










-
Retinal ischemia–reperfusion (RIR) injury is a prevalent cause of irreversible visual impairment[1]. It is inextricably implicated in the pathological processes of various vision-threatening ocular diseases, including glaucoma, diabetic retinopathy, and retinal artery occlusion[2,3]. Neuroinflammation and immune responses constitute the core pathological events driving RIR injury[4], encompassing oxidative stress, mitochondrial dysfunction, energy-related metabolic disturbances, and ultimately neuronal death[5]. However, current clinical interventions primarily focus on restoring blood perfusion or controlling intraocular pressure[6], and effective therapeutics that directly target the inflammatory and neurodegenerative cascades of RIR remain limited. Therefore, elucidating novel pathogenic mechanisms and identifying actionable therapeutic targets for RIR injury is of pressing clinical importance.
Microglia are the resident immune cells of the retina and play a pivotal role in orchestrating neuroinflammatory responses following ischemic injury[7]. In the context of RIR, the microglia undergo rapid activation, proliferation, and migration to the injury site, where they initiate neuroinflammation cascades via the release of various proinflammatory cytokines, including tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β)[8]. Sustained microglial activation exacerbates the inflammatory microenvironment and contributes directly to progressive degeneration and loss of retinal ganglion cells (RGCs)[9].
Single-cell RNA sequencing (scRNA-seq) technology has emerged as a powerful approach for dissecting cell-type-specific responses and complex intercellular signaling networks at a single-cell resolution, facilitating high-throughput analysis and enhancing the efficiency of data acquisition[10,11]. In this study, we applied scRNA-seq to systematically profile and compare the retinal transcriptomes of RIR model mice and normal controls. Notably, this analysis revealed a marked and selective upregulation of Lgals3 expression in the microglia following RIR injury.
Galectin-3, encoded by the Lgals3 gene, is a β-galactoside-binding lectin that has been recognized as a critical modulator of microglial activation and inflammatory signaling[12−14]. Previous studies have implicated Galectin-3 in the pathogenesis of diverse inflammatory and ischemic disorders[15]. Mechanistically, Galectin-3 can potentiate inflammatory responses by binding to the Toll-like receptor 4 (TLR4) and activating the downstream nuclear factor kappa B (NF-κB) signaling pathway in the microglia[12,16]. Given the crosstalk between NF-κB and hypoxia-inducible factor 1-alpha (HIF-1α) in ischemic conditions, Galectin-3 may also modulate the hypoxic response[17]. These findings suggest that blocking Galectin-3 could be a promising strategy to inhibit RIR-associated neuroinflammation.
GB1107 is a highly selective small-molecule inhibitor of Galectin-3 that has demonstrated robust anti-inflammatory and antifibrotic efficacy in multiple inflammatory conditions[18−20]. However, its therapeutic potential in ophthalmology and retinal ischemic injury remains unexplored. Therefore, this study integrated scRNA-seq-guided target identification with pharmacological intervention to evaluate the efficacy of GB1107 in RIR injury. Specifically, we investigated whether GB1107 can attenuate microglia-mediated neuroinflammation, preserve RGCs' survival, and ultimately mitigate retinal damage following ischemia–reperfusion. This work seeks to provide mechanistic insights and establish a promising therapeutic strategy for ischemic ocular diseases.
-
For our study, 6–8-week-old male C57BL/6 mice were obtained from Guangdong Medical Laboratory Animal Center (Foshan, China). All animal experiments were conducted in accordance with the Association for Research in Vision and Ophthalmology's Statement for the Use of Animals in Ophthalmic and Vision Research and received approval from the Institutional Review Board of Guangdong Provincial People’s Hospital (Guangzhou, China; approval ID: KY2025-140-03).
The RIR model was established as previously described[21]. The mice were first anesthetized with an intraperitoneal injection of 1% pentobarbital sodium. We then carefully inserted a 30-gauge needle, attached to a normal saline (NS) bottle, into the ocular anterior chamber. To induce elevated intraocular pressure (IOP), the NS bottle was raised to 150 cm, generating and sustaining an IOP of 110 mmHg for 1 h. In the Galectin-3 inhibition experiment, the mice were assigned to receive intraperitoneal injections of either GB1107 (5 mg/kg, MedChemExpress, HY-114409) or a vehicle control. Injections were given at three time points: immediately and at 24 and 48 h after the induction of RIR. The mice were euthanized on Day 3 post-RIR injury, and their retinas or entire eyeballs were harvested for analysis.
Hematoxylin and eosin staining
-
The eyeballs were collected on the third day following surgery and fixed in FAS Fixator (Servicebio, Wuhan, China) for 24 h at room temperature. Following fixation, the samples were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). To quantify the thickness of the inner plexiform layer (IPL), four cross-sectional areas within a 1-mm radius of the optic nerve head were selected for each retina. These sections were imaged with an Eclipse 80i microscope (Nikon, Tokyo, Japan), and thickness measurements were conducted with ImageJ software.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay
-
Apoptosis in the retinal cells was evaluated with a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) FITC Apoptosis Detection Kit (Vazyme Biotechnology, A111), following the supplied instructions. Paraffin-embedded mouse eye sections were dewaxed, rehydrated, and treated with proteinase K for permeabilization. Subsequently, the sections were equilibrated with a buffer for 20 min. Finally, they were stained by incubating with the TUNEL reaction mixture at 37 °C for 1 h. Stained retinal sections were imaged on a Nikon AX confocal microscope. For analysis, TUNEL-positive cells were counted manually. The total number of cells in the same fields was obtained by quantifying 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei using ImageJ software. The apoptotic cell percentage was then calculated as the ratio of TUNEL-positive cells to DAPI-positive cells.
Immunofluorescence staining
-
Eyeballs were enucleated and fixed in 4% paraformaldehyde (PFA) for 2 h. For whole-mount immunolabeling, retinas were carefully dissected and permeabilized with 0.5% Triton X-100. After blocking, they were incubated overnight at 4 °C with the following primary antibodies: Anti-RNA-binding protein with multiple splicing (RBPMS, 1:400, GeneTex, GTX118619), anti-TUJ1 (1:400, GeneTex, GTX631836), and anti-Iba1 (1:200, Cell Signaling Technology, 17198). The following day, samples were incubated with species-appropriate secondary antibodies for 2 h at room temperature. The retinas were then flat-mounted on glass slides and radially incised to create a four-leaf clover configuration for imaging. Images were acquired using a Nikon AX confocal microscope, and the average number of RBPMS-positive or Iba1-positive cells per retina was quantified.
Quantitative real-time polymerase chain reaction
-
Total RNA was isolated from mouse retinal tissue with the RNA-Quick Purification Kit (ESScience) according to the manufacturer's instructions, and its concentration was measured with an ND-1000 spectrophotometer (Thermo Fisher Scientific). The extracted RNA was then reverse-transcribed into complementary DNA (cDNA) using the HiScript II First Strand cDNA Synthesis Kit (Vazyme Biotechnology). Gene expression levels were assessed by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) with SYBR Green reagents (Vazyme Biotechnology). Data were analyzed via the comparative threshold cycle (CT) method, using GAPDH expression as the endogenous control for normalization. The primer sequences used for qRT-PCR are listed in Supplementary Table S1.
Western blot analysis
-
Retinal tissues were lysed in a radioimmunoprecipitation assay (RIPA) buffer (Beyotime) containing 1% of a protease inhibitor (Thermo Fisher Scientific). The lysates were centrifuged to clarify them, and the supernatant protein concentration was determined using a bicinchoninic acid (BCA) assay kit (Thermo Fisher Scientific). Equal amounts of protein were separated by 12% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto 0.22-μm polyvinylidene difluoride (PVDF) membranes (Millipore). After blocking with 5% nonfat milk for 1.5 h, the membranes were incubated overnight at 4 °C with primary antibodies against Galectin-3 (1:1,000, Cell Signaling Technology, 89572), Iba1 (1:1,000, Cell Signaling Technology, 17198), NF-κB (1:1,000, Cell Signaling Technology, 8242), phosphorylated NF-κB (1:1,000, Cell Signaling Technology, 3033), HIF-1α (1:1,000, Cell Signaling Technology, 36169), and β-actin (1:1,000, Proteintech, 66009). After washing, the membranes were incubated for 2 h with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (antirabbit or antimouse immunoglobulin G [IgG], 1:5,000, Cell Signaling Technology). Protein bands were visualized using an enhanced chemiluminescence kit (Beyotime Biotechnology), and their grayscale values were quantified with ImageJ software for expression analysis. All Western blot experiments were performed with at least three independent replicates.
