scale process of three WWTPs (Fig. 2a). A relatively large number of family-level viruses were detected in 88% of all samples, and seven viruses were persistent across all samples with 100% prevalence (families like Kyanoviridae, Casjensviridae, Herelleviridae, Peduoviridae, Triavirus, and subfamilies like Weiservirinae, Queuovirinae) (Fig. 2c). Viral abundance and diversity in various treatment units of AAO WWTP samples, were higher than in the other WWTP types (AAO-MBR and MBR) (Fig. 2a, b). Significant variability in treatment process conditions may account for differences in viral community composition (such as AAO, AAO-MBR, and MBR). Wastewater samples contained a higher diversity of viruses than sludge samples (Fig. 2b), consistent with our prior findings on pathogens[3]. Despite this difference, the viral Shannon diversity was comparable across the three WWTPs (Fig. 2d, p > 0.05). This suggests that the environmental selective pressures in different wastewater treatment plants drive similar viral diversity. The similarity may be attributed to the universal constraints imposed by conventional consistent treatment objectives, including organic matter removal (e.g., nitrogen and phosphorus) and the reduction of narrow indicator pathogens, such as Escherichia coli, which collectively drive the viral community toward a functionally stable coexistence state[38,39]. The viral diversity found in effluent samples from the three WWTPs increased slightly compared to their influent level. This further implies a convergent effect of the viral community under the environmental stress of different complex treatment processes. Wastewater treatment systems can effectively reduce microbial abundance but slightly increase diversity[3]. From the virus's community composition view, the top 20 most abundant viruses, with average