









-
The cultivated strawberry (Fragaria × ananassa Duch.) is a globally important perennial horticultural crop and one of the most widely consumed berry fruits. Its global annual production now exceeds nine million metric tons, with China, the United States, and Spain as the leading producers (FAOSTAT, 2023). Beyond its economic importance, strawberries are a rich source of essential nutrients, such as vitamin C and folate, and health-promoting phytonutrients, including anthocyanins and other phenolic compounds. These compounds have been associated with a reduced risk of cardiovascular diseases and certain cancers[1].
The exceptional agronomic traits of the cultivated strawberry are underpinned by a remarkably complex genetic background. This relatively young domesticated crop originated from an accidental hybridization in 18th-century Europe between two wild octoploid species: F. virginiana from North America and F. chiloensis from Chile[2]. Its allo-octoploid genome (2n = 8x = 56) comprises four distinct diploid subgenomes, designated A, B, C, and D, which remain incompletely characterized[3−5]. This intricate polyploid structure, coupled with the high heterozygosity typical of an outcrossing species, poses substantial challenges for genomic analyses and has markedly hindered progress in molecular breeding and functional genomics. Researchers frequently rely on the structurally simpler diploid ancestor, F. vesca, as a model system[6]. However, to fully elucidate complex biological phenomena in polyploids, such as chromosomal interactions, allelic expression patterns, subgenome stabilization, and divergence, studies must ultimately focus on the octoploid strawberry itself. Moreover, octoploid strawberries dominate current commercial cultivation, creating an urgent industry need to improve traits such as disease resistance, climate resilience, and fruit quality. Improving foundational genomic resources for the octoploid strawberry is therefore critical to overcoming this long-standing 'genomic bottleneck', thereby facilitating more precise and efficient research and breeding programs.
In recent years, significant advancements have been made in cultivated strawberry genomics. The release of the first chromosome-scale reference genome for the octoploid cultivar 'Camarosa' marked a pivotal milestone. Published by Edger et al., this assembly combined short-read sequencing with PacBio long-read sequencing for scaffolding[5]. It provided the first comprehensive insight into octoploid genome architecture, revealing a dominant subgenome derived from the F. vesca lineage that disproportionately influences key agronomic traits. Building on this foundation, and enabled by advances in sequencing technologies, particularly the widespread adoption of long-read sequencing and improved assembly algorithms, high-quality reference genomes have been developed for additional cultivars, including 'Yanli'[7], 'Benihoppe'[3,8], 'EA78'[9], 'Florida Brilliance'[10], 'Royal Royce'[11], 'Seolhyang'[12], 'Chulian'[13], and FL17.68–110[14], as well as for several wild progenitors, including F. vesca 'Ruegen'[15] and accessions of F. chiloensis and F. virginiana[16]. These genomes have helped elucidate fundamental biological questions related to subgenome structural variation, biased allelic expression, centromeric sequence expansion and divergence, and the genetic basis of variation and evolution in genes underlying critical agronomic traits.
Despite this progress, the corresponding gene annotations remain suboptimal. Most assemblies still define only one mRNA per locus and lack comprehensive untranslated regions. For example, current gene annotations for various strawberry genomes struggle to accurately delineate complete transcript boundaries, often resulting in missing or incorrectly predicted gene models. In the case of the 'Benihoppe' strawberry, its initial genome annotation achieved a benchmarking universal single-copy orthologs (BUSCO) completeness score of approximately 96%[3], indicating that a subset of conserved single-copy orthologous genes was either absent or fragmented. Subsequently, a haplotype-resolved telomere-to-telomere (T2T) genome of 'Benihoppe' was assembled, with BUSCO completeness scores of 98.0%–98.4%[8]. However, large-scale full-length transcript evidence, essential for capturing alternative splicing and complete UTRs, remains lacking. 'Benihoppe', a cultivar developed in Japan from a cross between 'Akihime' and 'Sachinoka', is one of the most widely cultivated varieties in China, and is frequently used as a parent in Asian breeding programs[17]. These imperfections in its structural annotation have impeded research relying on this cultivar and limited the application of efficient molecular-assisted selection and genomic selection breeding strategies.
