










-
Rice (Oryza sativa L.) is one of the most important food crops globally, providing essential nutrition for over half of the world's population[1]. As global food demand continues to rise due to population growth and changing dietary habits, improving rice yield and quality has become a critical focus of agricultural research and development[2,3]. Hybrid rice, which exploits heterosis or hybrid vigor, has played a significant role in increasing rice productivity over the past few decades[4]. However, the molecular mechanisms underlying the superior performance of hybrid rice, particularly during the crucial grain filling process, remain incompletely understood.
The grain filling process is a critical developmental stage that determines both yield and quality in rice. During this period, complex physiological and biochemical changes occur as the developing grain accumulates starch, proteins, and other nutrients[5]. Starch, a complex polymer of glucose, serves as the primary energy reserve for the germinating embryo and constitutes up to 90% of the dry weight of a mature rice grain[6]. The quantity and structure of this starch—specifically the ratio of amylose to amylopectin and the architecture of the amylopectin molecule—are the fundamental determinants of rice grain quality. Starch synthesis occurs within amyloplasts and involves a well-orchestrated enzymatic cascade. It is governed by a sophisticated network of enzymatic and non-enzymatic regulators[6]. The starch synthesis pathway initiates with ADP-glucose pyrophosphorylase (AGPase), which catalyzes the rate-limiting step of ADP-glucose production. Mutations in AGPase genes cause severe grain-filling defects, resulting in reduced grain size and weight, underscoring its indispensable role in carbon flux toward starch accumulation[7,8]. Downstream, granule-bound starch synthase I (GBSSI), encoded by the Waxy (Wx) locus[9,10], exclusively controls amylose synthesis. Recent studies reveal that GBSSI activity is modulated post-translationally: its oligomerization and functionality depend on redox states regulated by glutathione (GSH) levels. Knockdown of OsGCS (glutathione synthesis enzyme) elevates GBSSI activity, while disruption of its binding partner OsGBP enhances amylose content, grain length, and yield[11,12]. Amylopectin synthesis, essential for starch granule crystallinity, is orchestrated by isoforms of soluble starch synthase (SS). Notably, SSIIa deficiency alters amylopectin chain length distribution, leading to chalky grains with reduced weight[13,14]. In addition to the starch metabolic enzymes themselves, more non-enzymatic regulators, such as transcription factors, play crucial roles in starch synthesis[6]. More than five major types of transcription factors have been confirmed to be involved in regulating rice starch synthesis. Within the OsbZIP family, OsbZIP58 (also known as RISBZ1) binds to the GCN4 motif in the promoters of Wx and AGPS2b[15], as well as to the ACGT motif in the promoters of AGPL3 and SSIIa, thereby activating the transcription of starch synthesis genes[16]. Knockout of OsbZIP58 reduces starch content in rice endosperm by 30%[17,18]. OsbZIP76 interacts with the nuclear factor Y transcription factors OsNF-YB1 and OsNF-YB9, collectively regulating endosperm development and storage substance accumulation[19,20]. Genetic disruption of OsbZIP76 similarly decreases amylose content in rice grains. Among OsNAC family transcription factors, OsNAC025 binds to the OsAGPL2 promoter, activating its expression and enhancing starch biosynthesis[21]. OsNAC127 competes with OsbZIP58 for binding to the OsSSIIIa promoter, promoting expression of this starch synthase[22]. OsNAC20 and OsNAC26 alone could directly transactivate the expression of starch synthase I (SSI) to regulate starch synthesis[23]. Other transcription factors such as Ys (NF-Ys)[24−26], APETALA2/ethylene responsive element binding protein (AP2/EREBP)[27], and MADS-box were also reported to be regulators of rice endosperm development[28−30].
In recent years, the advent of high-throughput omics technologies has revolutionized the ability to study complex biological processes at a systems level. Among these, metabolomics and transcriptomics have emerged as powerful tools for elucidating the molecular mechanisms underlying various physiological processes in plants, including rice[31]. Metabolomics provides a comprehensive profile of small molecule metabolites in biological samples, offering insights into the biochemical status of an organism[32]. Transcriptomics, on the other hand, allows the global analysis of gene expression patterns, revealing the regulatory networks that control various biological processes.
The integration of metabolomics and transcriptomics data has been proven to be a powerful approach for unraveling complex biological phenomena in plants[33]. This integrative approach allows researchers to establish connections between gene expression patterns and metabolite profiles, providing a more comprehensive understanding of the molecular mechanisms underlying various physiological processes[34]. In the context of rice research, several studies have successfully employed this approach to investigate various aspects of its development and stress responses. For instance, integrated metabolomics and transcriptomics were used to study the defensive mechanisms of rice grains against environmental stressors[35]. Their findings revealed complex interactions between metabolic pathways and gene expression networks in response to stress, highlighting the potential of this approach for improving crop resilience. Similarly, transcriptomics and metabolomics were employed to investigate the effects of cold stress on rice microspores, providing valuable insights into the molecular basis of cold tolerance in rice[36].
In the context of grain filling and quality, several studies have demonstrated the utility of omics approaches in elucidating the molecular mechanisms underlying these processes. Ranjitha et al. used metabolomics to investigate biochemical differences among rice cultivars, revealing significant variations in metabolite profiles that contribute to grain quality traits[37]. Similarly, Zhang et al. employed an integrated metabolomics and transcriptomics approach to study the molecular basis of nutritional quality differences between landrace and cultivated rice varieties[38]. Their findings revealed key genes and metabolic pathways associated with the superior nutritional qualities of landrace varieties, providing valuable insights for rice breeding programs.
LY287 is a super hybrid rice, which is also an early-season rice with fine grain quality and high yield[39]. However, the molecular basis of this superiority, particularly high grain quality, remains incompletely understood. The present study aims to address this knowledge gap by employing an integrated metabolomics and transcriptomics approach to investigate the grain filling process and quality differences in hybrid rice. By analyzing metabolite profiles and gene expression patterns at various stages of grain development in LY287 and its parental lines, this study seeks to: (1) elucidate the dynamic changes in metabolite composition and gene expression during the grain filling process in hybrid rice; (2) Identify key metabolic pathways and regulatory networks associated with superior grain filling and quality traits in hybrid rice; and (3) Compare the metabolic and transcriptomic profiles of hybrid rice with its parental lines to reveal potential molecular mechanisms underlying heterosis in grain filling and quality traits.
-
The hybrid rice 'Liangyou 287' (LY287) possesses an indica rice background and was officially approved by the Hubei Provincial Crop Variety Approval Committee in 2005. LY287 and its male parent R287 (denoted as F7M in this study) were germinated and planted in the field in Xiaogan, Hubei Province, China (113.844166° E, 30.919245° N), starting from late March. All rice in the field is managed according to normal cultivation measures. At the initiation of rice flowering, the glume was marked as 'day 0' upon spikelet opening. Subsequently, seeds were gathered at 10, 15, and 20 d after pollination (DAP) and identified as 10, 15, and 20 DAP, respectively. Before being used, all the gathered seeds were kept at −80 °C.
Measurement of seed quality
-
Rice quality characteristics consist of processing quality, appearance, cooking properties, taste, and nutritional composition. This research is centered on agricultural methodologies and employs the Standardization Administration of the People's Republic of China (GB/T 50081- 2019, Cooking rice variety quality NY/T59-2021) to evaluate the processing quality of rice, which encompasses brown rice yield, polished rice yield, and whole polished rice yield. Additionally, the study assesses appearance quality by analyzing grain dimensions like length, width, thickness, and aspect ratio, along with examining cooking and taste quality through the determination of amylose content and gel consistency[40]. All the rice quality data are provided by third-party testing agencies, specifically the Food Quality Supervision, Inspection, and Testing Center of the Ministry of Agriculture in Wuhan.
Microscopic observation of starch granules of endosperm
-
The dry seed was halved, and the grain's cross-section was sputter-coated with gold. Starch granules from the rice endosperm were then examined using scanning electron microscopy (Hitachi SU8100, Tokyo, Japan), following established protocols[18].
RNA isolation and Illumina sequencing
-
Total RNA was extracted from 0.5 g of samples with CTAB and Adlai RN40 kit (Adlai, Beijing, China) according to the standard protocol. The concentration of extracted nucleic acid was detected with Nanodrop 2000 (Thermo, Waltham, MA, USA), and the integrity was detected with Agilent 2100, LabChip GX (Perkin Elmer LabChip GX, Waltham, MA, USA).
The mRNA was purified by mRNA Capture Beads from total RNA samples. The cDNA was synthesized by a cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer's instructions. cDNA library construction was done by Hieff NGS Ultima Dual-mode mRNA Library Prep Kit (Yeasen, Shanghai, China). Then, library products were sequenced with the Illumina NovaSeq 6000 platform (San Diego, CA, USA). Each sample had three biological replicates. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive[41] in the National/Genomics Data Center[42], China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA020201), which are publicly accessible at
https://ngdc.cncb.ac.cn/gsa .Metabolite extraction and detection
-
Ten seeds from each biological replicate were ground into powder using liquid nitrogen. Fifty milligrams of the resulting powder were then mixed with 1,200 μL of 70% pre-cooled methanolic aqueous solution. The mixture was vortexed 30 min and repeated six times. Following centrifugation at 12,000 rpm for 3 min, the supernatant was removed, and the sample was filtered through a 0.22 μm pore size microporous membrane before being stored in an injection vial for UPLC-MS/MS analysis. The metabolite identification was analyzed by Metware Biotechnology Co., Ltd. (Wuhan, China).