relative abundance across the full-scale treatment process, were significantly different (Fig. 3a, p = 0.001). Gracegardnervirinae, Straboviridae, and Peduoviridae were identified as the most abundant viruses in AAO, AAO-MBR, and MBR treatment plants, respectively (Fig. 3b–d). The Gracegardnervirinae family predominantly includes Mycobacterium phages, while Straboviridae and Peduoviridae harbor various phages targeting pathogenic bacteria such as Aeromonas, Pseudomonas, Klebsiella, and Escherichia. Their high abundance suggests a potential role in the functional removal of these pathogens during wastewater treatment. Moreover, the strong correlation between viral and microbial diversity suggests that these viruses may have a broad host range, encompassing diverse pathogenic bacteria[6]. The heatmap of viral abundance provided a detailed visualization of relative abundance changes at the family level across samples. It highlighted that certain viruses exhibited significantly different abundances across full-scale treatment units, indicating that the wastewater treatment process exerts selective pressure on the viral community. This result elucidates the diversity and distribution characteristics of viruses in WWTPs, laying a foundation for further exploration of the ecological functions of viruses in wastewater treatment systems.
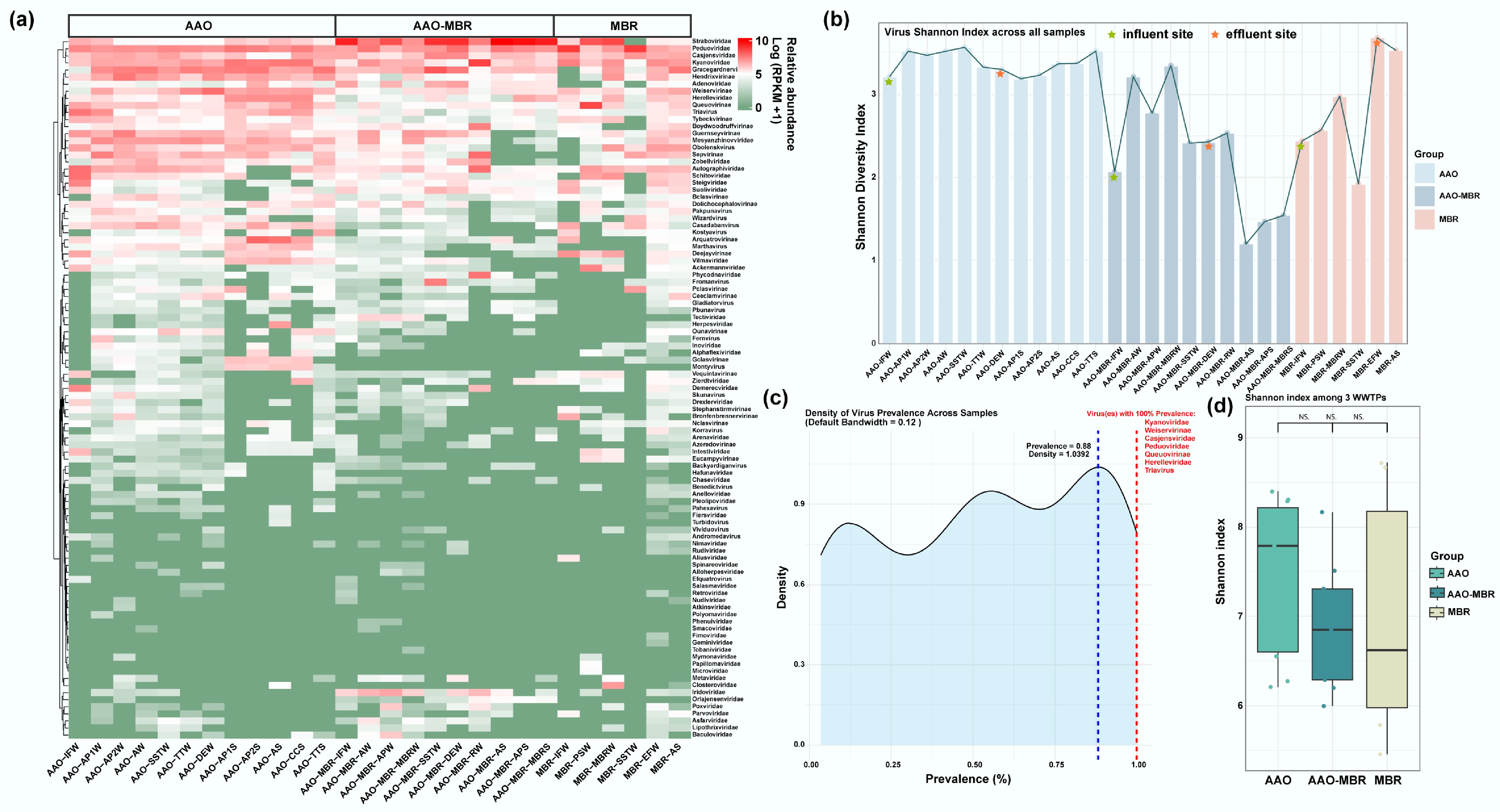
Figure 2.
Overview of virus distribution. (a) Relative abundance (log10 [RPKM + 1]) of identified viruses based on contigs along three full-scale WWTPs; (b) Shannon index of viruses across all samples. (c) Density curve of virus prevalence across samples (default bandwidth = 0.12), viruses denoted in red are persistent across full-scale with 100% prevalence. (d) Shannon diversity index among three WWTPs.
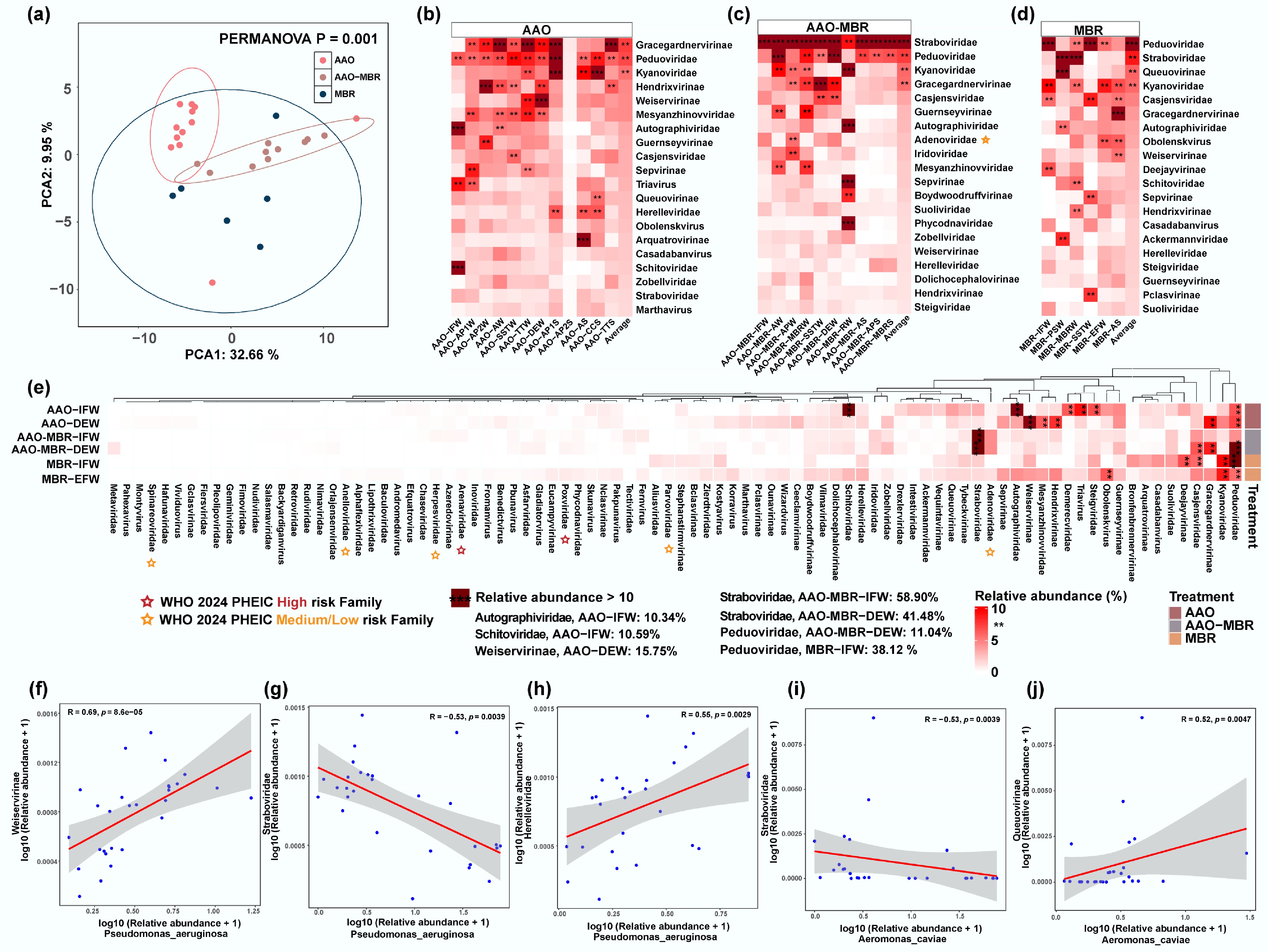
Figure 3.