Flow cytometry
-
Retinal tissues were harvested and enzymatically dissociated into single-cell suspensions. Cells were incubated with a viability dye to exclude dead cells, followed by surface staining with fluorochrome-conjugated antibodies against CD11b (Biolegend, #101205) and Ly6G (Biolegend, #127613). After washing, samples were analyzed by flow cytometry, and the data were processed by FlowJo software.
Single-cell sample preparation and sequencing
-
On Day 3 after establishment of the RIR model, the retinas from three mice in each group were pooled to generate one single-cell sample for sequencing. Mice were euthanized, and their retinas were rapidly dissected under sterile and ice-cold conditions. The isolated retinal tissues were enzymatically and mechanically dissociated into single-cell suspensions according to standard protocols. Cell suspensions were subsequently passed through a cell strainer to remove debris and cell aggregates, and cell viability was assessed prior to library preparation. Viable single cells were then subjected to single-cell RNA sequencing using the 10× Genomics Chromium platform, and libraries were sequenced on the Illumina NovaSeq 6000 platform following standard protocols.
Single-cell RNA-seq data preprocessing and quality control
-
Raw sequencing data were processed using the Cell Ranger pipeline (10× Genomics) to perform demultiplexing, read alignment, and gene–cell count matrix generation against the mouse reference genome. The resulting expression matrices were imported into R for downstream analysis using the Seurat package. Low-quality cells were excluded by standard quality control criteria, including cells with extremely low or high numbers of detected genes, as well as cells with excessive mitochondrial gene expression, which were indicative of poor cell integrity. Following quality filtering, the retained cells were normalized to correct for differences in sequencing depth across cells and were used for subsequent analyses.
Cell clustering and differential gene expression analysis
-
Following normalization, highly variable genes were selected for reducing the dimensionality. Principal component analysis was performed, and cell clusters were identified using a graph-based clustering method and visualized by uniform manifold approximation and projection (UMAP). Differentially expressed genes (DEGs) were determined using Wilcoxon's rank-sum test, with genes meeting the criteria of |log fold change| > 0.25, adjusted p < 0.05, and expression in more than 5% of cells were considered to be significant.
Functional enrichment analysis
-
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the Metascape online platform (
https://metascape.org ). DEGs were uploaded to Metascape and analyzed using the default parameters. Enriched GO terms and KEGG pathways with a p value < 0.05 after multiple testing correction were considered to be statistically significant. The enrichment results were visualized using bar plots or enrichment networks.Cell–cell communication analysis
-
Cell–cell communication analysis was performed using the CellChat package in R. Normalized single-cell expression data were used to infer potential ligand–receptor interactions between different cell populations on the basis of a curated database. Communication probability and signaling pathway activity were computed according to the standard CellChat workflow. The inferred intercellular communication networks were visualized to compare signaling interactions among distinct cell types.
Metabolic pathway analysis
-
Cellular metabolic pathway activity was assessed using the scMetabolism package in R. As the curated metabolic gene sets implemented in scMetabolism are primarily based on human genes, mouse DEGs were first converted to their corresponding human orthologs prior to analysis. The transformed gene expression data were then used to quantify metabolic pathway activity at the single-cell level according to the standard scMetabolism workflow, enabling a comparative analysis of metabolic alterations across different cell populations.
Gene set scoring and visualization
-
Gene set-based scoring was performed to evaluate pathway activity at the single-cell level. Gene sets were obtained from the Reactome database and applied to normalized expression data using the AddModuleScore function implemented in the Seurat package. The resulting module scores were calculated for each cell and used for downstream comparisons among different cell populations. UMAP-based visualization of gene sets' scores was generated using the scplotter R package.
Identification of core genes
-
Core genes were identified on the basis of the DEGs obtained from the preceding analyses. These genes were imported into Cytoscape software to construct gene interaction networks, and key genes were determined according to the EcCentricity algorithm according to the network topology analysis.
Statistical analysis
-
Statistical analyses were performed with Prism 9.3 software (GraphPad). Data are expressed as the mean ± standard deviation (SD). Differences between two groups were assessed using Student's t-test. For comparisons involving three groups, one-way analysis of variance (ANOVA) was applied, followed by Bonferroni's post hoc test for multiple comparisons. A p-value of less than 0.05 was considered to be statistically significant.
-
To dissect the cellular heterogeneity in RIR injury, we conducted scRNA-seq on retinal tissues from both the RIR model and normal control (NC) groups. Figure 1a outlines the study's design, aiming to identify potential therapeutic targets in the RIR model. After rigorous quality control and filtering, data integration was performed using Harmony to correct for batch effects. Unsupervised clustering and UMAP[22] revealed 11 unique cellular subpopulations, clearly demarcating the primary retinal lineages into separate clusters (Fig. 1b).
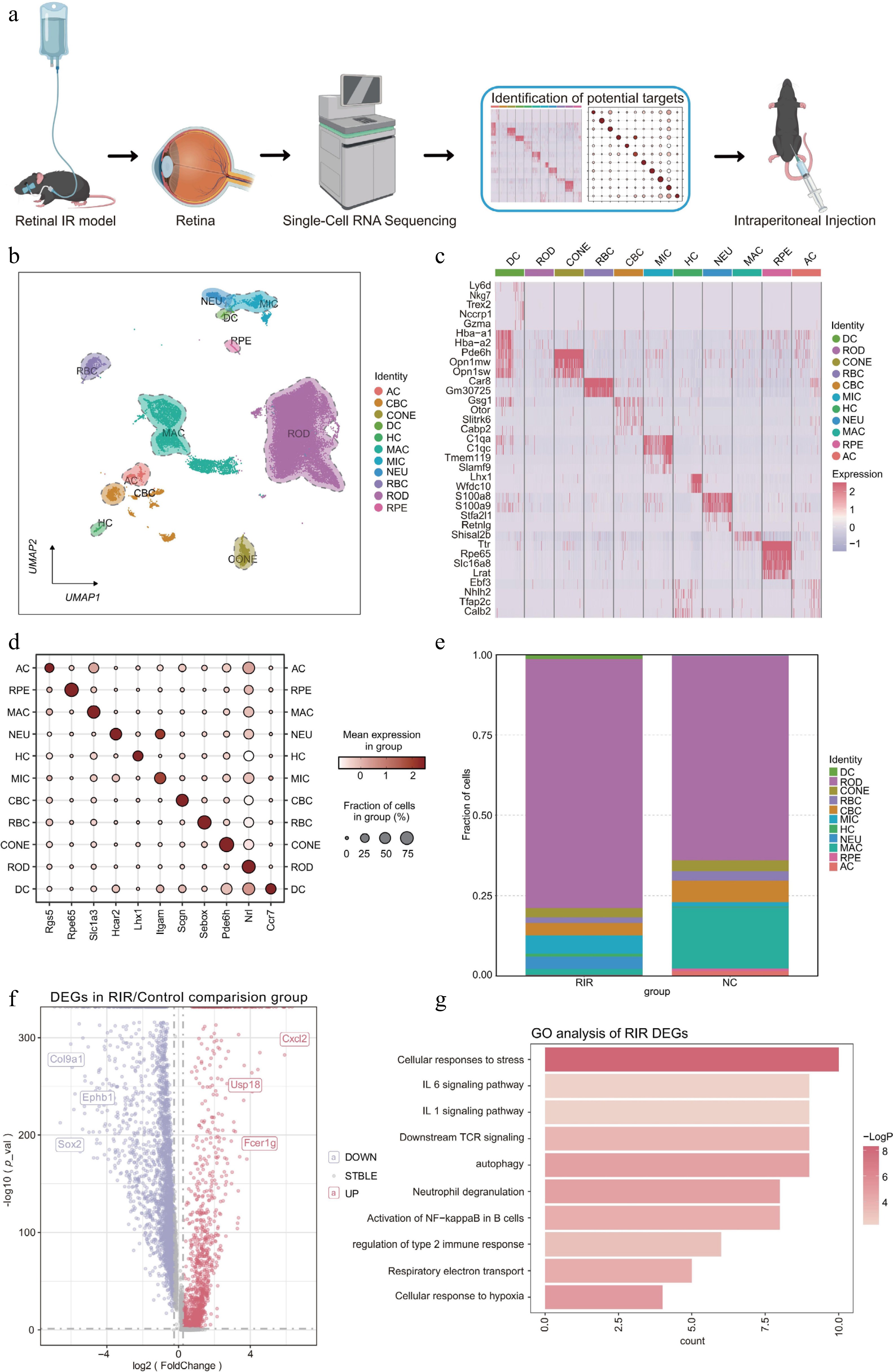
Figure 1.