All samples were obtained using the LC-MS system according to machine protocols. The analytical parameters were as follows: UPLC system with a Waters ACQUITY UPLC HSS T3 1.8 µm, 2.1 mm × 100 mm column, operating at 40 °C with a flow rate of 0.40 mL/min and an injection volume of 4 μL. The solvent system consisted of water (0.1% formic acid) and acetonitrile (0.1 % formic acid). Sample analysis was conducted using a gradient program starting at 95% A, 5% B. This composition linearly transitioned to 35% A, 65% B in 5 min, followed by a linear gradient to 1% A, 99% B within 1 min, which was maintained for 1.5 min. The composition was then adjusted to 95% A, 5.0% B within 0.1 min and held for 2.4 min. During UPLC system operation, data acquisition was performed in information-dependent acquisition (IDA) mode using Analyst TF 1.7.1 Software (Sciex, Concord, ON, Canada). The source parameters were configured as follows: ion source gas 1 (GAS1) at 50 psi; ion source gas 2 (GAS2) at 60 psi; curtain gas (CUR) at 35 psi; temperature (TEM) at 550 °C; declustering potential (DP) at 80 or –80 V in positive or negative modes, respectively; and ion spray voltage floating (ISVF) at 5,500 or –4,500 V in positive or negative modes, respectively. The TOF MS scan settings were: mass range of 50–1,250 Da; accumulation time of 200 ms; and dynamic background subtract enabled. For the product ion scan, the parameters were set as follows: mass range of 50–1,250 Da; accumulation time of 40 ms; collision energy at 30 or –30 V in positive or negative modes, respectively; collision energy spread at 15; resolution set to UNIT; charge state from 1 to 1; intensity at 100 cps; exclusion of isotopes within 4 Da; mass tolerance of 50 mDa; and a maximum of 12 candidate ions to monitor per cycle.
The original LC-MS data file was converted to mzXML format using ProteoWizard software. Subsequently, peak extraction, alignment, and retention time correction were conducted using the XCMS program. Peak area correction was implemented using the 'SVR' method. Peaks with a detection rate below 50% in each sample group were excluded. Metabolic identification details were then acquired through searches in the laboratory's proprietary database (Metware Biotechnology Co., Ltd.). The raw data have been deposited in MetaboLights with the identifier MTBLS11706 (
www.ebi.ac.uk/metabolights/MTBLS11706 )[43].RNA extraction and gene expression analysis
-
Total RNA was extracted from rice leaf and seed using the FastPure Universal Plant Total RNA Isolation Kit (Vazyme Biotech, Jiangsu, China). HiScript II Q RT SuperMix (Vazyme Biotech, Jiangsu, China) was used for the first-strand cDNA synthesis. The expression of each gene was quantified by RT-qPCR using the ChamQ Blue Universal SYBR qPCR Master Mix (Vazyme Biotech, Jiangsu, China) on the CFX Connect Real-Time PCR Detection System (Bio-Rad, California, USA). The rice Actin gene (Os03g0718100) as used as the internal control. All experiments were performed in three biological replicates. The relevant primers used in RT-qPCR assays are presented in Supplementary Table S1.
Apparent amylose and total starch content measurement
-
Rice seed samples from various developmental stages were ground into powder using liquid nitrogen. Then, 0.1 g of the powder was weighed and added to the starch extraction buffer for the extraction of total starch and amylose. Detailed operational steps can be referred to in the Amylopectin-Amylose-Total Starch Content Assay Kit manual (Beijing Boxbio Science & Technology Co., Ltd, Beijing, China).
Paraffin sectioning and starch granule staining
-
Rice seed samples without husk from various developmental stages were fixed in FAA solution, which consisted of 5% glacial acetic acid, 5% formaldehyde, and 70% ethanol, and stored until further processing. The samples subsequently underwent dehydration and infiltration steps before being embedded in paraffin. After sectioning, Periodic Acid-Schiff (PAS) staining, and microscopic observation were performed following previously described methods[44]. For each sample, at least three biological replicates were fixed and examined.
Data analysis
-
In BMK Cloud (Beijing, China,
www.biocloud.net ), RNA sequencing data processing, gene expression calculation, identification of differentially expressed genes (DEGs), gene ontology (GO) enrichment analysis[45,46], and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment were carried out[47,48]. The significance of gene expression differences between samples was assessed based on a false discovery rate (FDR) < 1% and an absolute log2FC (fold change) value > 1. The raw RNA sequencing data were annotated with the reference genome stored at Rice Information Gateway (http://rice.hzau.edu.cn/rice_rs3 ).By utilizing the secondary spectral information stored in the MWDB (Metware database), the substance was qualitatively identified within Metware Cloud, a free online platform for data analysis (Wuhan, China,
https://cloud.metware.cn ). Significantly regulated metabolites between groups were determined by variable importance in projection (VIP) ≥ 1 and an absolute log2FC (fold change) value > 1. The identified metabolites were annotated utilizing the KEGG Compound database, followed by mapping these annotated metabolites to the KEGG Pathway database[47,48]. Subsequently, pathways containing significantly regulated metabolites were input into the Metabolite Sets Enrichment Analysis (MSEA), with their significance assessed based on the p-values derived from the hypergeometric test. -
LY287 is a two-line early-cropping super hybrid rice with superior grain quality. Its male parent, R287 (denoted as F7M in this study) was bred through pedigree selection by crossing Ezao 17 (serving as the female parent) with Guinong 07P6. The female parent of LY287 is HD9802S, an early-maturing indica thermosensitive genetic male sterile variety cultivated through the hybridization of Huda 51 and Hongfuzao, followed by rigorous low-temperature selection and evaluation in an artificial climate chamber (Supplementary Fig. S1). In the rice-growing areas of Hubei, LY287 is sown in late March or early April. It heads (flowers) between June 18 and 23, and matures between July 18 and 20, with a total growth duration of 110 to 114 d. The female parent exhibits sterility when planted at the same time. Given the thermosensitive male sterility of the female parent line during synchronous cultivation, the experimental design incorporated seeds solely from the paternal line and hybrid combinations.
Seeds were gathered at 10, 15, and 20 d after pollination (DAP) and identified as 10, 15, and 20 DAP respectively (Fig. 1a). During grain filling, the color of the grain transitions from green to light yellow, while the weight of the grain undergoes a significant increase in the male parent F7M and hybrid LY287 (Fig. 1b). Microscopic examination reveals that LY287 exhibits a more compact and larger starch crystal structure in the endosperm (Fig. 1c). Mature rice seeds of male parent F7M and hybrid LY287 were sent to third-party testing agencies, specifically the Food Quality Supervision, Inspection, and Testing Center of the Ministry of Agriculture in Wuhan, China. The detailed rice quality data are shown in Table 1. There are no significant differences in most rice quality parameters between male parent F7M and hybrid LY287, as shown in the table. However, notable distinctions are evident in the head rice percentage, chalkiness degree, amylose content, and gel consistency. The total starch and amylose content in seeds of various developmental stages were detected by staining and kit-based assays in the laboratory. The results indicated that starch had begun to accumulate substantially at 5 DAP, although it had not yet filled the entire endosperm tissue (Fig. 1d). Notably, the starch content was lower in the peripheral regions of the endosperm compared to the central area. By the 15 DAP, starch had largely filled the whole endosperm. These observations are consistent with the measured total starch content, which showed no significant difference between the 15 and 20 DAP (Fig. 1e). For amylose, the content remained relatively low at approximately 1% on the 5 DAP. It increased sharply to around 19% by the 10 DAP and remained at this level until the 20 DAP. Throughout the later stages of development, the amylose content of LY287 was significantly lower than that of F7M (Fig. 1e).
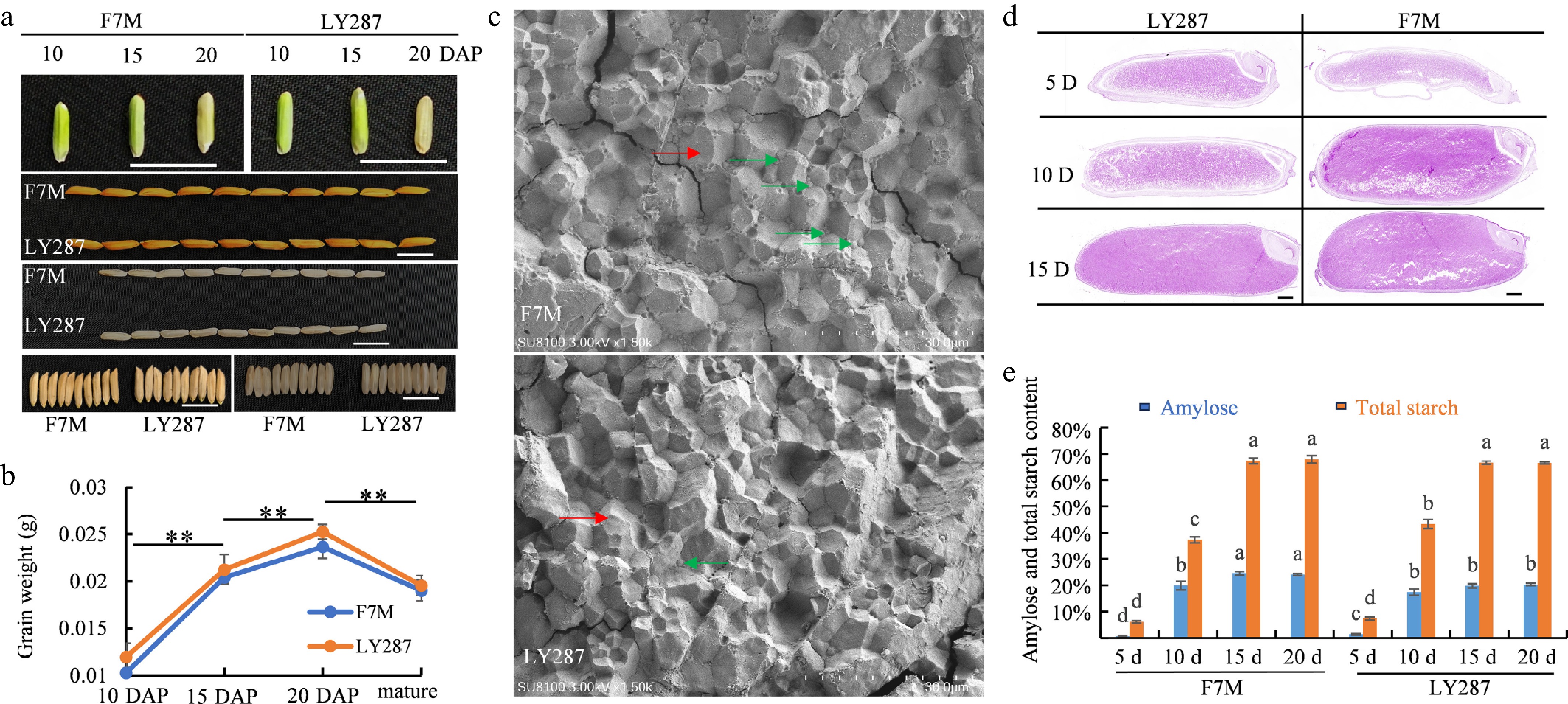
Figure 1.