(a) PCA analysis of viruses among three WWTPs. Composition and abundance of the top 20 most abundant (average abundance) viruses along the three full-scale WWTPs. (b) AAO WWTP. (c) AAO-MBR WWTP. (d) MBR. (e) A total of 88 viruses were detected in the influent and effluent of three WWTPs, and the abundance heatmap. Viruses with relative abundance > 10% were highlighted with ***, and ** refers to relative abundance between 5% and 10%. The risk family on the WHO 2024 PHEIC list was marked in red (high risk), and orange (medium/low risk). (f)–(j) Linear regression analyses between representative pathogen and virus with highest prevalence and abundance—(f)–(h) P. aeruginosa; (i), (j) A. caviae.
Potential functional and indicative viruses persist in high prevalence and abundance throughout the full-scale treatment process
-
Although viral diversity and abundance vary across WWTP units, the most significant differences are observed between the influent and effluent. Intriguingly, Peduoviridae and Casjensviridae were consistently observed among the top 20 viruses in influent and effluent samples from all three WWTPs (Fig. 3e, Table 1), indicating that they exhibit remarkable environmental adaptability and cross-plant activity stability. This phenomenon is likely closely related to their ability to tolerate the complex physicochemical conditions encountered during wastewater treatment. Peduoviridae is one of the dominant functional populations in aerobic biological reaction processes[40], associated with nitrogen and carbon removal functions in the treatment process[41]. Casjensviridae harbors the ability to inactivate carbapenem-resistant Klebsiella pneumoniae[42]. This suggests they could serve as potential bioindicators or functional phages for assessing or improving wastewater treatment efficiency. In addition, five out of the 20 most abundant viruses (Steigviridae, Straboviridae, Tybeckvirinae, Kyanoviridae, and Suoliviridae) were consistently detected in all influent samples, and seven (Weiservirinae, Gracegardnervirinae, Hendrixvirinae, Mesyanzhinovviridae, Guernseyvirinae, Autographiviridae, and Dolichocephalovirinae) were present in all effluent samples. Steigviridae, with high prevalence in all influent samples, is a member of the crAssphage group (crAss-like phages), which are highly abundant and predominantly found in the human gut microbiome. Due to their host specificity and prevalence, Steigviridae phages have been proposed as promising indicators of human fecal contamination in wastewater environments[43]. The high prevalence of Mesyanzhinovviridae in the effluent, which typically infects Pseudomonas aeruginosa[44], aligns with previous work reporting a high abundance of P. aeruginosa in the effluent sections[3].
Table 1. The top 20 relatively abundant viruses of influent and effluent in three WWTPs
Ranking AAO-IFW AAO-DEW AAO-MBR-IFW AAO-MBR-DEW MBR-IFW MBR-EFW 1 Schitoviridae *Weiservirinae *Straboviridae Straboviridae **Peduoviridae Kyanoviridae 2 Autographiviridae *Gracegardnervirinae Adenoviridae **Peduoviridae *Kyanoviridae Obolenskvirus 3 Triavirus *Hendrixvirinae **Peduoviridae *Gracegardnervirinae Deejayvirinae **Peduoviridae 4 *Steigviridae **Peduoviridae **Casjensviridae **Casjensviridae **Casjensviridae *Hendrixvirinae 5 **Peduoviridae Mesyanzhinovviridae Gracegardnervirinae Adenoviridae Casadabanvirus **Casjensviridae 6 Demerecviridae Kyanoviridae *Suoliviridae *Guernseyvirinae Bronfenbrennervirinae *Autographiviridae 7 Guernseyvirinae *Guernseyvirinae Iridoviridae Zobellviridae *Suoliviridae *Gracegardnervirinae 8 Obolenskvirus Sepvirinae Hendrixvirinae Suoliviridae Arquatrovirinae *Weiservirinae 9 Queuovirinae *Autographiviridae Autographiviridae *Weiservirinae *Steigviridae Sepvirinae 10 Deejayvirinae Ceeclamvirinae *Steigviridae Iridoviridae Weiservirinae *Mesyanzhinovviridae 11 *Straboviridae Zobellviridae Guernseyvirinae Steigviridae *Tybeckvirinae Steigviridae 12 *Tybeckvirinae **Casjensviridae *Kyanoviridae Schitoviridae Kostyavirus Herelleviridae 13 Intestiviridae Wizardvirus Zobellviridae Queuovirinae *Straboviridae Schitoviridae 14 Ackermannviridae Triavirus Obolenskvirus *Dolichocephalovirinae Herelleviridae *Guernseyvirinae 15 **Casjensviridae Ounavirinae Mesyanzhinovviridae Metaviridae Aliusviridae *Dolichocephalovirinae 16 *Kyanoviridae Herelleviridae *Tybeckvirinae *Mesyanzhinovviridae Queuovirinae Boydwoodruffvirinae 17 Vequintavirinae Queuovirinae Weiservirinae Vilmaviridae Stephanstirmvirinae Korravirus 18 *Suoliviridae *Dolichocephalovirinae Schitoviridae Boydwoodruffvirinae Parvoviridae Suoliviridae 19 Drexlerviridae Marthavirus Fernvirus *Hendrixvirinae Triavirus Tybeckvirinae 20 Arquatrovirinae Pclasvirinae Tectiviridae *Autographiviridae Boydwoodruffvirinae Vilmaviridae ** Peduoviridae, ** Casjensviridae are two of the top 20 viruses that persistently exist in six treatment units with the highest prevalence, marked in pink. Species marked in green and * are the viruses that persistently exist in influent sections, and species marked in grey and * are persistent in effluent. Significant selective pressures were observed on the composition of viral families across different