Recent technological advancements in sequencing, particularly the synergistic integration of long- and short-read RNA sequencing (RNA-seq), provide a powerful approach to substantially enhance genome annotation quality by capturing full-length transcripts and defining complete gene structures[18−21]. This study has two primary objectives: (1) to generate a substantially improved genome annotation (FxaBHv1.0.a2) for the economically important octoploid strawberry cultivar 'Benihoppe' by integrating 257 short-read and 33 long-read (PacBio/Nanopore) RNA-seq datasets that collectively span various tissues, developmental stages, and biotic/abiotic stresses; and (2) to leverage this high-fidelity genomic resource for an in-depth analysis of the transcriptional regulatory networks governing fruit development and ripening. By applying Mfuzz clustering and constructing a causal inference network using gene network inference with an ensemble of trees (GENIE3), key master regulators orchestrating this critical agronomic process were identified. This research not only delivers an essential updated resource for the strawberry research community but also provides novel insights into the molecular regulatory mechanisms underlying fruit development and maturation.
-
To construct a comprehensive transcriptomic atlas for the cultivated strawberry, 290 available RNA-seq datasets were compiled and curated (Supplementary Table S1). These datasets encompass nine distinct strawberry cultivars and cover major plant organs, including fruits, leaves, roots, flowers, stolons, and crowns. A significant portion of the data (56%) focuses on fruit, capturing key developmental and ripening stages, such as green, white, turning, full red, and over-ripe. Additionally, the dataset includes samples subjected to various external treatments, including the application of key plant hormones, notably abscisic acid (ABA) and auxin[22,23], as well as exposure to abiotic stresses such as drought[24], salinity[25], and biotic stress from Botrytis cinerea[26]. To harness the advantages of long-read RNA-seq, we incorporated 21 PacBio Iso-Seq libraries and 12 Oxford Nanopore PromethION libraries from our group, derived from various strawberry tissues, including fruit at different developmental stages[10,11,27,28]. Each experimental condition for short-read data is typically represented by two or three biological replicates, ensuring robust resources for downstream analyses.
Reannotation of the strawberry genome using a custom pipeline
-
To generate a high-quality reannotation of the 'Benihoppe' genome, a custom pipeline integrating comprehensive sequencing data was developed (Fig. 1). Initially, PacBio Iso-Seq and Oxford Nanopore PromethION long-read RNA-seq data were aligned to the reference genome using minimap2 (v2.28-r1209; parameters: -ax splice:hq -uf for Iso-Seq and -ax splice for Nanopore data). To ensure consistency with the subsequent network analysis, the genome reported by Song et al.[3] was chosen for use. Concurrently, bulk short-read RNA-seq data were aligned using HISAT2 (v2.2.1) with default parameters. The resulting BAM alignment files were used for genome-guided transcriptome assembly with Trinity (v2.8.5). In parallel, an initial set of gene predictions was generated from the genome sequence via ab initio prediction using Helixer (v0.3.4), which employs deep neural networks and hidden Markov models trained on thousands of existing genomes[29]. These ab initio models were subsequently refined through two iterative rounds of the PASA pipeline[30], using both short- and long-read alignments as evidence to improve gene structures, particularly untranslated regions (UTRs) and intron boundaries. To ensure accuracy, all resulting isoforms were quantified against the bulk RNA-seq data using Kallisto (v0.46.1). Only alternative isoforms with expression levels exceeding 10% of the total isoform expression at a given locus were retained to create a high-confidence gene model set. This set was further manually curated using IGV-GSAman (v0.46.1) with RNA-seq alignment evidence to produce the final gene structural annotations.