Characteristics of grain appearance and structure of starch in Liangyou 287 (LY287), and male parent F7M. (a) Grain appearance traits of LY287 and male parent F7M at 10, 15, and 20 d after pollination (DAP), bar = 1 cm. (b) Grain weights of LY287 and male parent F7M at 10, 15, 20 DAP, and the mature stage. Significant differences were determined by Student's t-test (** p < 0.01). (c) Scanning electron microscope, SU8100, was used to visualize the starch structure of grains, bar = 30 μm, as shown above. The red arrow points to the large starch crystal structure, while the green arrow points to the small starch crystal structure. (d) Paraffin sectioning and starch granule Periodic Acid–Schiff (PAS) staining, bar = 500 μm. (e) Apparent amylose and total starch content measurement. Significant differences were determined by ANOVA (p < 0.05).
Table 1. Rice quality of LY287 and its male parent F7M.
Brown rice percentage
(%)Head rice percentage
(%)Grain length (mm) Grain width (mm) Length-width ratio (%) Translucency (%) Chalkiness degree (%) Amylose
(%)Gel consistency (mm) Alkali spreading value LY287 80.4 ± 1.2 65.3 ± 0.7 ** 7.3 ± 0.1 2.1 ± 0.2 3.5 ± 0.5 1 ± 0.2 1.0 ± 0.2 * 19.5 ± 1.3 * 61.0 ± 0.5 ** 5.3 ± 1.5 F7M 80.8 ± 0.8 62.8 ± 1.3 7.4 ± 0.2 2.2 ± 0.1 3.4 ± 0.7 1 ± 0.5 1.6 ± 0.5 20.3 ± 1.6 56.8 ± 0.4 5.6 ± 1.4 Brown rice percentage: Shell rice is huller-milled to produce brown rice. The brown rice percentage is calculated by the complete brown rice number to the shell rice number. Head rice percentage: Polished rice is derived from brown rice through the milling conducted by a rice mill. Head rice percentage is calculated by complete polished rice number to the brown rice number. Length-width ratio: The ratio of grain length to grain width. Translucency: The percentage of transparent or semi-transparent rice grains among all rice grains. Chalkiness degree: The projected area of the white opaque portion within the rice endosperm equates to the total projected area of the rice grains being studied. Amylose: The ratio of amylose weight to total starch weight. Gel consistency: The flow length of rice gel is observed after alkali gelatinization under specified conditions upon cooling. Alkali spreading value: The erosion degree of intact rice grains caused by an alkaline solution. Asterisk means significant differences were determined by Student's t-test (* p < 0.05, ** p < 0.01). A higher head rice percentage in hybrid LY287 leads to a higher output of shell grain from hybrid LY287, reflecting an enhanced quality of rice processing in hybrid LY287. The lower chalkiness degree in hybrid LY287 contributes to the aesthetic quality of rice, thereby increasing its market price. There is a strong correlation between the amylose content and gel consistency of rice and its cooking characteristics and flavor profile. Hybrid rice LY287 is shown to outperform its parental counterparts based on the quality assessment conducted using the aforementioned indicators.
Differential metabolite analysis throughout the developmental stages of rice seeds
-
In 18 samples, a total of 1,935 metabolites were identified, with 1,331 metabolites detected in positive mode and 604 in negative mode in the mass spectrometer (Supplementary Table S2). Excluding the unknown metabolites, amino acids and derivatives, organic acids, benzene, and substituted derivatives, lipids, and flavonoids are the five classes of metabolites with the highest content (Fig. 2a). The examination of the TIC overlaps plot, CV distribution plot, and QC plot for each sample group analyzed via mass spectrometry indicated the high reproducibility and reliability of the data obtained in this study (Supplementary Fig. S2). The analysis of data in positive mode through Principal Component Analysis (PCA) demonstrated variances of 43.65% and 15.1% in PC1 and PC2, respectively. Notably, the separation of the same variety across the three time periods was evident along PC1, while PC2 showed distinct separation of different varieties within the same period. Additionally, tight clustering of the three biological replicates within each group was observed. Remarkably, consistent results were obtained from the analysis conducted in negative mode (Fig. 2b).
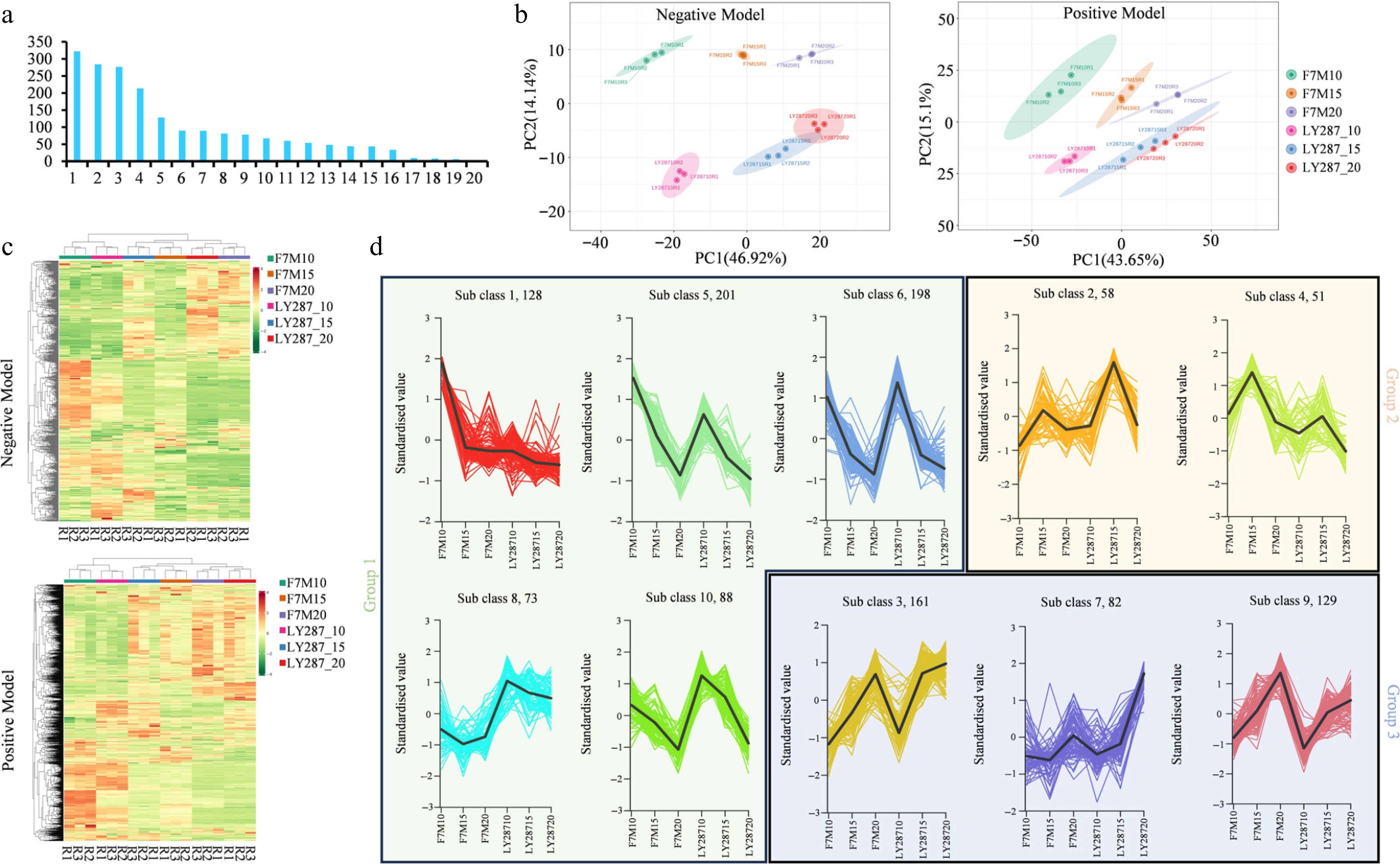
Figure 2.
Metabolites analysis of male parent F7M and hybrid LY287. (a) Categorization of metabolites discovered in rice grain. 1 to 20 means amino acids and derivatives, the unknown metabolites, organic acids, benzene and substituted derivatives, lipids, flavonoids, alcohol and amines, alkaloids, glycoprotein, heterocyclic compounds, phenolic acids, nucleotides and derivatives, terpenoids, fatty acids, glycerolipids, lignin-s and coumarins, saccharolipids, steroids, quinones, tannins. (b) Principal Component Analysis (PCA) of identified metabolites in positive and negative modes. (c) Clustering heat map of identified metabolite content. R1, R2, and R3 represent three biological samples. (d) Clustering analysis of differentially accumulated metabolites (DAMs) using k-means. These DAMs can be categorized into three major groups based on their overall trends.
Across all 18 samples, 1,451 and 648 metabolites were quantified in positive and negative modes, respectively (Fig. 2c). A total of 1,169 differentially accumulated metabolites (DAMs) were identified across all 18 comparison groups using the equations |log2(FC)| > 1 and VIP ≥ 1 (Supplementary Table S3). After conducting k-means clustering, the DAMs were classified into ten subgroups, which could be consolidated into three main groups based on the variations in metabolite abundance (Fig. 2d). The overall abundance of metabolites decreases, indicating that metabolites are plentiful in the early stages of rice grain development before being synthesized into storage substances like starch. Amino acids and derivatives, organic acids, benzene and substituted derivatives are the three classes of metabolites with the highest content in all three groups. However, nucleic acids and derivatives, flavonoids, and lipids rank as the fourth most abundant substances in groups 1, 2, and 3, respectively. It suggests that nucleic acids are primarily biosynthesized in the early stage, and the intensity of nucleic acid metabolism diminishes progressively as seed development advances. Meanwhile, the peak period of flavonoid metabolism and lipids was observed in the middle stage and the later stage, respectively (Supplementary Fig. S3).