wastewater treatment processes[38]. Specifically, in the effluent samples from the AAO-MBR WWTP, Straboviridae and Peduoviridae exhibited high relative abundances of 58.90% and 11.04%, respectively, indicating a high degree of enrichment. Peduoviridae showed a dominant presence with a relative abundance of 38.12% in the effluent sample of MBR WWTP (Fig. 3e). Straboviridae, an important component of the gut virome[45], may signal the continuous input of fecal pollutants, similar to Steigviridae, in terms of its high abundance. Moreover, Poxviridae, Arenaviridae, Adenoviridae, Parvoviridae, Herpesviridae, Anelloviridae, and Spinareoviridae were identified in the influent and effluent samples and were included in the 2024 PHEICs (Public Health Emergencies of International Concern) Risk Family List released by WHO (World Health Organization)[46] (Fig. 3e). This highlights the necessity for establishing a comprehensive monitoring system for high-risk pathogens throughout the full-scale treatment process. Furthermore, as a conventional indicator of wastewater treatment, E. coli did not exhibit abundance dynamics that mirrored those of the highly abundant viruses observed across the treatment process (p > 0.05) (Supplementary Fig. S3a–S3j). In contrast, P. aeruginosa showed significant correlations with the abundance patterns of Weiservirinae (*** p < 0.001), Straboviridae (** p < 0.01), and Herelleviridae (** p < 0.01) throughout the full-scale treatment (Fig. 3f–h). Similarly, Aeromonas caviae effectively indicated the abundance variations of Straboviridae (** p < 0.01) and Queuovirinae (** p < 0.01) across the same continuum (Fig. 3i, j). Notably, both P. aeruginosa and A. caviae were also moderately associated with the dynamics of Kyanoviridae, Hendrixvirinae, and Triavirus (Supplementary Fig. S3l–S3o, * p < 0.05).
These findings collectively underscore the limitations of relying solely on traditional bacterial indicators and highlight the urgent need to expand the repertoire of microbial indicators for biological monitoring of wastewater reclamation processes. Furthermore, from the perspective of wastewater treatment process optimization, future efforts should shift from macro-level regulation to micro-level manipulation, with feedback mechanisms implemented through the modulation of individual treatment units to guide system-wide adjustments. Therefore, this study reveals the richness and complexity of viral communities throughout wastewater treatment, offering critical scientific insights to support the refinement of treatment technologies and the effective mitigation of viral contamination risks.
Virus-encoded AMGs in WWTPs as a double-edged sword: enhancing contaminants degradation but promoting antimicrobial resistance
-
While the core community structure and dynamic patterns within wastewater treatment systems have been characterized, the functional roles of viruses, particularly at the level of metabolic genes, remain to be elucidated. A total of 117 AMGs were identified, spanning 12 distinct metabolic pathways, with their prevalence showing notable variation across sampling sites (Supplementary Fig. S4a, S4b). In both the AAO and AAO-MBR WWTPs, viral AMGs were significantly more abundant in wastewater samples than in sludge samples. Previous research focused mainly on activated sludge[19], and this study has enriched our understanding of the full-scale process. Conversely, in the MBR WWTP, elevated AMG abundances were primarily detected in the secondary sedimentation tank wastewater (MBR-SSTW), and the returned activated sludge (MBR-AS). Disinfected effluent wastewater (DEW) from the AAO-MBR WWTP showed a statistically significant decline in AMG detection values compared with upstream units. In contrast, no such reduction was observed in AAO's effluent wastewater (DEW). Amino acid metabolism, carbohydrate metabolism, and metabolism of cofactors and vitamins related to AMGs were primarily distributed across three WWTPs (Fig. 4a), and were detected with the highest gene counts (Fig. 4f) across all the samples. Consistent trends in AMG distribution were observed across different WWTPs (Fig. 4b, c): (1) wastewater samples were found to exhibit significantly higher AMG diversity and abundance than sludge samples; (2) biological treatment stages were characterized by greater metabolic pathway complexity; and (3) AMGs in biological treatment stages harbor a higher similarity than others. Furthermore, regarding the distribution of AMGs within bioreactor sections, no significant differences were observed among the three WWTPs (Fig. 4d, p > 0.05). However, a pronounced difference was observed between wastewater and sludge (Fig. 4e, *** p < 0.01), with the number of AMGs identified in wastewater samples substantially higher than in sludge samples. This suggests that the distribution patterns of viral AMGs may be synergistically influenced by wastewater treatment processes, while being predominantly enriched and functionally active in the wastewater phase of the bioreactor sections. These findings further demonstrate the critical role of the biological treatment units within the overall wastewater treatment system.