Changes in the single-cell transcriptomic landscape and cellular composition in the retina following RIR injury. (a) Schematic workflow illustrating the experimental design. (b) UMAP visualization revealing 11 distinct cell clusters in the integrated retinal dataset. (c) Heatmap displaying the expression profiles of the top marker DEGs across identified cell clusters. Rows represent genes, and columns represent individual cells sorted by cluster identity. Color intensity indicates relative gene expression levels (red: high; purple: low). (d) Dot plot showing the expression of canonical marker genes used to annotate each cell type. The size of the dot represents the percentage of cells expressing the marker, and the color intensity indicates the average expression level within that cluster. (e) Stacked bar chart illustrating the proportional shifts in cell populations between the NC and RIR groups. (f) Volcano plot of DEGs in the RIR group vs. NC group. Red and blue dots represent significantly upregulated and downregulated genes, respectively. (g) GO enrichment analysis of upregulated genes in the RIR group. Data for single-cell RNA sequencing were derived from pooled retinas of three mice per group.
We determined the biological identity of each cluster on the basis of the expression of canonical marker genes (Fig. 1c, d). The analysis revealed major neuronal and structural populations, including rod photoreceptors (ROD), cone photoreceptors (CONE), rod bipolar cells (RBC), cone bipolar cells (CBC), amacrine cells (AC), horizontal cells (HC), and retinal pigment epithelium (RPE). Furthermore, we successfully captured the infiltration of distinct immune subsets, specifically microglia (MIC), macrophages (MAC), dendritic cells (DC), and neutrophils (NEU).
A comparison of the cell type composition demonstrated a dramatic shift in the immune landscape of the RIR group. We observed an expansion of immune lineages – particularly MIC, NEU, and DC – accompanied by a relative decline in the proportions of MAC and visual neuron cells compared with the controls (Fig. 1e). To explore the molecular drivers of this pathology, we performed DEG analysis, which highlighted the significant upregulation of inflammatory mediators, most notably Cxcl2 (Fig. 1f), a potent chemokine that has been documented for its role in promoting neutrophil recruitment and exacerbating tissue injury in various inflammatory diseases[23]. Moreover, GO enrichment analysis of the upregulated genes in the RIR group indicated robust activation of inflammatory and stress-related cascades, including 'cellular response to hypoxia', 'autophagy', 'neutrophil degranulation', and 'activation of NF-κB in B cells' (Fig. 1g).
In summary, we generated a comprehensive transcriptional atlas encompassing diverse neuronal and immune cell subpopulations, providing a detailed cellular roadmap to elucidate the dynamic alterations in the retina following RIR injury.
Integrated analysis reveals inflammatory signaling networks and metabolic reprogramming in RIR injury
-
To characterize transcriptomic alterations induced by RIR injury, we first profiled DEGs across major retinal cell populations. Quantitative analysis demonstrated marked transcriptional remodeling, defined by extensive gene repression in neuronal populations (e.g., 2,375 downregulated genes in the cones) and robust transcriptional activation in glial and immune lineages (e.g., 1,240 upregulated genes in the microglia) (Fig. 2a). Pathway enrichment analysis consistently identified inflammatory stress-related pathways, including Fc epsilon receptor I (FCERI)-mediated NF-κB activation, as dominant features throughout the retina (Fig. 2b). Strikingly, the microglia exhibited a highly specialized activation signature characterized by leukocyte activation and regulation of an immune effector process. These findings suggest that the microglia, as resident immune effector cells, undergo intense functional reprogramming to counteract RIR-induced tissue damage.
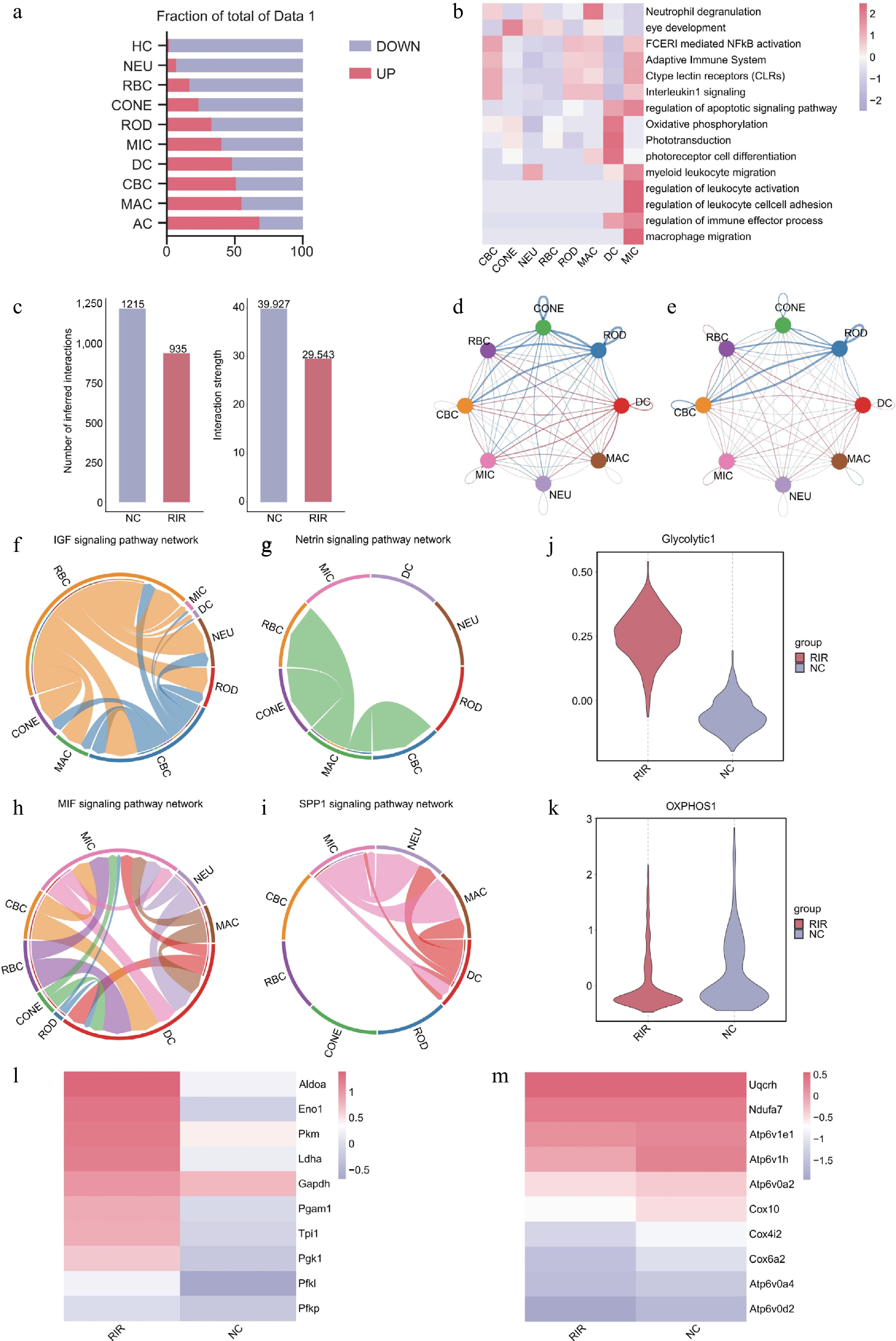
Figure 2.