In Group 1, a total of 688 DAMs displayed consistent expression patterns between LY287 and its male parent F7M. These 688 DAMs exhibited peak abundance at 10 d after pollination (DAP), followed by a decrease at 15 and 20 DAP (Fig. 2d). There are five subclasses within Group 1, including subclasses 1, 5, 6, 8, and 10. Among these, the abundance of metabolites in subclasses 1 and 5 was higher in paternal rice grains than in hybrid rice grains at different developmental stages. An abundance of metabolites in subclasses 6 and 10 showed no significant differences between paternal and hybrid rice grains across different developmental stages. Abundance of metabolites in subclass 8 was lower in paternal rice grains than in hybrid rice grains at different developmental stages. In Group 2 and Group 3, 109 and 372 DAMs, respectively, demonstrated the highest abundance at 15 and 20 DAP. Among the top four most abundant substance categories-amino acids and derivatives, benzene and substituted derivatives, organic acids, and others-42%, 38%, 38%, and 42% of their constituent metabolites, respectively, decreased during seed development. Conversely, 31%, 31%, 31%, and 37% of metabolites in these categories increased. The remaining metabolites exhibited an initial increase followed by a decrease. This suggests that individual metabolites are produced at distinct developmental stages, even within the same chemical category (Supplementary Fig. S4).
The variations in metabolites between male parent F7M and hybrid LY287 are less pronounced compared to the inherent differences within the variety across various developmental stages
-
Upon detailed comparison, it was observed that the quantity of upregulated metabolites was lower than that of downregulated metabolites in two rice cultivars throughout the developmental stages of rice grain (Fig. 3). In male parent F7M, 529 and 793 DAMs were identified at 15 and 20 DAP compared to 10 DAP (Fig. 3a). Likewise, in LY287, 556, and 745 DAMs were detected at 15 and 20 DAP relative to 10 DAP (Fig. 3b). This suggests that metabolic activity is highest at 10 DAP, and more metabolites degrade gradually or are utilized in the synthesis of other macromolecule substances during the later stages of development (Figs 2d, 3a–c). In comparing the total number of DAMs between male F7M and LY287 at 10, 15, and 20 DAP, a lower count than that found at various developmental stages was also observed in either male F7M or LY287 (Fig. 3d). It indicates that the difference in metabolite changes between the parent and hybrid is less significant compared to that observed during seed development. When comparing the metabolites in the grain at 15 DAP to those at 10 DAP, significant enrichment was observed in pathways related to alkaloid biosynthesis, oxidative phosphorylation, carbon fixation in photosynthetic organisms, beta-alanine metabolism, photosynthesis, and the citrate cycle in the male parent F7M using KEGG enrichment analysis (Fig. 4a). Similarly, metabolites associated with phenylalanine, tyrosine, and tryptophan biosynthesis, alkaloid biosynthesis, ascorbate and aldarate metabolism, valine, leucine, and isoleucine biosynthesis, photosynthesis, and the citrate cycle were found to be mostly enriched in the grain when comparing 15 to 10 DAP in LY287 (Fig. 4b). Metabolites involved in glucosinolate, phenylalanine, tyrosine, and tryptophan biosynthesis, along with isoquinoline alkaloid and various alkaloid biosynthesis pathways, exhibited higher enrichment levels in grain at 20 DAP relative to 10 DAP in F7M (Fig. 4c). In contrast, grain at 20 DAP compared to 10 DAP in LY287 showed enrichment solely in metabolites associated with ascorbate and aldarate metabolism (Fig. 4d). Moreover, enriched pathways such as glucosinolate, phenylalanine, tyrosine, tryptophan biosynthesis, and isoquinoline alkaloid biosynthesis were observed in grain when comparing 20 to 10 DAP in F7M. Conversely, these pathways were identified in grain when comparing 15 to 10 DAP in LY287. This discrepancy highlights a significantly higher metabolic activity of these substances in LY287 seeds compared to F7M seeds.
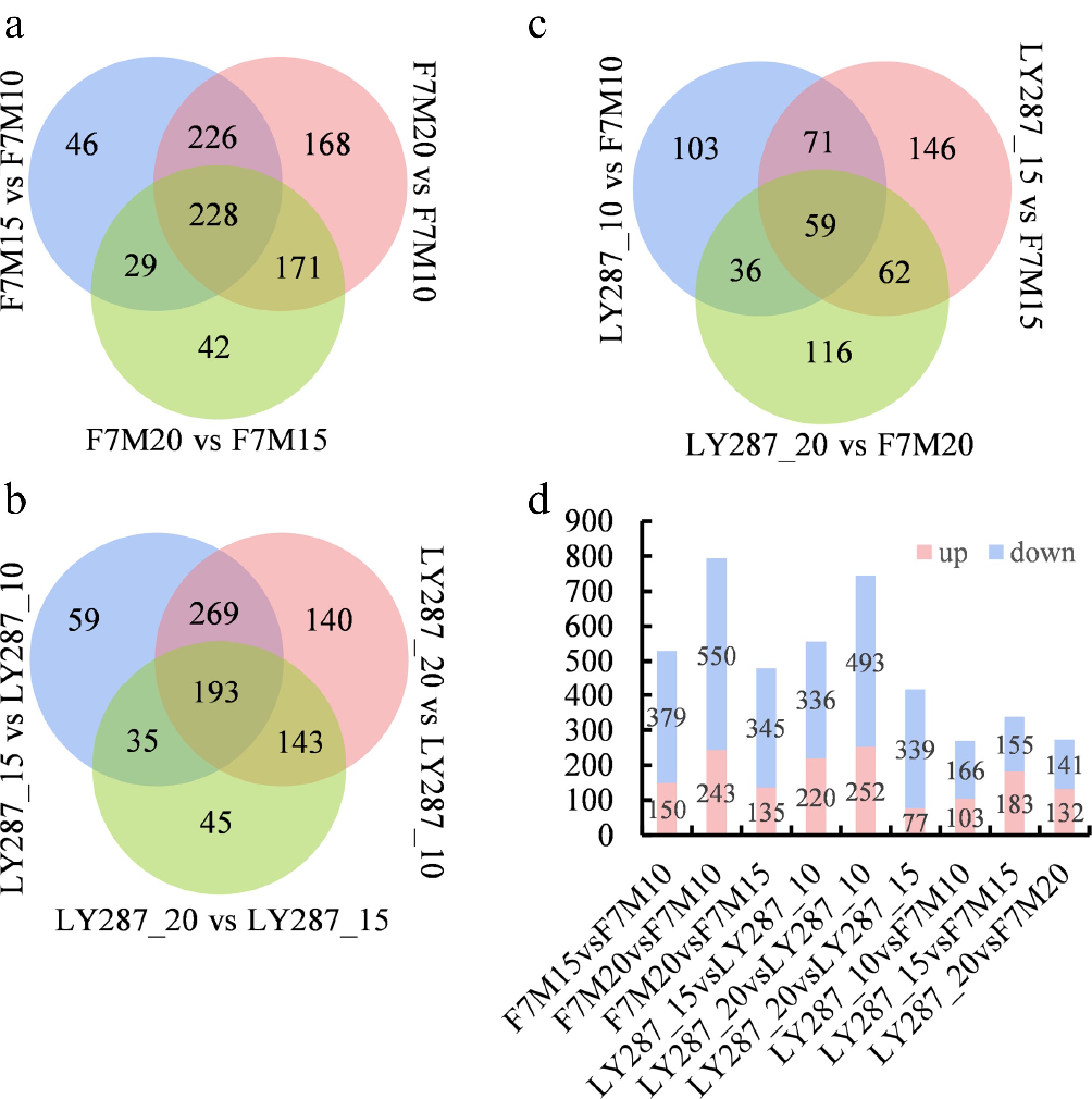
Figure 3.
Statistical analysis of differentially accumulated metabolites (DAMs). (a) Venn analysis of DAMs in F7M at three stages of grain filling. (b) Venn analysis of DAMs in LY287 at three stages of grain filling. (c) Venn analysis of DAMs between F7M and LY287 at three stages of grain filling. (d) Number of DAMs in different comparisons.
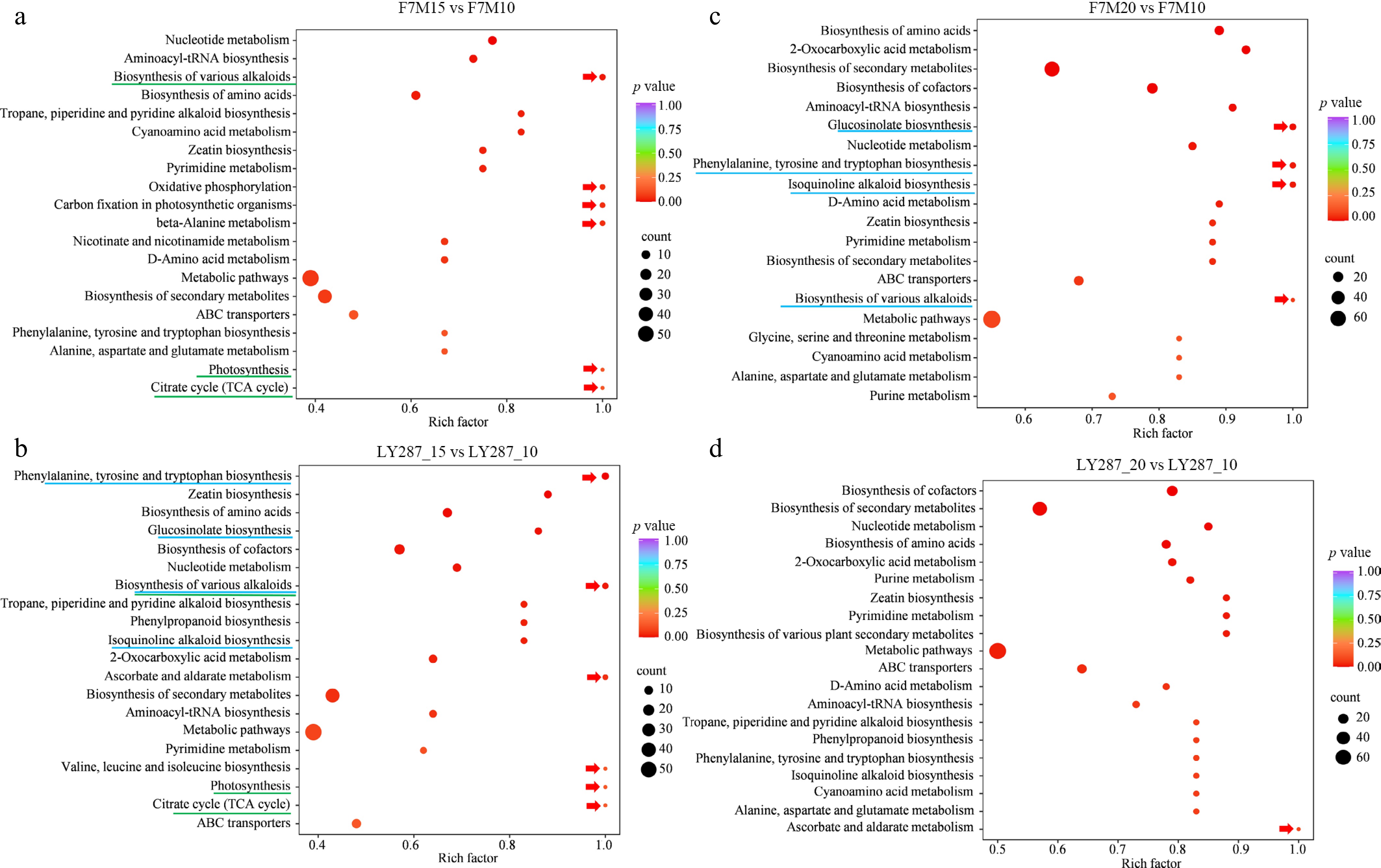
Figure 4.