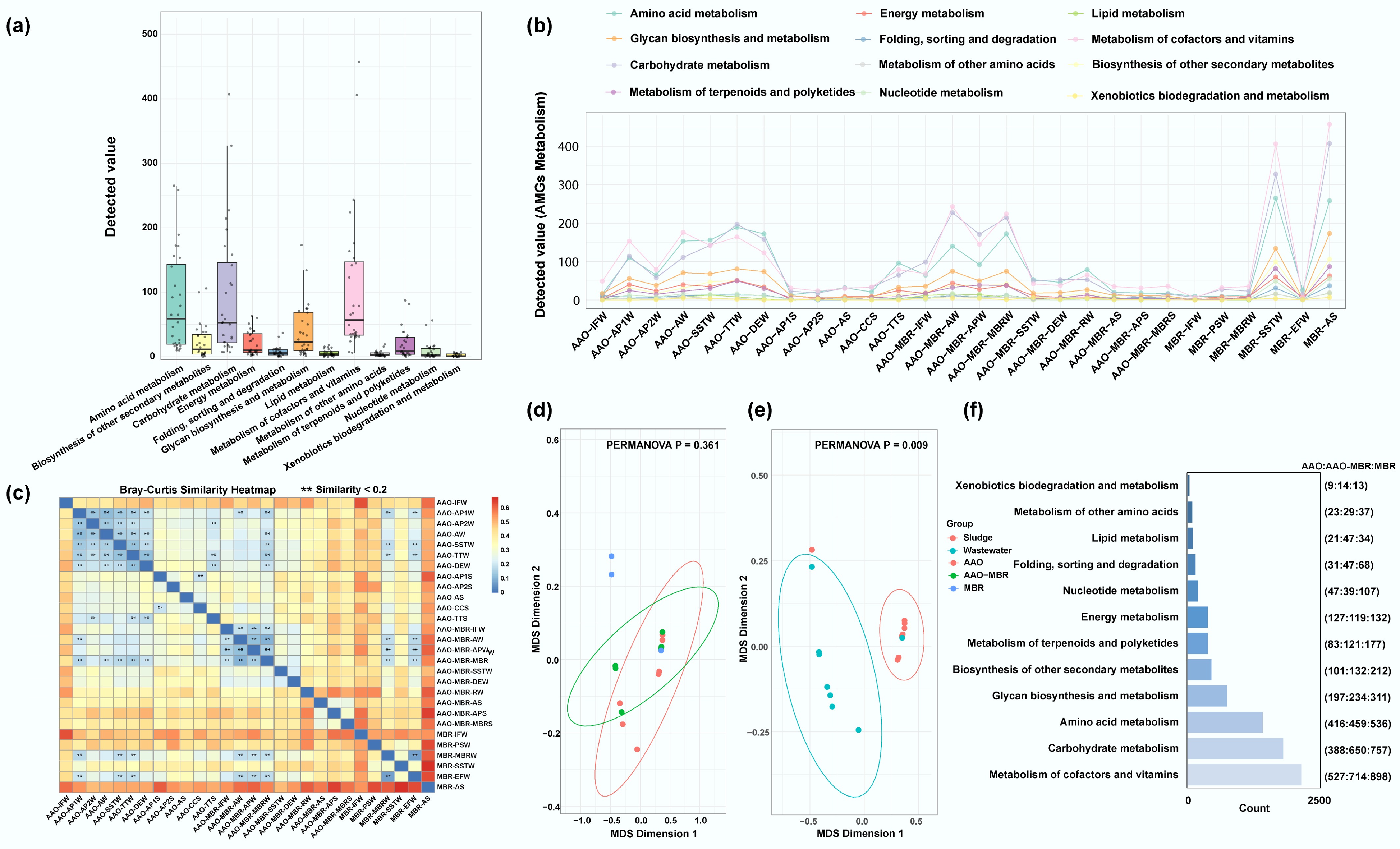
Figure 4.
Occurrence of AMGs and their full-scale regulation in three WWTPs. (a), (b) Detected 12 metabolism AMGs value across all samples and their distribution along the full-scale process, respectively. (c) Bray-Curtis similarity heatmap of AMGs across all the samples, ** refers to that similarity < 0.2. (d), (e) MDS analyses of AMGs in bioreactor sections among three WWTPs (ns. p > 0.05), and between wastewater and sludge (** p < 0.01), respectively. (f) The left bar plot represents the number of detected metabolism-related genes, while the numbers in parentheses indicate the number of corresponding AMGs identified in each WWTP.