Integrated analysis reveals inflammatory signaling networks and metabolic reprogramming in RIR injury. (a) Bar graph quantifying upregulated (red) and downregulated (blue) DEGs across retinal cell types. (b) Heatmap of pathway enrichment analysis of significantly upregulated DEGs in the RIR group across eight major cell clusters. (c) Comparison of total cell–cell interactions and interaction strength between NC and RIR groups. Chord diagrams visualizing the differential number of interactions (d) and interaction strength (e). Circle plots of specific signaling pathways. Homeostatic pathways like IGF (f) and netrin (g) characterize the NC group, whereas inflammatory pathways like MIF (h) and SPP1 (i) dominate the RIR landscape. Box plots showing single-cell scores for glycolysis (j) and oxidative phosphorylation (OXPHOS) (k). Heatmaps displaying the expression of key genes involved in glycolysis (l) and OXPHOS (m).
We next interrogated intercellular communication dynamics using CellChat[24]. Whereas the RIR group displayed a global attenuation in total interaction strength—likely attributable to extensive neuronal loss and structural disruption (Fig. 2c)—differential network analysis revealed a profound reorganization of signaling hierarchies. In contrast to the diminished neuronal connectivity, signaling activity originating from myeloid-derived cells, particularly the microglia and dendritic cells, was greatly amplified in the RIR retina (Fig. 2e). This network remodeling supports the notion that, despite overall tissue compromise, immune lineages emerge as central regulatory hubs by intensifying their intercellular communication within the injured microenvironment.
Further dissection of group-specific signaling pathways uncovered a fundamental shift in retinal microenvironmental function. Pathways unique to the NC group featured insulin-like growth factor (IGF) and netrin signaling (Fig. 2f, g), which are documented to play essential neuroprotective roles in maintaining neuronal survival and synaptic homeostasis[25,26]. Conversely, the RIR signaling landscape was dominated by the MIF (macrophage migration inhibitory factor) and SPP1 (osteopontin) pathways (Fig. 2h, i), which are established drivers of myeloid cell activation, recruitment, and pro-inflammatory polarization[27,28]. This transition from homeostatic neurotrophic support to an injury-induced inflammatory drive underscores the central contribution of immune cells to RIR's pathogenesis.
Transcriptomic analysis suggested that immune cell activation might be coupled with metabolic reprogramming. Consistent with prior evidence that proinflammatory (M1-like) polarization necessitates a metabolic switch from oxidative phosphorylation to glycolysis (the Warburg effect)[29,30], gene set enrichment analysis revealed that RIR retinas exhibited significantly elevated glycolytic scores alongside concomitantly reduced oxidative phosphorylation (OXPHOS) scores (Fig. 2j, k). This predicted shift was further corroborated by the coordinated upregulation of key glycolytic enzymes, including Aldoa, Eno1, Pkm, and Pgam1 (phosphoglycerate mutase 1) (Fig. 2l), alongside the downregulation of OXPHOS genes (Fig. 2m). Pgam1 has been identified as a critical mediator linking metabolic reprogramming to inflammatory activation in neuroinflammation[31−33]. These transcriptional signatures suggest a potential metabolic adaptation that supports sustained immune activation in the injured retina.
Microglia drive the neuroinflammatory response in RIR via a Galectin-3-centered regulatory network
-
To delineate the cellular drivers of the postischemic inflammatory surge, we first quantified the inflammatory state within the retinal immune microenvironment. Compared with the controls, the RIR group exhibited a global elevation in inflammatory scores (Fig. 3a). Subsequent stratification of these scores across individual cell clusters identified microglia as the primary contributors to this inflammatory burden, with significantly higher scores than other immune populations, including NEU and DC (Fig. 3b). This observation was further reinforced by UMAP visualization (Fig. 3c), where the microglia consistently localized to the most prominent inflammatory hotspots. Notably, the intensity of the inflammatory signal was markedly enhanced in the RIR group, indicating that RIR selectively induces a hyperinflammatory state within the microglial compartment.
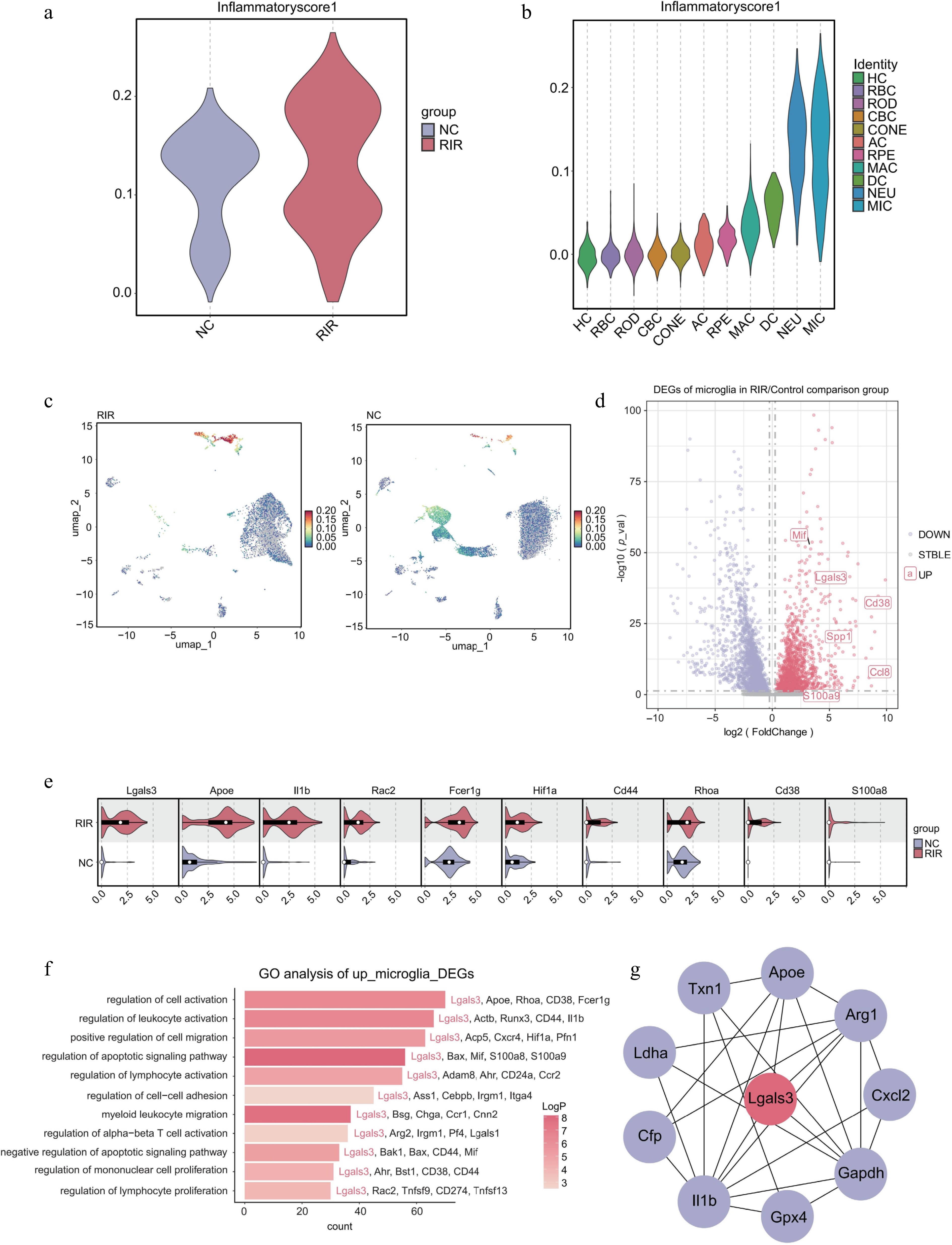
Figure 3.
Microglia drive the neuroinflammatory response via a Galectin-3-centered regulatory network. (a) Violin plot comparing the global inflammatory score between NC and RIR groups. (b) Stratified violin plot displaying inflammatory scores across different retinal cell clusters. (c) Feature plots visualizing the distribution of inflammatory scores on the UMAP. (d) Volcano plot of DEGs in the microglia (RIR vs. NC). (e) Split violin plots validating the upregulation of specific genes in RIR-associated microglia. (f) GO enrichment analysis of upregulated DEGs in RIR microglia. (g) PPI network analysis of microglial DEGs.