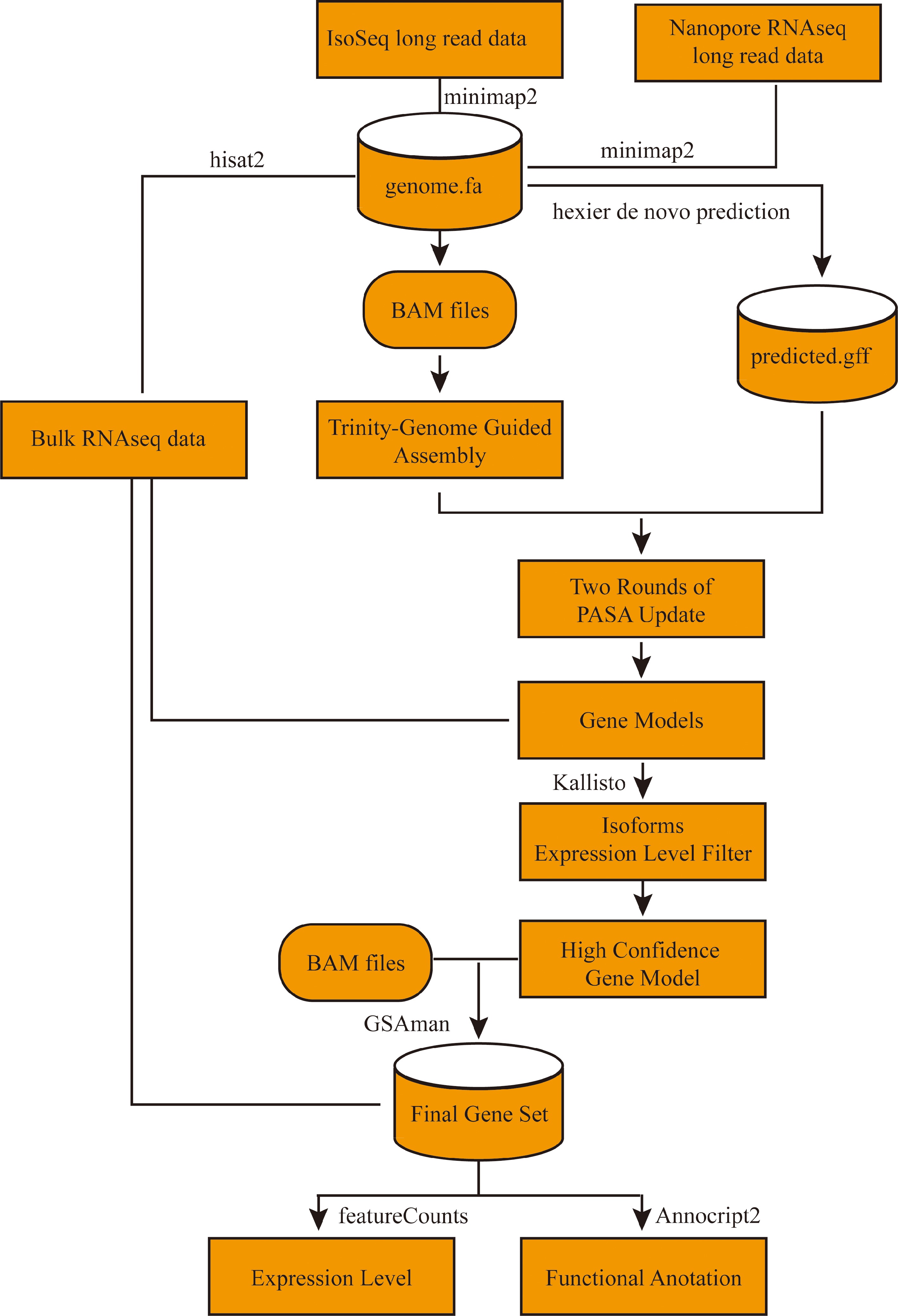
Figure 1.
Workfolw for the update annotation of octoploid strawberry 'Benihoppe'.