High expression of amino acid metabolism-related AMGs is beneficial for methanogenesis in wastewater[47]. Carbohydrate metabolism-related accessory AMGs can enhance the competitive advantage of their hosts in conditions of carbon starvation by carrying genes associated with carbon metabolism. Additionally, these elements may facilitate the dissemination of ARGs[48]. The metabolism of cofactors and vitamins can promote the growth of microalgae in WWTPs[49,50]. Mutations in metabolic genes play a crucial role in facilitating bacterial hosts to evade the toxic effects of diverse antibacterial agents, contributing to the emergence and propagation of bacterial resistance. However, the promotion of host metabolic activity may serve as a potent mechanism for enhancing the capacity to degrade antibiotics[51]. In addition, viruses may enhance their survival by encoding AMGs for xenobiotic biodegradation and metabolism, allowing them to acquire energy through the degradation of exogenous compounds[52], including benzoate, xylene, and chlorocyclohexane/chlorobenzene degradation pathways. These metabolisms demonstrated remarkable activation in viral contigs, potentially serving as core targets for microbial enhancement of pollutant degradation. The identified AMGs primarily participate in carbohydrate processing, amino sugar and nucleotide sugar metabolism, and sulfur metabolism-related pathways (Supplementary Fig. S4c, S4d)—key processes for optimizing the removal efficiencies of conventional pollutants (carbon, nitrogen, and sulfur compounds) through host metabolic augmentation[19]. Additionally, it was reported that specific phages can promote microplastic degradation via AMGs[53]. They will encode more AMGs to overcome the microplastic stress, enhancing the metabolism and adaptability of the host and carbon storage[54,55]. This discrepancy may reflect differences in treatment strategies and the selective pressures they impose on viral-host dynamics. The observed distribution patterns are likely driven by synergistic interactions among site-specific operational parameters (e.g., redox conditions, hydraulic retention time), microbial community composition, and the ecological roles of viral AMGs in enhancing pollutants transformation. These results suggest that viruses (phages) in wastewater not only regulate microbial populations via direct infection. However, they may also confer ecological advantages to their hosts by encoding AMGs that support microbial adaptation to complex environments. Through these mechanisms, viral AMGs may contribute to the degradation of conventional pollutants, recalcitrant organic compounds, and emerging contaminants.
The most prevalent pathogen phylum serves as the most widespread viral host in WWTPs
-
Using machine learning methods to predict potential hosts for viral contigs in each WWTP, where 30, 23, and 32 potential hosts were predicted in AAO, AAO-MBR, and MBR WWTPs, respectively (Table 2). The most frequently identified host phylum was Pseudomonadota, which represents one of the predominant phyla encompassing pathogenic bacteria, many of which harbor pathogenicity and ARGs. Several pathogens in this phylum, such as A. caviae, P. aeruginosa, and Pseudomonas putida, were frequently observed to persist throughout the full-scale treatment processes in our previous studies[3,56]. The number of potential viral host types identified in the MBR WWTP was markedly higher than that in the AAO and AAO-MBR systems (Supplementary Fig. S5). Moreover, wastewater samples consistently exhibited significantly greater predicted host diversity than sludge samples across the entire treatment process. A total of 541 MAGs were recovered from three WWTPs, including 185 from AAO, 282 from AAO-MBR, and 74 from MBR WWTP, respectively, spanning 29, 23, and 13 phyla (Supplementary Fig. S6a–S6c). Among these, 17, 14, and nine species based on MAGs corresponded to the predicted host taxa based on viral contigs, respectively. This correspondence suggests that these taxa represent actual viral hosts present within the WWTP environments (Table 2, species marked in red and * denote those for which corresponding MAGs were recovered). MBR WWTP possessed a significantly richer and more taxonomically diverse array of potential host populations (n = 32); however, the actual hosts represented by recovered MAGs were relatively limited (n = 9, predicted/potential actual = 9/32). Given the substantial proportion of unclassified viral sequences previously identified in the samples, it is plausible that unknown viral hosts may serve as reservoirs that facilitate infection by diverse potential novel viruses[57]. In contrast, the predicted host populations from AAO-MBR WWTP closely matched the actual host categories reflected by MAGs (predicted/potential actual = 14/17), suggesting a stable viral host within a limited number of microbial phyla.