We next explored the molecular basis of this activation through DEG analysis. The volcano plot (Fig. 3d) revealed a profound transcriptional reprogramming in RIR microglia, highlighted by the prominent enrichment of genes such as Lgals3, Spp1, and Mif. Of particular interest, Lgals3 (encoding Galectin-3) emerged as one of the most significantly upregulated transcripts. Given the established role of Galectin-3 as a critical regulator in a broad spectrum of inflammatory pathologies, its selective induction in RIR microglia implicates a potential pathogenic mechanism. The violin plots further verified a robust expression of inflammation- and hypoxia-related genes, including Lgals3, Apoe, Il1b, and Hif1α, in the RIR group (Fig. 3e). In line with this transcriptional signature, GO enrichment analysis (Fig. 3f) demonstrated significant associations with 'myeloid leukocyte migration', 'regulation of lymphocyte activation', and 'regulation of cell activation'. Taken together, these findings indicate that RIR microglia are not merely passive responders to ischemic stress but actively engage inflammatory programs, facilitating immune recruitment and activation.
To identify the core regulator governing this complex inflammatory network, we performed a protein–protein interaction (PPI) analysis using the Eccentricity algorithm in Cytoscape. This topological analysis identified Lgals3 as the principal hub gene (Fig. 3g), exhibiting the highest centrality and connectivity within the interaction landscape. The central positioning of Lgals3 across multiple signaling axes suggests that it functions as a critical regulatory node, potentially bridging upstream ischemic stress to the sustained inflammatory output characteristic of RIR's pathology.
In conclusion, these data demonstrate that the microglia drive the initiation and amplification of neuroinflammation in RIR injury through the selective upregulation of Lgals3 (Galectin-3) and its associated regulatory network.
Pharmacological inhibition of Galectin-3 with GB1107 attenuates ischemia-induced upregulation in the retina
-
To validate Lgals3 (Galectin-3) as a potential therapeutic target identified in our preliminary screening, we systematically characterized its protein expression levels and spatial distribution in the retina following RIR injury. Western blot analysis demonstrated a marked upregulation of Galectin-3 protein in the RIR group compared with the NC group (Fig. 4a, b). Interestingly, although our scRNA-seq data indicated that Lgals3 transcripts were predominantly restricted to activated microglia, immunofluorescence (IF) staining revealed a broader distribution of Galectin-3 protein. Fluorescence signals were predominantly localized to the ganglion cell layer (GCL) and inner nuclear layer (INL) (Fig. 4c, d). As previous studies have established that Galectin-3, as a protein secreted in the retina, is predominantly expressed by activated microglia, whereas homeostatic microglia express negligible levels[34−37], the observed broad staining pattern in the GCL and INL likely results from the secretion of Galectin-3 by activated microglia and its subsequent binding to the surface of RGCs or Müller glia. Consistent with this spatial distribution, Galectin-3 may act as a potential key regulator of the postischemic neuroinflammatory microenvironment in the retina.
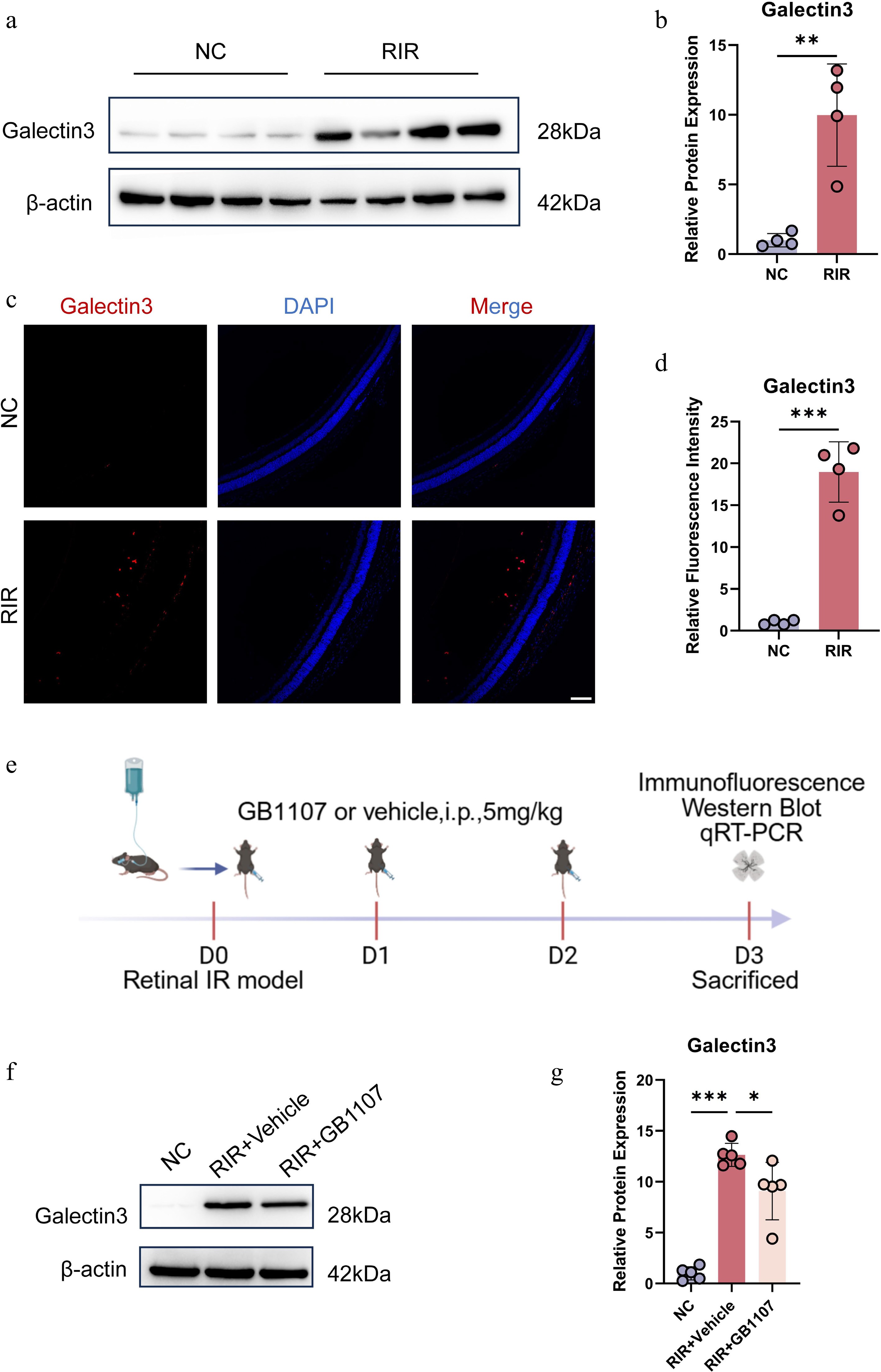
Figure 4.
Pharmacological inhibition of Galectin-3 with GB1107 attenuates its ischemia-induced upregulation. Representative Western blot images (a) and quantitative analysis (b) of Galectin-3 protein expression in retinal tissues. (c) Immunofluorescence image labeling Galectin-3 with red is shown. Cell nucleuses were stained with DAPI (blue). Scale bar: 100 µm. (d) Quantification of Galectin-3 fluorescence intensity. (e) Experimental timeline. Representative Western blots (f) and quantification (g) demonstrating that GB1107 treatment suppresses RIR-induced Galectin-3 protein elevation. Data are mean ± SD (n = 5 per group). One-way ANOVA with Bonferroni's post hoc test was used. *p < 0.05, **p < 0.01, ***p < 0.001.
In light of the aberrant upregulation of Galectin-3 following RIR injury, we next investigated whether pharmacological inhibition of Galectin-3 could mitigate this pathological response. We used GB1107, a highly selective Galectin-3 inhibitor with favorable bioavailability[20]. The experimental paradigm is illustrated in Fig. 4e. Immediately after RIR induction (Day 0), mice received intraperitoneal injections of GB1107 (5 mg/kg) or a vehicle, followed by additional administrations on Day 1 and Day 2. Retinal tissues were collected on Day 3 for subsequent molecular and histological analyses.