KEGG enrichment analysis of DAMs. (a) Enriched pathways in F7M were discovered through the use of DAMs obtained from comparing 15 to 10 DAP. (b) Enriched pathways in LY287 were discovered through the use of DAMs obtained from comparing 15 to 10 DAP. (c) Enriched pathways in F7M were discovered through the use of DAMs obtained from comparing 20 to 10 DAP. (d) Enriched pathways in LY287 were discovered through the use of DAMs obtained from comparing 20 to 10 DAP. Red arrows indicate the most enriched pathways in each comparison.
Furthermore, this study identifies the metabolites that were enriched in the pathways discussed earlier through KEGG enrichment analysis for subsequent clustering analysis. Similar to the k-means clustering of DAMs in group 1, the abundance of most metabolites identified from KEGG enrichment analysis has the highest abundance at 10 DAP, and then gradually decreases at 15 and 20 DAP in both LY287 and F7M (Supplementary Fig. S5). At 10 DAP, nucleotides and their derivatives are the predominant metabolites, whereas organic acids dominate at 20 DAP. This shift indicates a transition in the primary metabolism from nucleotide processing to sugar and lipid metabolism. Metabolites that exhibit an increase in content at 20 DAP appear significantly earlier in the LY287 compared to the F7M. Consequently, these substances also sustain elevated levels in the LY287 for an extended duration (Supplementary Fig. S5).
Investigation into the differential expression of genes in the course of rice seed maturation
-
In 18 samples, over 11 thousand genes with FPKM value > 1 was identified in this study, and more genes were identified at 20 DAP than 10 or 15 DAP (Fig. 5a, Supplementary Table S4). PCA and correlation analysis of gene expression levels results indicated that three biological replicates of the same sample exhibit high consistency. It also showed that the grain has the most similarity between F7M and LY287 at 10 DAP, and they have the minimum similarity at 20 DAP (Fig. 5b, c).
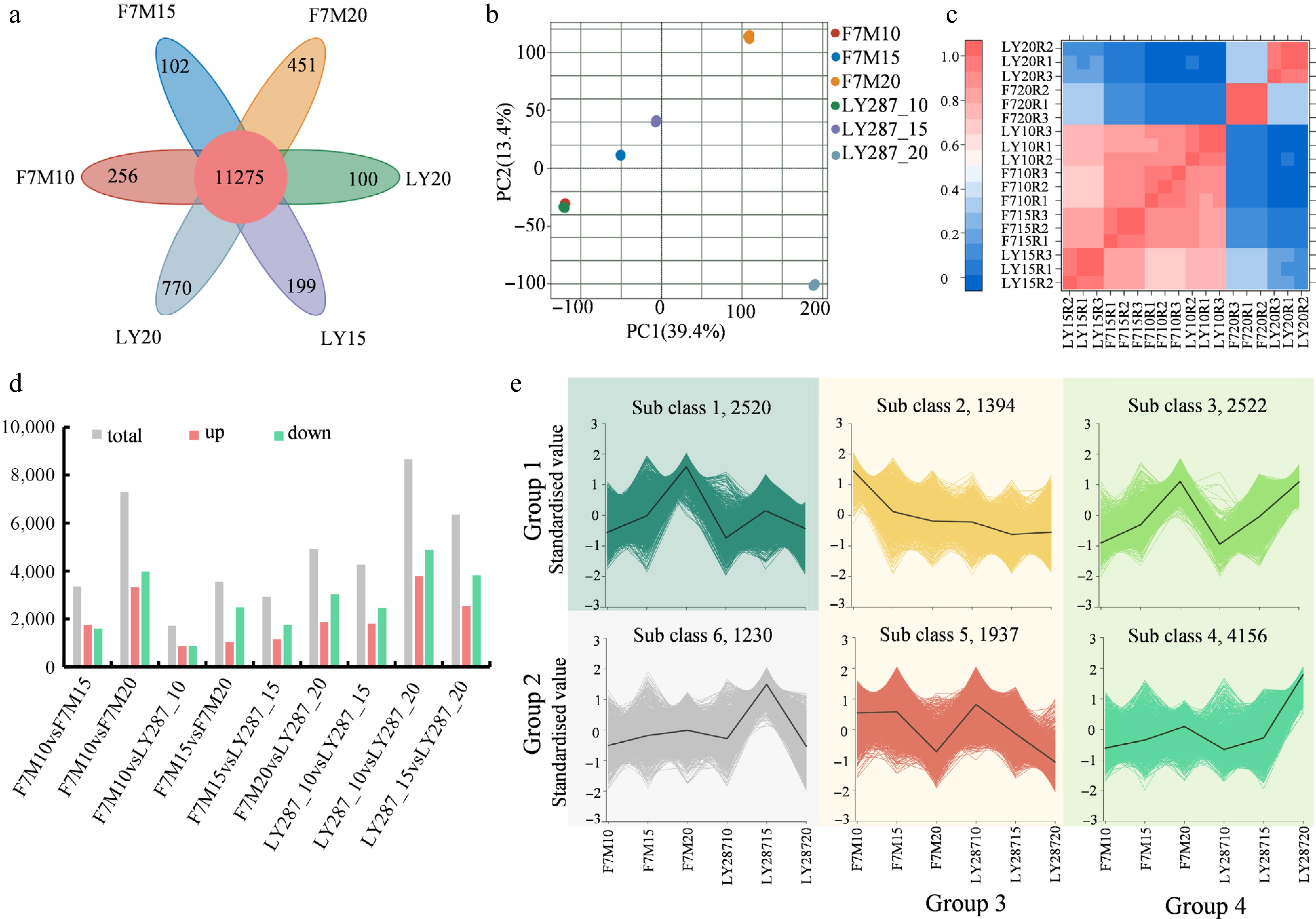
Figure 5.
Statistical analysis of differentially expressed genes (DEGs). (a) Venn analysis of DEGs in F7M and LY287at three stages of grain filling. (b) Principal component analysis (PCA) of identified genes in rice grain. (c) A heatmap illustrating the correlation among samples as determined by the Pearson correlation coefficient. A correlation coefficient approaching one indicates a stronger resemblance in expression patterns between samples. (d) Number of DEGs in different comparisons. (e) Clustering analysis of DEGs using k-means. These DEGs can be classified into four main groups according to their overall patterns.
A total of 3,362, 3,532, and 7,301 differentially expressed genes (DEGs) were identified in comparison groups 10 vs 15 DAP, 15 vs 20 DAP, and 10 vs 20 DAP in male F7M, respectively, using the equations |log2(FC)| > 1 and FDR ≤ 0.01. Similarly, a total of 4,262, 6,355, and 8,664 DEGs were identified in comparison groups 10 vs 15 DAP, 15 vs 20 DAP, and 10 vs 20 DAP in LY287 (Fig. 5d). Generally speaking, these DEGs could be clearly divided into four main groups (including six sub classes) according to the gene expression in F7M and LY287 at 10, 15, and 20 DAP (Fig. 5e). A total of 2,520 DEGs in group 1 exhibited a sustained increasement throughout seed development in F7M, while they were initially up-regulated and then down-regulated in LY287. In group 2, 1,230 DEGs showed a peak expression level in LY287 at 15 DAP, but no significant change in F7M. In group 3, a total of 3,331 DEGs in both F7M and LY287 exhibited a consistent downregulation trend during three time periods of 10, 15, and 20 DAP. In the last group, there were 6,678 DEGs that exhibited a consistent increase throughout seed development in both F7M and LY287 (Fig. 5e).
GO enrichment analysis was conducted to explore the roles of the DEGs. In group 1, these DEGs are primarily associated with the biosynthesis and metabolic processes of small molecules, carbohydrates, organic acids, and lipids (Supplementary Fig. S6a). The expedited accumulation of primary metabolites during seed development in LY287 is suggested by the earlier attainment of maximum expression levels by these genes compared to F7M. Within group 2, the DEGs are predominantly involved in cell wall organization and biogenesis, along with the response to abiotic stimuli, specifically heat response (Supplementary Fig. S6b). At 15 DAP, the expression levels of these DEGs were at their highest in LY287, while showing no significant changes in F7M. The discrepancy points towards a more significant cell wall development process and a swifter temperature response in LY287 when contrasted with F7M. In group 3, the DEGs exhibited a consistent decrease during seed development in both F7M and LY287. These genes primarily participate in metabolic processes and regulation of metabolic processes, including regulation of carbohydrate metabolic process and polysaccharide metabolic process, and regulation of gene transcription (Supplementary Fig. S6c). The downregulation of metabolic pathway-regulating genes during this period suggests a predominance of synthesis, while the simultaneous downregulation of transcription-regulating genes implies early activation of gene expression during plant development. In contrast to group 3, the DEGs in group 4 exhibited a consistent increase during seed development in both F7M and LY287. The main functions of these genes revolve around biosynthesis processes, encompassing macromolecule and protein biosynthesis, along with defunct cellular metabolic processes (Supplementary Fig. S6d). KEGG enrichment analysis revealed comparable findings, indicating that starch biosynthesis-related pathways were initially enriched on the 20th day of development in the male parent rice grains, whereas they were enriched on the 15th day in hybrid rice (Supplementary Fig. S7). Taken together, it suggests that synthesis is the predominant activity in the seeds during this period, with some substances that have already been functional undergoing conversion into storage compounds.