Table 2. The quantity of predicted host contigs across all sampling sites of three WWTPs
AAO WWTP Contigs predicted AAO-MBR WWTP Contigs predicted MBR WWTP Contigs predicted *Acidobacteriota 18 *Acidobacteriota 14 *Acidobacteriota 9 *Actinomycetota 560 Actinomycetota 254 Actinomycetota 159 Aenigmatarchaeota 4 − − − − − − Armatimonadota 2 Armatimonadota 1 Asgardarchaeota 3 Asgardarchaeota 1 Asgardarchaeota 7 Atribacterota 4 Atribacterota 0 Atribacterota 0 *Bacillota 137 Bacillota 54 Bacillota 118 *Bacteroidota 356 *Bacteroidota 503 *Bacteroidota 297 − − − − Bipolaricaulota 1 Caldisericota 1 − − Caldisericota 3 *Campylobacterota 10 *Campylobacterota 2 *Campylobacterota 16 *Chloroflexota 88 *Chloroflexota 20 *Chloroflexota 24 *Cyanobacteriota 34 Cyanobacteriota 41 Cyanobacteriota 27 − − − − Deinococcota 2 *Dependentiae 3 *Dependentiae 6 Dependentiae 1 − − Desulfobacterota 2 *Desulfobacterota 5 Elusimicrobiota 5 − − − − Fibrobacterota 1 − − − − *Gemmatimonadota 20 Gemmatimonadota 1 Gemmatimonadota 11 − − Goldbacteria 0 Goldbacteria 1 *Halobacteriota 1 Halobacteriota 1 Halobacteriota 4 Hydrogenedentota 4 − − − − Iainarchaeota 1 − − − − Krumholzibacteriota 32 − − Krumholzibacteriota 3 Methanobacteriota 2 Methanobacteriota 16 Methanobacteriota 8 − − Methylomirabilota 1 − − − − − − Muirbacteria 2 *Myxococcota 93 *Myxococcota 56 Myxococcota 26 Nanoarchaeota 6 − − Nanoarchaeota 2 *Nitrospirota 26 Nitrospirota 22 Nitrospirota 12 Omnitrophota 15 − − Omnitrophota 4 *Patescibacteria 47 *Patescibacteria 35 *Patescibacteria 33 *Planctomycetota 67 Planctomycetota 23 Planctomycetota 26 *Pseudomonadota 1,675 Pseudomonadota 855 Pseudomonadota 637 Spirochaetota 4 − − *Spirochaetota 2 Sumerlaeota 1 − − Sumerlaeota 2 − − Thermoplasmatota 1 Thermoplasmatota 10 − − Thermoproteota 1 Thermoproteota 4 *Verrucomicrobiota 28 *Verrucomicrobiota 29 Verrucomicrobiota 17 Taxonomies identified from recovered MAGs are highlighted in red. Utilizing the pathogen-virus co-occurrence network to further investigate the potential virus-host relationship (Supplementary Text S2, Supplementary Fig. S7, and Supplementary Table S2). Differences observed in pathogen-virus coexistence networks not only reflect the distinct microbial community control strategies employed by WWTPs but also provide profound implications for environmental health. As hotspots for environmental pathogens, microbial evolution during wastewater treatment and its subsequent environmental impact, especially regarding the survival and dissemination of multidrug-resistant pathogens[58], could pose significant threats to adjacent water bodies and ecosystem health[59]. However, highly connected viral nodes may serve as potential tools for regulating pathogens during wastewater treatment[16], and their ability to infect pathogens could be enhanced through artificial interventions[60]. Additionally, several ARGs were identified across viral contigs (Supplementary Text S3), indicating that horizontal transfer of ARGs mediated by bacteriophage vectors may exacerbate the expansion of environmental resistomes[61], posing potential public health risks upon their discharge into the aquatic environment.
The observed phenomenon suggests that the treatment processes in the AAO-MBR wastewater treatment plant may strongly suppress specific microbial populations. As the most advanced treatment system included in this study, the AAO-MBR process exerts considerable selective pressure, potentially leading to convergence in host community composition. Given the complex coexistence patterns among viral hosts and pathogens, it is advisable to regularly monitor viral community composition and dynamics using molecular tools, such as viral metagenomics. A significant increase in viral markers associated with specific host populations, such as multidrug-resistant bacteria that can serve as an early warning signal, prompting operational adjustments (e.g., to sludge retention time [SRT] or dissolved oxygen levels) to disrupt potential pathogen enrichment cycles. For persistent host–virus complexes, further optimization of treatment technologies is necessary. Beyond routine physical and chemical cleaning, targeted disinfection procedures should be explored. In this context, mitigation strategies addressing virus–host interactions should be incorporated into the optimization of water reclamation processes. Strengthened monitoring and management of emerging pathogens, particularly multidrug-resistant strains, is essential to reducing their risks to environmental and public health.