To evaluate the in vivo efficacy of this intervention, Galectin-3 protein levels were assessed by Western blotting. Although RIR injury resulted in a significant elevation of Galectin-3 expression in the vehicle-treated group, administration of GB1107 substantially attenuated this ischemia-induced increase (Fig. 4f, g). Collectively, these results demonstrate that the selected dosage and treatment regimen of GB1107 effectively suppress pathological Galectin-3 upregulation in the ischemic retina, thereby establishing a robust foundation for subsequent investigations into its functional role in RIR-associated retinal injury.
GB1107 alleviates RIR-induced retinal damage and confers neuroprotection
-
We next examined the neuroprotective potential of Galectin-3 inhibition by systematically analyzing the effects of GB1107 on retinal histopathology. H&E staining revealed that compared with the NC group, retinas subjected to RIR exhibited severe structural disruption 3 days after reperfusion injury. This damage was characterized by marked thinning and vacuolization, particularly within the IPL, accompanied by cellular disorganization[38,39]. In contrast, GB1107 treatment significantly ameliorated these pathological changes. Retinas in the treated group showed improved structural integrity and a significant attenuation of IPL thinning (Fig. 5a, b), suggesting that targeted inhibition of Galectin-3 is critical for maintaining the retinal architecture following ischemic injury.
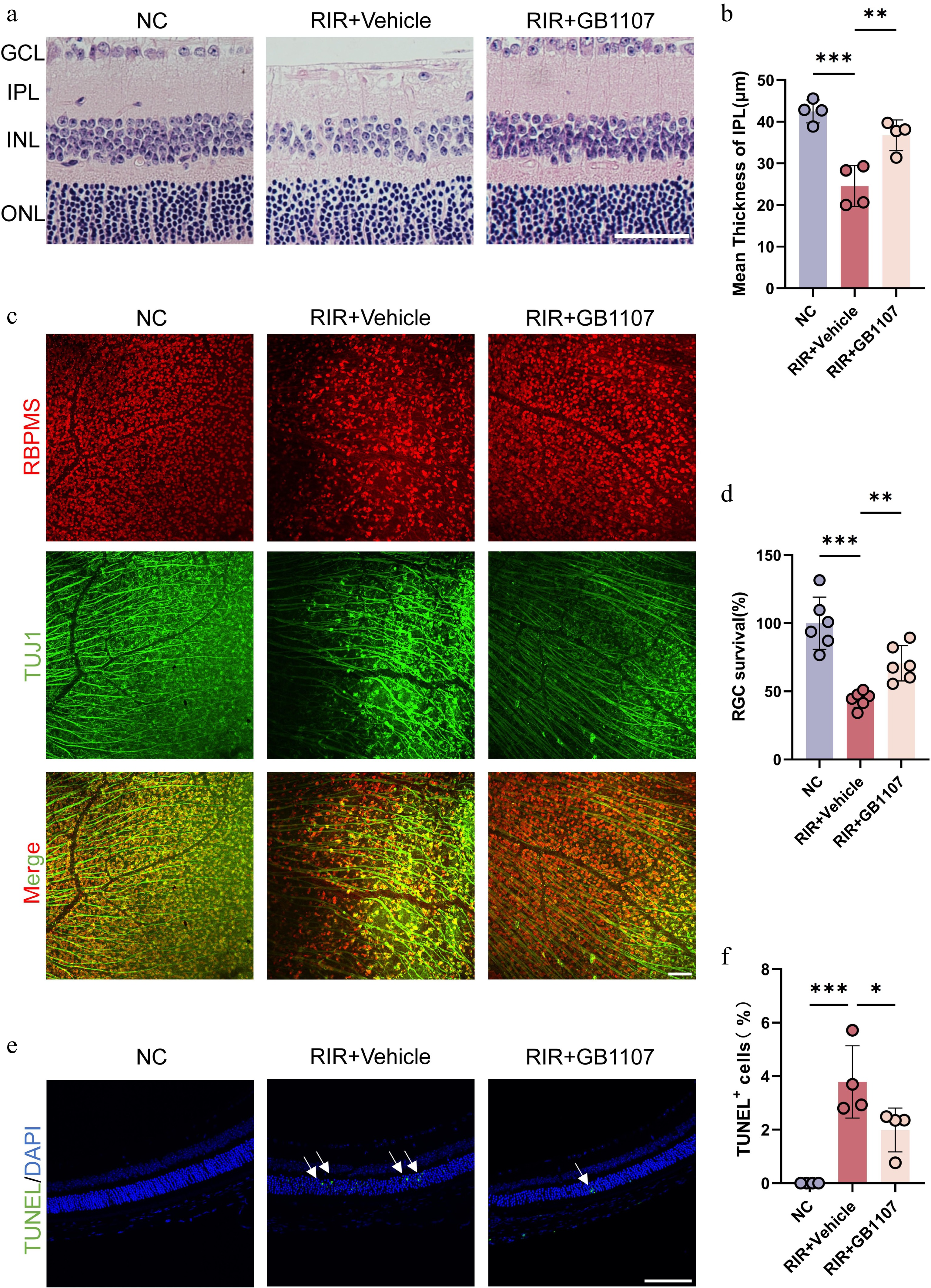
Figure 5.
GB1107 alleviates RIR-induced retinal structural damage and confers neuroprotection. (a) Representative H&E staining of retinal sections. Scale bar: 50 µm. (b) Quantification of IPL thickness across groups (n = 4 per group). (c) Immunofluorescence image labeling RGCs in the retina with RBPMS (red) and TUJ1 (green), and the merged image of two channels is shown above. Scale bar: 100 µm. (d) Quantification of RBPMS-positive RGC density (n = 6 per group). (e) Representative TUNEL staining (green) assessing retinal apoptosis. Nuclei are stained with DAPI (blue). Scale bar: 100 µm. (f) Quantification of TUNEL-positive cells (n = 4 per group). All data are presented as the mean ± SD. One-way ANOVA with Bonferroni's post hoc test was used for statistical analyses. * p < 0.05, ** p < 0.01, *** p < 0.001.
Given that RGCs are the neuronal population most vulnerable to ischemia–reperfusion injury, we assessed their survival using retinal flat mounts immunolabeled with RBPMS (a specific marker for RGC somas) and TUJ1 (a marker for neuronal axons). The results demonstrated a dramatic loss of RGCs in the RIR group compared with the NC controls. Furthermore, the vehicle-treated retinas exhibited fragmented and disorganized axonal bundles. Conversely, administration of GB1107 significantly mitigated this RIR-induced neuronal loss, preserving a higher density of RGCs and maintaining the continuity and overall integrity of the axonal network (Fig. 5c, d). These findings indicate that Galectin-3 inhibition effectively protects RGCs from degeneration and axonal damage.
Programmed cell death is a key driver of neuronal loss in ischemic retinopathy[40,41]. To further investigate the mechanism of protection, we performed a TUNEL assay to evaluate the extent of apoptosis in the retina. RIR injury triggered a substantial increase in the number of TUNEL-positive cells, indicating heightened apoptotic activity. Strikingly, GB1107 treatment significantly reduced the proportion of apoptotic cells compared with the vehicle group (Fig. 5e, f).
To sum up, these data provide direct evidence that GB1107-mediated inhibition of Galectin-3 confers robust neuroprotection against RIR injury by preserving retinal structure, promoting RGCs' survival, and suppressing apoptosis.
GB1107 suppresses microglial activation, neutrophil infiltration, and downregulates NF-κB/HIF-1α signaling
-
Activated microglia represent the primary immune cells and key drivers of neuroinflammation following RIR injury. To determine whether Galectin-3 inhibition influences the activation state of the microglia, we analyzed the expression of Iba1, a specific microglial marker. In the NC group, Iba1+ cells were relatively sparse, were uniformly distributed, and exhibited a homeostatic morphology. In contrast, RIR injury induced pronounced morphological alterations in the microglia, characterized by hypertrophic cell bodies and shortened, thickened processes, accompanied by a substantial increase in Iba1+ cell density, indicating enhanced microglial activation (Fig. 6a, b). Western blot analysis further confirmed a robust elevation in Iba1 protein levels in the vehicle-treated group. Notably, GB1107 treatment markedly reduced both Iba1+ cell density and protein abundance, while partially restoring resting microglial features (Fig. 6c, d), indicating that inhibiting Galectin-3 effectively restrains RIR-induced microglial activation.