Hybrid rice initiates starch and sucrose metabolism earlier than its parental counterparts
-
Given the variations in starch granule morphology observed between hybrid rice and its parental lines, a comparative analysis of gene expression profiles was conducted at various developmental stages. In KEGG enrichment analysis by DEGs, various pathways were significantly enriched in comparing combinations of three different periods (Fig. 6a–c). DEGs involved in plant-pathogen interaction, MAPK signaling pathway and phenylpropanoid biosynthesis are the top three pathways in comparison groups LY287_10 vs F7M10 at 10 DAP. While in comparison groups, LY287_15 vs F7M15 at 15 DAP, plant hormone signal transduction replaced phenylpropanoid biosynthesis as one of the top three metabolic pathways. In comparison groups LY287_20 vs F7M20 at 20 DAP, starch and sucrose metabolism replaced plant-pathogen interaction as the top metabolic pathways. The shift of pathway related to starch and sucrose metabolism ranking from fourth to first place between the 10th and 20th day of development was particularly striking (Fig. 6a–c). Although starch metabolism pathways were identified in all three combinations, it is noteworthy that the quantity of DEGs related to starch metabolism pathways escalates as developmental time progresses (Fig. 6d, e). Among the 115 DEGs associated with starch metabolism pathways, nine genes showed exclusive differential expression at 10 DAP, 21 genes at 15 DAP, and 49 genes at 20 DAP when comparing LY287 to F7M (Fig. 6d).
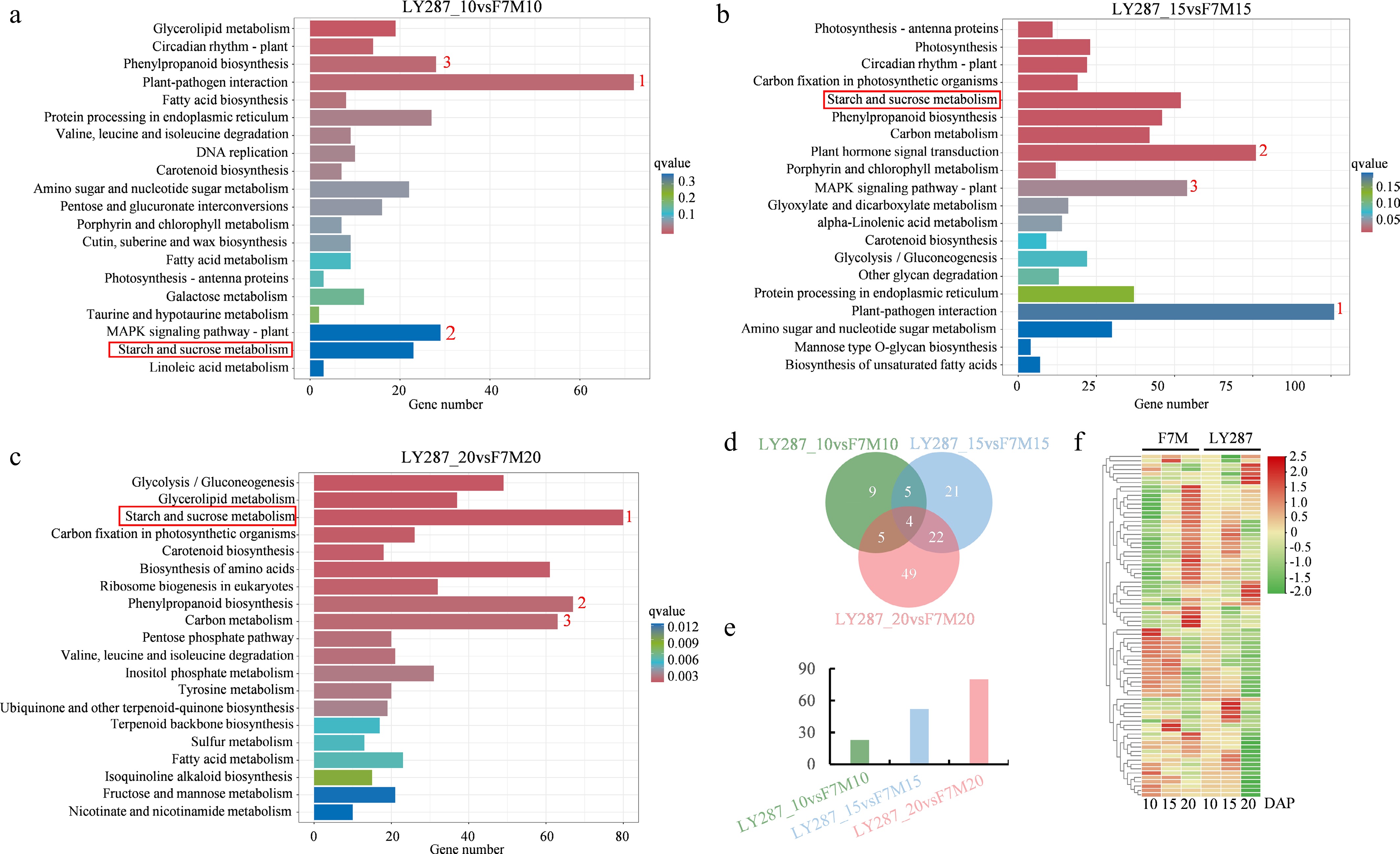
Figure 6.
Comparative KEGG analysis of F7M and LY287. (a) Enriched KEGG pathways were discovered through the use of DEGs obtained from comparing F7M and LY287 at 10 DAP. (b) Enriched KEGG pathways were discovered through the use of DEGs obtained from comparing F7M and LY287 at 15 DAP. (c) Enriched KEGG pathways were discovered through the use of DEGs obtained from comparing F7M and LY287 at 20 DAP. The red number represents the stop three enriched pathways, and the red box highlights the starch and sucrose metabolism pathways. (d) Venn analysis of DEGs related to starch and sucrose metabolism in three comparisons. (e) The number of DEGs that are specifically identified during individual comparisons. (f) Heat map showing abundance of DEGs that are specifically identified during individual comparisons (same as the DEGs in Fig. 6e).
To better understand the role of these 79 DEGs (nine genes at 10 DAP, 21 genes at 15 DAP, and 49 genes at 20 DAP) which showed exclusive differential expression at 10 DAP, 15 DAP, and 20 DAP, the transcription level of these genes were showed in heatmap (Fig. 6f, Supplementary Table S5). In male parent F7M, approximately half of the genes associated with starch and sucrose metabolism exhibit high expression levels during the initial stage of grain development, while the remaining half demonstrate heightened expression during the later stages of grain development. However, in the hybrid LY287, the majority of genes exhibit high expression levels during the early stage of grain development, while only a small number of genes are highly expressed during the late stage of grain development. It suggests that starch and sucrose metabolism commence earlier in hybrid rice compared to the parental lines, exhibiting a reduced duration of gene expression in the former.
Integrated analysis of metabolic and transcriptomic results indicates that Abscisic acid (ABA) regulates grain filling by regulating the expression of starch-synthesis-related genes
-
ABA, as a plant hormone, promotes the grain filling process during the maturation of rice grains. In this study, ABA was the only phytohormone detected in grain by a widely targeted metabolome (Fig. 7a). The abundance of ABA in LY287 grains is higher than that in the parental grains from 10 to 20 DAP during grain filling (Fig. 7a). Multiple key enzymes are involved in the ABA biosynthesis process. Among them, 9-cis-epoxycarotenoid dioxygenase (NCED) serves as the rate-limiting enzyme in the ABA synthetic pathway. The expression of NCED1 was higher in LY287 than that in male parent, which is consistent with the ABA content levels in the grains (Fig. 7b, Supplementary Fig. S8). Previous study has demonstrated that the majority of ABA in rice grains is primarily derived from the leaves, revealing the molecular mechanism through which the MATE transporter DG1 regulates its long-distance transport[49]. In mature flag leaf, both DG1 and NCED1 expression levels were higher in LY287 than in its paternal line, and the ABA content in LY287 was also higher, as expected (Fig. 7c, d, Supplementary Fig. S8). Consistent with previous studies, the high levels of ABA in the grain led to increased expression of SLRL2, thereby suppressing the expression of the GBSSI gene and ultimately resulting in reduced amylose content (Figs 7e, f and 1d, e, Supplementary Fig. S8). In addition, transcription factors NF-YB1 and RISBZ1 also promote the expression of the GBSSI gene, thereby facilitating amylose synthesis. The expression levels of these two genes in LY287 were higher than those in F7M during the early stages of seed development, but became significantly lower than those in F7M in the later stages. Correspondingly, the amylose content in LY287 grains was significantly higher than that in F7M on 5 DAP but lower than that in F7M during the later developmental stages (Fig. 7g, h, Supplementary Fig. S8).
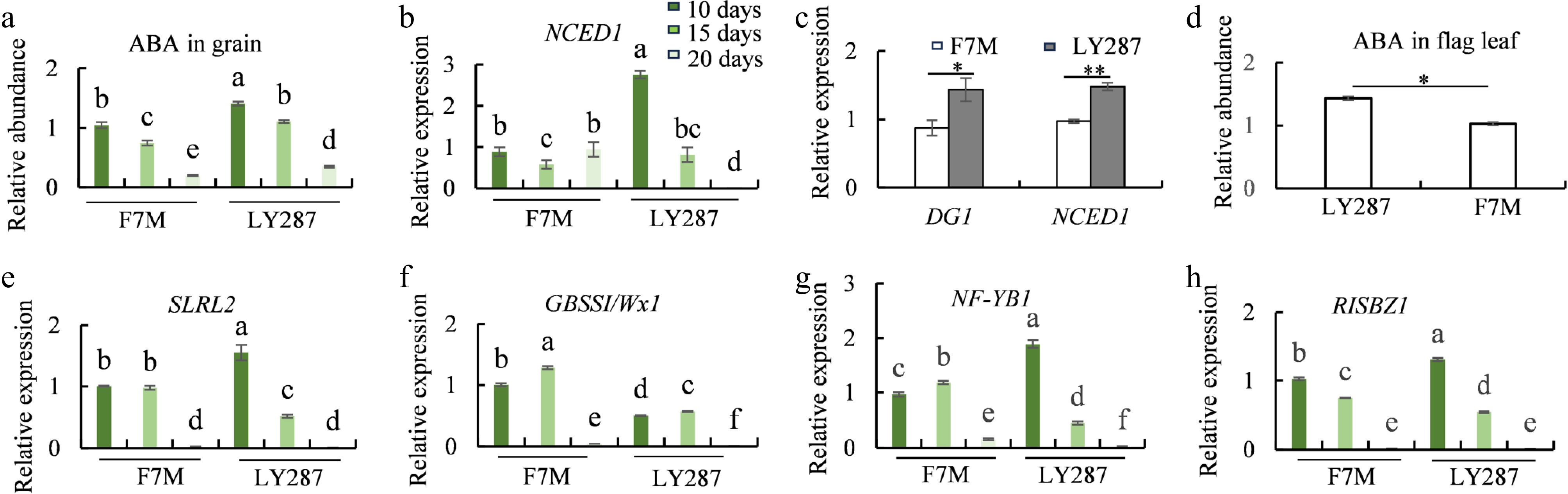
Figure 7.