-
This study systematically elucidates the dynamic characteristics of virus communities in full-scale wastewater treatment systems and their mechanistic interactions with process-mediated regulations. Selective pressures across treatment stages significantly reshaped virus community structures, although specific virus taxa (e.g., Peduoviridae and Casjensviridae, etc.) demonstrated remarkable cross-environmental adaptability and robust bio-indicator potential for treatment performance. Furthermore, P. aeruginosa and A. caviae showed strong correlations with the fluctuations in the abundance of persistent virus taxa throughout the full-scale treatment process. These species may serve as alternative indicators to the conventional bio-indicator, Escherichia coli. Pseudomonadota, identified as the predominant host phylum encompassing numerous multidrug-resistant pathogens, accounted for the highest proportion of predicted viral hosts, exceeding the number of hosts identified from captured MAGs. This disparity suggests the potential existence of unclassified novel viral hosts and undocumented cross-species genetic transmission mechanisms. Beyond direct infection-mediated horizontal transfer of ARGs, viral communities may facilitate the dissemination of ARGs by serving as mobile genetic elements for cross-environmental propagation, and by enhancing host metabolic capabilities through virus-encoded AMGs. Disinfection processes exhibited plant-specific inhibitory effects on AMGs, with a marked reduction observed in the disinfected effluent from AAO-MBR, suggesting that disinfection technologies may selectively remodel viral functional genomes. These findings necessitate prioritized investigation into the ecological impacts of disinfection modalities on viral communities. While conventional treatment objectives drive viral communities toward functional convergence across plants, the coupling of host diversity and process-specific selection creates opportunities for targeted phage-based interventions and resistance blockade strategies. However, the inherent risk of ARGs' dissemination during pathogen removal warrants critical attention. Given the insufficiency of E. coli as a representative bio-indicator for persistent viral and pathogenic populations, a comprehensive monitoring framework incorporating newly identified bio-indicative microbes should be implemented throughout treatment processes. Moreover, process optimization through microecological regulation and advanced tertiary treatment, for instance, further development of new materials and coupling processes was deemed necessary to achieve the synergistic control of chemical and biological risk factors[62], is essential for achieving synergistic control of chemical and biological risks, thereby enhancing the sustainability of wastewater reuse systems.
-
It accompanies this paper at: https://doi.org/10.48130/biocontam-0025-0015.
-
Not applicable.
-
The authors confirm their contributions to the paper as follows: conceptualization: Zhao Y, Huang F, Fan L, Gao SH; visualization: Zhao Y; writing − original draft: Zhao Y; Wang X; writing − review and editing: Zhao Y, Wang X, Huang F, Gao R, Fan L, Liang B, Wang A, Gao SH; supervision: Fan L, Liang B, Wang A, Gao SH; funding acquisition: Wang A, Gao SH. All authors reviewed the results and approved the final version of the manuscript.
-
All raw sequence data have been submitted to the National Omics Data Encyclopedia (NODE, www.biosino.org/node) database at Bio-Med Big Data Center (BMBDC) affiliated with the Shanghai Institute of Nutrition and Health (SINH), Chinese Academy of Sciences (CAS), with a project ID: OEP00005633. The data collected from public references were downloaded from NCBI (Accession No.: PRJNA438174. A total of six samples were included: SRR6837553, SRR6837554, SRR6837557, SRR6837558, SRR6837571, and SRR6837572).
-
The study was financially supported by the National Natural Science Foundation of China (grants No. 52293443, 52321005, 52091545, and 52230004), the Natural Science Foundation of Guangdong Basic and Applied Basic Research Foundation (Grant No. 2024A1515010085), Shenzhen Science and Technology Program (Grant Nos GXWD20231127195344001 and JCYJ20241202123735045), and Shenzhen Overseas High-level Talents Research Startup Program (Grant No. 20200518750C).
-
The authors declare that there are no conflicts of interest.
-
Full list of author information is available at the end of the article.
- The supplementary files can be downloaded from here.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhao Y, Wang X, Huang F, Gao R, Liang B, et al. 2025. Decoding pathogen-virus-metabolic gene networks in full-scale wastewater treatment: from virus diversity to hosts interaction. Biocontaminant 1: e013 doi: 10.48130/biocontam-0025-0015 

Decoding pathogen-virus-metabolic gene networks in full-scale wastewater treatment: from virus diversity to hosts interaction
- Received: 10 October 2025
- Revised: 05 November 2025
- Accepted: 18 November 2025
- Published online: 04 December 2025
Abstract: Viruses, metabolically interdependent communities that interact with their hosts, are abundant in wastewater treatment plants (WWTPs), posing an emerging threat to global health. However, the complex interactions between pathogens and viruses remain poorly understood. In this study, 99 family-level viruses were identified from 28 samples collected across three WWTPs. Peduoviridae and Casjensviridae exhibited consistently high abundance throughout the full treatment processes. The highly abundant and prevalent viruses across the treatment process showed strong correlations with Pseudomonas aeruginosa and Aeromonas caviae, which effectively represent the removal of viruses after treatment and can serve as potential bioindicators. Virus-encoded auxiliary metabolic genes (AMGs) analysis revealed that specific metabolic functions, particularly those related to carbohydrate metabolism and xenobiotic biodegradation, may act as a double-edged sword by enhancing pollutant degradation while concurrently facilitating the spread of antibiotic resistance genes (ARGs) among pathogens via enhancing competitiveness of their hosts. Additionally, Pseudomonadota, a pathogen-associated phylum encompassing many frequently detected functional pathogens, emerged as the predominant viral host group in these systems, further highlighting the ecological relevance of virus–host interactions in wastewater environments. Collectively, these findings underscore the need to expand the current biological monitoring indicators and optimize treatment processes to mitigate the potential health risks posed by viruses in WWTPs.
-
Key words:
- Wastewater treatment plants /
- Pathogens /
- Viruses /
- Auxiliary metabolic genes