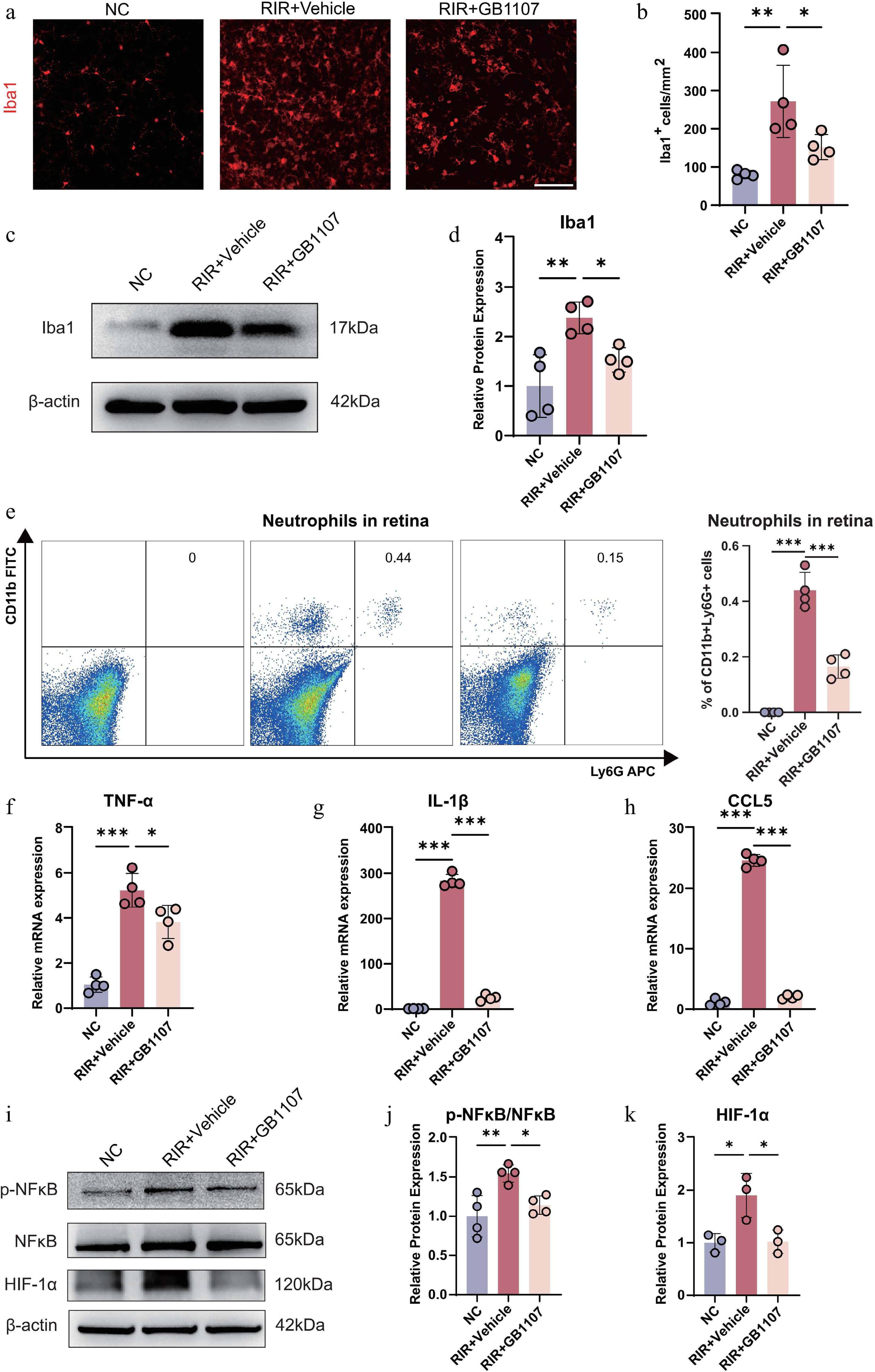
Figure 6.
GB1107 suppresses microglial activation and neuroinflammation associated with the downregulation of NF-κB and HIF-1α signaling. (a) Immunofluorescence showing microglial morphology (Iba1, red). Scale bar: 100 µm. (b) Quantification of Iba1-positive cell density. (n = 4 per group). (c) Western blot analysis and (d) quantification of Iba1 protein levels, confirming reduced microglial activation with GB1107 treatment. (n = 4 per group). (e) Representative flow cytometry plots and quantification of retinal neutrophils (CD11b+Ly6G+). (n = 4 per group). The mRNA expression levels of pro-inflammatory cytokines TNF-α (f) and IL-1β (g), and chemokine CCL5 (h) were measured with qRT-PCR (n = 4 per group). (i) Western blot analysis of p-NF-κB, NF-κB, and HIF-1α, with (j) ratio quantification of p-NF-κB/NF-κB (p65) (n = 4 per group) and (k) protein quantification of HIF-1α (n = 3 per group). All data are presented as the mean ± SD. One-way ANOVA with Bonferroni's post hoc test was used for statistical analyses. * p < 0.05, ** p < 0.01, *** p < 0.001.
ScRNA-seq analysis revealed a significant expansion of neutrophils in the RIR retina (Fig. 1e). Galectin-3 is known to modulate neutrophil recruitment[42], so we also investigated the effect of GB1107 on neutrophil infiltration. Flow cytometry analysis using the CD11b and Ly6G markers showed a substantial influx of neutrophils (CD11b+Ly6G+) into the retina following RIR injury compared with the NC group. However, GB1107 treatment significantly attenuated this infiltration (Fig. 6e). These results suggest that GB1107 exerts its anti-inflammatory effects by simultaneously suppressing microglial activation and blocking neutrophil recruitment.
Microglial activation is typically coupled with the release of proinflammatory cytokines and chemokines. To further assess the impact of GB1107 on the retinal inflammatory microenvironment, we quantified the mRNA levels of key pro-inflammatory cytokines (TNF-α, IL-1β) and the chemokine CCL5 by qRT-PCR. RIR injury triggered a dramatic transcriptional upregulation of TNF-α, IL-1β, and CCL5 in the retina. However, GB1107 treatment significantly suppressed the expression of these inflammatory mediators (Fig. 6f–h), suggesting a dampened inflammatory response following Galectin-3 inhibition.
To elucidate the molecular mechanisms underlying these protective effects, we examined the NF-κB signaling pathway and the hypoxia-related regulator HIF-1α. Western blot analysis revealed that RIR injury markedly increased the p-NF-κB/NF-κB ratio, reflecting enhanced NF-κB signaling activity. Concurrently, the expression of HIF-1α, a pivotal transcription factor in the cellular response to ischemia, was significantly upregulated. In contrast, GB1107 treatment significantly downregulated both NF-κB phosphorylation levels and HIF-1α protein expression (Fig. 6i–k). These findings suggest that the anti-inflammatory and neuroprotective effects of GB1107 are closely correlated with suppression of the NF-κB–HIF-1α signaling axis.
-
By integrating scRNA-seq with a targeted pharmacological intervention, this study systematically elucidated the cellular and molecular mechanisms underlying RIR injury. RIR is a critical pathological process leading to irreversible visual impairment[6]. Our results explicitly identify the microglia as the primary drivers of the postischemic neuroinflammatory response[43] and establish Lgals3 (encoding Galectin-3) as a pivotal molecular hub regulating microglial inflammatory activation. Crucially, we demonstrate that the Galectin-3 inhibitor GB1107 effectively alleviates microglial-mediated neuroinflammation, which is accompanied by the suppression of the NF-κB pathway, thereby revealing a novel therapeutic avenue for ischemic retinal diseases.
Our scRNA-seq analysis revealed a dramatic remodeling of the retinal immune microenvironment following RIR injury. Among all immune cell populations, the microglia exhibited the most robust transcriptional response, characterized by the highest inflammatory scores and significant upregulation of proinflammatory genes, including Lgals3, Spp1, and Mif. These findings suggest that the microglia serve as the central regulators of retinal neuroinflammation, undergoing a decisive phenotypic shift from a homeostatic state to a proinflammatory profile under ischemia–reperfusion stress[44,45].