ABA content and the expression of starch-synthesis-related genes. (a) ABA content in grain of male parent F7M and hybrid LY287 at different developmental stages. (b) Expression levels of NCED1 in rice grain of F7M and LY287 at different developmental stages. (c) Expression levels of DG1 and NCED1 in mature flag leaf of F7M and LY287. (d) ABA content in mature flag leaf of F7M and LY287. (e−h) Expression levels of SLRL2, GBSSI, NF-YB1, and RISBZ1 in rice grain of F7M and LY287 at different developmental stages.
In the starch synthesis pathway, the expression of GBSSI, responsible for amylose synthesis, peaks in early grain development and declines significantly to its lowest point at 20 DAP. The decline in expression level of GBSSI indicates a potential reduction or halt in amylose synthesis, aligning with the finding that grains achieve full maturity by 20 DAP (Fig. 1a). Transcription factors like SERF1, RISBZ1/bZIP58, NAC20, NAC26, and RPBF, which regulate GBSSI expression, mirror its expression pattern (Fig. 8). In LY287, the expression level of GBSSI is lower compared to F7M, and it decreases more rapidly as seed development progresses. This finding implies that the duration of amylose synthesis in LY287's endosperm is shorter than in F7M, leading to a reduced amylose content in LY287 relative to F7M (Table 1). Amylopectin synthesis, involving enzymes like soluble starch synthase (SS) and starch branching enzyme (SBE), is intricate. Transcriptome analysis reveals varied expression patterns within the soluble starch synthase family, with some peaking early and others late in grain development. Starch branching enzyme gene expression remains relatively stable, peaking early and declining gradually. Notably, the rate of decline is faster in LY287 compared to F7M. Based on the findings from metabolomics and transcriptomics analyses, it is hypothesized that LY287 triggered the biosynthesis of storage compounds earlier than its male parent, resulting in a rapid starch biosynthesis process in LY287, thereby enhancing grain filling efficiency.
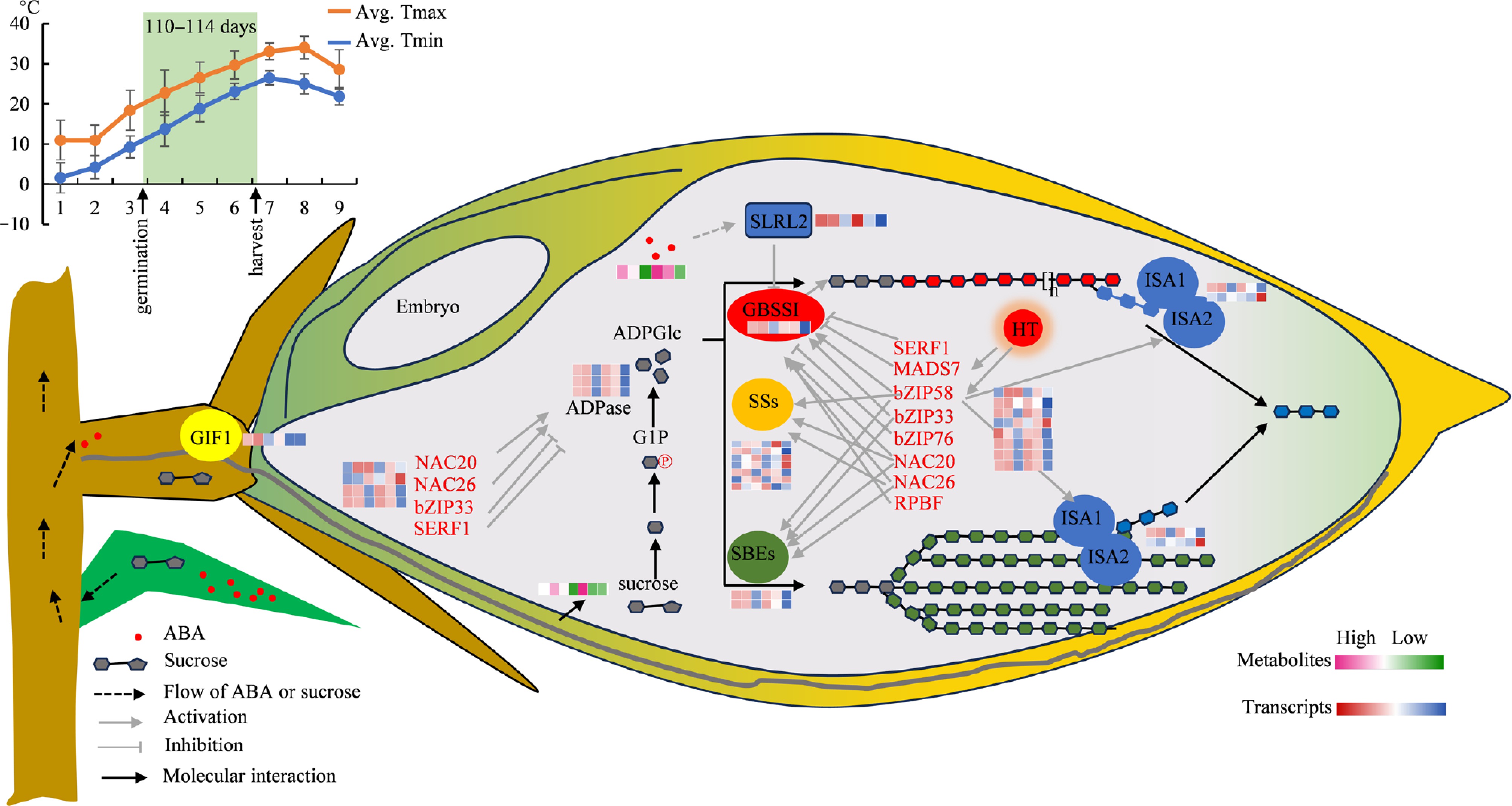
Figure 8.
An analysis diagram illustrating the principal metabolic pathways of key DEGs and DAMs involved in grain filling. LY287 starts the biosynthesis of storage compounds earlier than its male parent, which results in its superior grain quality. In spring, LY287 and its parents were germinated on Mar. 25th, and the growth cycle of rice, from germination to harvest, spans a total of 110–114 d. The line chart displays the temperatures of rice planting regions in 2023. As the rice transitions from the grain filling stage to the final harvest, the temperature steadily increases. Avg. Tmax means the average daily maximum temperature within a month, and Avg. Tmin means the average daily minimum temperature within a month. LY287 has the ability to kickstart the production of starch storage substances earlier than its male parent, which helps minimize the negative effects of high temperatures on the starch composition in seeds, leading to an enhancement in the overall quality of rice. GIF1, grain incomplete filling 1; GBSSI, granule-bound starch synthase I; SSs, soluble starch synthase; SBEs, starch branching enzyme; ISA, debranching enzyme; HT, high temperature. Six closely arranged colored square grids are used to illustrate the relative abundance of DEGs (blue, white, and red) and DAMs (green, white, and pink). From left to right, six square grids mean F7M seeds with 10, 15, 20 DAP, and LY287 seeds with 10, 15, 20 DAP, respectively.
-
LY287 was officially approved by the Hubei Provincial Crop Variety Approval Committee in 2005. As a super-hybrid rice, it has high yield and good grain quality. With exceptional rice quality, it distinguishes itself among the few early-maturing rice varieties. The reasons behind the advantages of traditional rice hybrid breeding have not been extensively explored by experts. The current study aims to bridge a knowledge gap by utilizing an integrated metabolomics and transcriptomics approach to investigate the grain filling process and quality disparities in hybrid rice. This study delves into the potential roles of crucial DEGs and DAMs in the regulation of grain filling. Additionally, the factors contributing to the quality distinctions were investigated and observed between LY287 and its parental lines.
Rapid transition from primary metabolism to storage compound biosynthesis contributes to its superior grain filling and quality in hybrid rice LY287
-
Cereal seeds are predominantly composed of endosperm, which is rich in starch granules. During grain filling, the plant transports assimilates (carbohydrates generated through photosynthesis) to the endosperm, where sucrose is enzymatically converted into starch[50]. Consequently, grain filling is intimately connected to the translocation of sugars, their metabolism and the synthesis of starch. Despite the critical role that sugar and starch metabolism play in grain filling, it is important to note that other primary metabolites, including amino acid metabolism, lipid metabolism, and secondary metabolites, also exert influence on this process. In the current study, overall metabolic activity peaks during early grain development (10 DAP) in both the hybrid LY287 and its male parent F7M, gradually declining in later stages (15 and 20 DAP) (Figs 2d, 3d, Supplementary Fig. S5). The data clearly indicate that the majority of identified metabolites exhibit peak abundance during the early stage of grain filling, implying a decrease in metabolic activity as seed development progresses. Similar results were extensively reported in wheat[51,52], rice[53−55]. It suggests that metabolic activity is vigorous during the early stages of grain filling, and it subsequently declines as the stored substances are fully accumulated.
In addition, in the comparison between 20 and 10 DAP (F7M20 vs F7M10) in F7M, the KEGG enrichment analysis highlighted the most prominent pathways as glucosinolate, phenylalanine, tyrosine, and tryptophan biosynthesis, in addition to quinoline alkaloid and various alkaloid biosynthesis pathways. Interestingly, these pathways, which were similarly enriched in hybrid rice LY287, exhibited earlier activation in the 15 vs 10 DAP comparison (LY287_15 vs LY287_10). During the late stage of seed development, metabolic activities related to the biosynthesis of storage substances are heightened. This metabolic activity commences at a faster rate in hybrid rice than in the parental lines, indicating a superior metabolic conversion rate in hybrid rice compared to that in the parents. Throughout the growth period of early rice, the temperature gradually increases (Fig. 8). Particularly during the seed filling stage, high temperatures shorten the rice filling process, leading to inadequate synthesis and accumulation of storage substances like starch. This alteration in starch composition and structure ultimately reduces rice yield and impairs its appearance and taste[18, 56,57]. In the current study, hybrid rice LY287, with its more compact and larger starch crystal structure, has a lower chalkiness rate (Table 1, Fig. 1). For the early rice cultivar LY287, rapid accumulation of storage substances is crucial and advantageous in mitigating the stress induced by high temperatures. Hybrid rice benefits from the rapid accumulation of storage substances, as it helps the plant withstand high temperatures, resulting in the formation of a starch structure that promotes the development of superior rice quality.
The early onset of starch and sucrose metabolism in hybrid rice LY287 is facilitated by the premature expression of genes governing these metabolic pathways
-
The process of grain filling in rice is characterized by dynamic changes in grain weight throughout seed development, directly impacting the final yield and quality of grains. This process is intricately connected to seed size and setting rate[58,59]. Throughout the grain filling stage, starch predominantly accumulates in the starchy endosperm located at the center of the caryopsis, alongside a minor fraction of storage proteins[60,61]. In contrast to most metabolites in the preceding metabolome that decrease steadily during the filling process, a gradual increase was observed in the expression levels of the majority of genes as seed filling progressed (Figs 2d, 5e). Additionally, the GO functional enrichment analysis of these genes highlighted their predominant roles in synthesizing various substances, with a particular emphasis on macromolecules such as starch, proteins, lipids, and nucleic acids (Supplementary Fig. S5). During the progression of grain filling, a significant decrease is also observed in the expression levels of many genes associated with carbohydrate and polysaccharide breakdown metabolism (Supplementary Fig. S5). It is suggested that the products of photosynthesis in leaves are predominantly either directly transferred to grains or reallocated to reserve pools in vegetative tissues[6, 62].
In addition, an analysis of differentially expressed genes during grain filling stages in hybrid rice and its parental lines demonstrated a gradual rise in the number of genes related to sugar metabolism and starch synthesis (Fig. 6). Particularly, the expression levels of these genes were found to be elevated during the initial stages of grain filling in hybrid rice in comparison to its parental varieties (Fig. 6f). Several genes, including various members of the glycosyl hydrolases family (glycosyl hydrolase family 1, 9, 17, 20), exhibit low expression levels in parental plants until the 20 DAP. However, in hybrid rice, these genes show high expression levels from 10 to 15 DAP, followed by downregulation starting on 20 DAP. Existing studies highlight the involvement of glycosyl hydrolase family members in immune responses and response to abiotic stress[63,64]. Unfortunately, no studies have yet explored the function of glycosyl hydrolase family members during the rice grain filling stage. The results of our study imply that specific genes in glycosyl hydrolases could play a pivotal role in rice grain filling and quality, necessitating further research to substantiate these observations.
Appropriate level of ABA in rice grain of LY287 promotes its grain-filling speed and grain quality
-
Rice seed development, particularly grain shape and quality, is governed by dynamic phytohormone levels. ABA levels in seeds correlate not only with seed dormancy but also play a critical role in developmental regulation[20, 65]. Regarding grain filling, ABA influences assimilate transport and distribution, enhances the activity of starch-synthesizing enzymes, and thereby promotes the translocation of photosynthetic products to grains and starch accumulation. Studies indicate that moderately increasing ABA levels during early grain filling enhances grains' competitive ability for assimilates, directing more photosynthetic products toward grains; in later stages, ABA maintains the activity of key metabolic enzymes in grains. This ensures smooth progression of the filling process and prevents premature grain aging[20, 49, 66]. Within the same rice panicle, grains exhibit differences in grain filling rates. Grains with faster filling rates are termed superior grains, while those with slower rates are called inferior grains. During grain filling, these two types of grains also show considerable differences in the trend of endogenous ABA content changes. Superior grains have higher ABA content, and their ABA level reaches its peak earlier. In contrast, inferior grains show the opposite pattern.
High ABA content promotes the expression of the transcription factor SLR2, which in turn suppresses the expression of the amylose synthesis enzyme GBSSI, ultimately resulting in lower amylose content in LY287 compared to F7M. Appropriate amylose content plays a decisive role in determining rice quality. Besides, transcription factors like SERF1, RISBZ1/bZIP58, NAC20, NAC26, and RPBF, which regulate GBSSI expression as well, mirror its expression pattern (Fig. 8). High temperatures typically lead to a significant increase in ABA levels in plants through a dual strategy of upregulating ABA biosynthesis and inhibiting its catabolism. Therefore, in this experiment, the growth of LY287 may face high-temperature stress during the grain-filling stage. These early-season rice varieties might respond by appropriately elevating ABA levels to ultimately regulate amylose content, or they may modulate starch synthesis through other high-temperature-responsive transcription factors, such as RISBZ1, thereby maintaining their excellent rice quality. Of course, further validation experiments under temperature-controlled cultivation conditions will be a key focus of future work.
-
This study integrated metabolomic and transcriptomic analyses to elucidate the molecular mechanisms underlying the formation of high-quality traits in the grains of the hybrid rice variety LY287. LY287 exhibited higher metabolic activity during the early grain-filling stage, initiating the synthesis of storage substances such as starch earlier than its parental line. Transcriptome analysis revealed that genes related to starch synthesis were activated earlier and showed a clustered expression pattern in LY287. Notably, the starch and sucrose metabolism pathways were significantly enriched during the middle and late grain-filling stages. Hormonal analysis indicated that ABA content was significantly higher in LY287 than in the paternal parent. ABA regulates the expression of GBSSI through the ABA-SLRL2-Wx1 pathway, thereby reducing amylose content and improving rice quality. In addition, transcription factors such as RISBZ1 and NAC were also involved in regulating key genes related to starch synthesis. In summary, LY287 enhances grain-filling efficiency and develops high-quality grains by initiating the accumulation of storage substances earlier and optimizing the starch metabolic regulatory network and hormone signaling pathways. This study provides important theoretical insights and genetic resources for improving the quality of hybrid rice.
This work was supported by the Hubei Provincial Key Research and Development Projects (Grant No. 2024BBB001), the Natural Science Foundation of Hubei Province (Grant No. 2023AFA016), Open Project Funding of the Key Laboratory of Evaluation and Utilization of Grain Crop Genetic Resources, Ministry of Agriculture and Rural Affairs (Grant No. SAGC-2023001), and Hubei Provincial Natural Science Foundation of China (Grant No. 2023AFB614). Professor Ju Chaoming and Professor Xu Guocheng are acknowledged for their pivotal contributions to the creation of LY287 hybrid rice and the management of rice bases.
-
The authors confirm contribution to the paper as follows: study conception and design: Yang P; methodology, data curation, and data analysis: Li M, Fu D; funding acquisition and resources: Lü S, Wu W, Yang P; draft manuscript preparation: Li M; manuscript review and editing: Li M, Fu D, Wu W, Lü S, Yang P; supervision: Yang P. All authors reviewed the results and approved the final version of the manuscript.
-
The raw sequence data can be downloaded for free from the download link in the materials and methods section. The rice resource generated during and/or analyzed in the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Ming Li, Dong Fu
- Supplementary Table S1 Primers used in this study.
- Supplementary Table S2 All identified metabolites and abundance.
- Supplementary Table S3 All DAMs and abundance.
- Supplementary Table S4 All identified genes and abundance.
- Supplementary Table S5 Genes involved in starch and sucrose metabolism.
- Supplementary Fig. S1 This is a brief diagram of the breeding process for hybrid rice LY287(a) and a field photo of LY287(b).
- Supplementary Fig. S2 Metabolite quality control.
- Supplementary Fig. S3 Categorization of DAMs.
- Supplementary Fig. S4 The distribution of each categorization of DAMs in ten subclasses of DAMs.
- Supplementary Fig. S5 Categorization and Clustering analysis of DAMs (the DAMs originate from the most enriched pathway identified in the KEGG analysis).
- Supplementary Fig. S6 GO enrichment analysis of DEGs.
- Supplementary Fig. S7 KEGG Analysis of DEGs in different comparisons.
- Supplementary Fig. S8 Validation of the expression levels of starch synthesis-related genes (identified from transcriptome data) by RT-qPCR.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li M, Fu D, Wu W, Lü S, Yang P. 2025. Mechanistic insights into hybrid rice Liangyou 287's superior performance: transcriptomic and metabolic evidence for rapid starch biosynthesis during grain filling. Seed Biology 4: e026 doi: 10.48130/seedbio-0025-0024 

Mechanistic insights into hybrid rice Liangyou 287's superior performance: transcriptomic and metabolic evidence for rapid starch biosynthesis during grain filling
- Received: 13 March 2025
- Revised: 28 September 2025
- Accepted: 24 October 2025
- Published online: 24 December 2025
Abstract: Hybrid rice has contributed significantly to the world's food security because of its high yield. Recently, the grain quality of hybrid rice has become increasingly of concern. Liangyou 287 (LY287) has emerged as a notable early-season hybrid rice cultivated in South China, distinguished by its superior grain quality. Using metabolomics and transcriptomics, this study has identified shifts in the abundance of 1,169 metabolites and over 11,000 genes throughout rice grain filling. The findings revealed metabolic activity peaks early in grain filling. Hybrid rice initiates starch biosynthesis earlier than its parental varieties and modulates grain filling rate and amylose content through the ABA-SLRL2-Wx1 pathway, ultimately leading to superior grain quality. These findings offer valuable insights for future breeding strategies aimed at improving the performance and quality of hybrid rice.
-
Key words:
- Starch biosynthesis /
- Grain quality /
- Metabolism /
- Transcriptome /
- Grain filling /
- Early‑season hybrid rice /
- Liangyou 287