A key discovery of this study is the identification of Galectin-3 as a central hub within the activated microglial inflammatory network. In RIR-associated microglia, Lgals3 was not only significantly upregulated but also exhibited high centrality and connectivity within the PPI network, underscoring its essential role in inflammatory regulation. Studies have shown that Galectin-3 in the retina is predominantly expressed by activated microglia, whereas microglia in a homeostatic state express little to no Galectin-3[34−37]. The broad Galectin-3 staining observed in the GCL and INL, which partially overlaps with the location of RGCs and Müller glia, is consistent with the known biology of Galectin-3 as a secreted protein. Upon injury or inflammation, activated microglia secrete Galectin-3, which contributes to neuronal degeneration through multiple mechanisms, including direct cytotoxic effects on RGCs, autocrine/paracrine activation of the microglia, and activation of other cell types such as astrocytes[15,46,47]. Thus, Galectin-3 serves as a critical node connecting initial ischemic injury to sustained inflammatory signaling.
Beyond intracellular transcriptional changes, intercellular communication analysis further highlighted the transition of the retinal signaling network following RIR injury. We observed a significant attenuation of neuroprotective pathways, such as IGF and netrin, and the emergence of characteristic signaling axes represented by MIF and SPP1, which are closely associated with myeloid cell activation and immuno-inflammation. This shift reflects a pathological reprogramming of the retinal microenvironment from a neuroprotective to an immune-driven state[48], further validating the microglia as the core regulators of the post-RIR response.
Metabolic reprogramming represents another hallmark of microglial activation. Our transcriptomic profiling indicated a potential metabolic shift in the microglia following RIR injury, characterized by gene signatures associated with enhanced glycolysis and diminished oxidative phosphorylation[49−51]. This predicted metabolic profile aligns with the characteristics of proinflammatory microglia and likely sustains persistent inflammatory responses and cytokine production. However, since these findings are based on gene expression, future functional assays, such as lactate production measurements or metabolic flux analyses, are required to definitively confirm this metabolic reprogramming. Given recent advances regarding the role of Galectin-3 in immunometabolic regulation, it is plausible that Galectin-3 participates not only in inflammatory signal transduction but also in modulating metabolic reprogramming of the microglia under RIR conditions, a hypothesis that warrants further investigation.
Importantly, pharmacological inhibition of Galectin-3 provided direct functional validation of its pathogenic role. GB1107, a targeted inhibitor of Galectin-3, significantly reduced Galectin-3 expression in the RIR retina, improved structural integrity, promoted the survival of RGCs, and markedly decreased apoptosis. Although GB1107 was administered systemically, its robust retinal accumulation and therapeutic efficacy are likely facilitated by the pathological breakdown of the blood–retinal barrier (BRB) induced by ischemic injury, which enhances the permeability of small-molecule inhibitors[52,53]. Furthermore, the significant reduction in microglial activation and the suppressed expression of proinflammatory cytokines demonstrate that Galectin-3 inhibition can effectively block the inflammatory cascade triggered by RIR that leads to neuronal degeneration at both the cellular and molecular levels.
Mechanistically, our findings suggest that the neuroprotective effects of GB1107 are closely associated with the downregulation of key inflammatory and hypoxic signaling pathways. NF-κB is a master regulator of inflammatory genes' transcription and is a known downstream effector of Galectin-3[54]. Furthermore, HIF-1α is a critical transcription factor driving ischemic injury, and its expression is often transcriptionally regulated by the NF-κB pathway during inflammation[17]. The concurrent decrease in NF-κB phosphorylation levels and HIF-1α following GB1107 treatment provides a plausible molecular mechanism linking Galectin-3 inhibition to the mitigation of neuroinflammation.
This study is not without limitations. First, our research primarily focused on the acute phase of RIR injury; the long-term effects of Galectin-3 inhibition on chronic inflammation, retinal remodeling, and neuroregeneration remain to be explored. Second, although GB1107 is a highly selective inhibitor, the use of microglia-specific Lgals3 conditional knockout mice in future studies would more precisely exclude systemic interference and further validate the cell-intrinsic mechanisms identified here. Despite these constraints, this study has systematically constructed a mechanistic framework connecting microglial activation, Galectin-3 signaling, and neuroinflammation. Future research focusing on therapeutic windows, dosing strategies, and assessments of visual function recovery will likely enhance the clinical translation potential of Galectin-3 targeted therapy.
-
To sum up, this study constructed a high-resolution single-cell landscape of RIR injury and identified the microglia–Galectin-3–NF-κB axis as a crucial pathogenic pathway driving postischemic neuroinflammation. Targeted inhibition of Galectin-3 by GB1107 exerts neuroprotective effects by effectively suppressing inflammatory signals, preserving the retinal structure, and promoting neuronal survival. These findings provide a solid theoretical foundation for developing therapeutic strategies centered on immunomodulatory targets for the treatment of ischemic ocular diseases.
-
All animal experiments were conducted in accordance with the Association for Research in Vision and Ophthalmology's Statement for the Use of Animals in Ophthalmic and Vision Research and received approval from the Institutional Review Board of Guangdong Provincial People's Hospital (Guangzhou, China; approval ID: KY2025-140-03; approval date: 2025-12-05).
-
The authors confirm their contributions to the paper as follows: study conception and design: Zeng H, Chen B, Geng X, Zuo X, Yu H, Huang Z; data collection: Zeng H, Chen B, Geng X, Wang Z, Fang Y, Wu Y, Huang Z; analysis and interpretation of results: Zeng H, Chen B, Geng X, Wu Y, Huang Z; draft manuscript preparation: Zeng H, Chen B, Yu H, Huang Z. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed in the current study are available from the corresponding author on reasonable request.
-
This work was supported by the National Natural Science Foundation of China (NSFC) (U24A20707, 82271125, 82301260, 82301205, and 82301246), Guangdong Basic and Applied Basic Research Foundation (2023B1515120028), China Postdoctoral Science Foundation (2025M781945, 2024T170185), Brolucizumab Efficacy and Safety Single-Arm Descriptive Trial in Patients with Persistent Diabetic Macular Edema (2024-29), and the launch fund of Guangdong Provincial People's Hospital for NSFC (8227041127).
-
The authors declare that they have no conflict of interest.
-
Authors contributed equally: Hao Zeng, Beier Chen, Xingchen Geng
- Supplementary Table S1 Primer sequences for qRT-PCR.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zeng H, Chen B, Geng X, Zuo X, Wang Z, et al. 2026. GB1107 attenuates microglia-mediated neuroinflammation by targeting Galectin-3 in retinal ischemia–reperfusion injury. Visual Neuroscience 43: e009 doi: 10.48130/vns-0026-0010 

GB1107 attenuates microglia-mediated neuroinflammation by targeting Galectin-3 in retinal ischemia–reperfusion injury
- Received: 26 January 2026
- Revised: 15 February 2026
- Accepted: 25 February 2026
- Published online: 13 March 2026
Abstract: Retinal ischemia–reperfusion (RIR) injury is a common cause of irreversible visual impairment, yet its pathogenic mechanisms remain incompletely understood. In this study, we used single-cell RNA sequencing to analyze a mouse RIR model and explore novel mechanisms underlying RIR injury. Single-cell RNA sequencing revealed a significant increase in the proportion of microglia in the RIR model group, and differential gene enrichment analysis indicated a marked activation of inflammatory signaling pathways. Further analysis suggested that the inflammatory score of the microglia was elevated, with the differentially expressed gene Lgals3 showing significant changes and involvement in multiple inflammatory pathway activations. The results of immunofluorescence staining and Western blotting also confirmed that the expression of Galectin-3, the protein encoded by Lgals3, was significantly upregulated in the RIR model. To verify the role of Galectin-3 in RIR injury, we used its specific inhibitor, GB1107, to intervene in RIR model mice. The results demonstrated that inhibiting Galectin-3 effectively alleviated the retinal damage caused by RIR, including restoring the thickness of the inner plexiform layer, increasing the survival of retinal ganglion cells (RGCs), and reducing the number of apoptotic cells. A further mechanistic investigation revealed that after inhibiting Galectin-3, microglial activation was reduced, the expression levels of corresponding inflammatory factors decreased significantly, and the expression of phosphorylated nuclear factor kappa B (NF-κB) was downregulated. This study confirms that Galectin-3 is a key target in RIR injury and that inhibiting Galectin-3 can partially mitigate the loss of RGCs and the neuroinflammatory activation caused by RIR, providing a new therapeutic target for related retinal diseases.