Functional annotation was performed using Annocript (v2.0)[31] through sequential homology searches against the Swiss-Prot (release 2025_01) and UniRef90 (release 2025_01) databases using DIAMOND (v0.9.51) with an E-value threshold of 10-7. InterProScan (v5.75) was employed to identify known protein functional domains. Redundant annotations were removed, and the results were integrated. KEGG pathway assignments were obtained via the online KAAS server (accessed January 10, 2025, using BBH mode). The final annotation table, encompassing functional annotations, pathway information, Gene Ontology (GO) terms, domain structures, and non-coding RNA evidence, was compiled into a single SQL database. The FxaBHv1.0.a2 annotation files are publicly available at
https://doi.org/10.6084/m9.figshare.30271885.v1 and have been deposited in the Genome Database for Rosaceae (GDR,www.rosaceae.org ) under Accession No. tfGDR1089.RNA-seq data analysis
-
RNA-seq data from fruit and achene tissues at various developmental stages were selected for downstream analysis. Paired-end reads were quality-controlled using fastp (v0.23.4), which removed adapters and primers, filtered out reads with a mean base quality below Q20, and trimmed low-quality bases from both ends. Only reads with a remaining length of at least 50 base pairs were retained. Prior to mapping, the reference genome was indexed using the updated FxaBHv1.0.a2 annotation file to define splice junctions. The processed paired-end clean reads were then mapped to the reference genome using STAR (v2.7.11b) in two-pass mode. The resulting BAM alignment files were used to quantify gene expression levels with featureCounts (v2.0.1). The raw read counts obtained were used for all subsequent analyses.
Regulatory network inference and master transcription factors mediating fruit development and maturation
-
To identify key transcriptional regulators of strawberry fruit development and maturation, an integrative workflow combining co-expression and causal network inference was employed. First, genes were identified by detecting those with dynamic expression profiles across developmental stages (green, white, turning, and red) using a likelihood ratio test (LRT) implemented in DESeq2 (adjusted p-value < 0.01)[32]. These genes were subsequently clustered based on their temporal expression patterns using the soft-clustering method Mfuzz[33], in fruit receptacles and achenes separately. Clusters exhibiting clear up- or down-regulation trends in fruit receptacles during development were selected. Next, a combined achene–receptacle co-expression network (unsigned) was constructed using the weighted gene co-expression network analysis (WGCNA) package[34], focusing on a curated list of expressed transcription factors (TFs), and all dynamically expressed genes. By correlating module eigengenes with traits such as developmental stage and tissue type, modules of interest were identified, including one specifically active in early-stage achenes, and another in late-stage receptacles.
To identify high-confidence master regulators of strawberry fruit receptacle maturation, a transparent, evidence-layered weighted scoring system was implemented that integrated four independent lines of evidence: (1) Significant temporal co-expression with the maturation-related gene set in receptacle (+1 point); (2) Differential expression in receptacle between green and ripe stages (FDR < 0.05 and |log2FC| > 1) (+1 point); (3) Strong hub status in the combined achene and receptacle WGCNA network (module membership |kME| > 0.8 in the maturation-associated module) (+2.5 points); (4) Ranked among the top 50 regulators of the maturation gene set in the fruit-specific and cross tissue (achene and receptacle) GENIE3 inference[35] (+3 points). This scoring system put higher weight in the network infrastructure rather than on a single gene status. The final total score for each transcription factor was calculated as the sum of the above weights. The TFs with the highest scores were selected as top candidate master regulators. This integrative approach enabled the robust identification of high-confidence TFs predicted to control strawberry fruit development and maturation.
Validating the accuracy of the updated annotation
-
To experimentally validate the accuracy of the revised gene models, four representative loci exhibiting major structural updates were selected. Locus-specific primers were designed to produce amplicons of distinct lengths depending on whether the original or the updated annotation was correct. PCR reactions (20 μL) contained 1 μL each of forward and reverse primer (Supplementary Table S2), 1 μL of cDNA synthesized from total RNA of small green 'Benihoppe' fruit (with achenes), and 10 μL of Premix Ex Taq II (Takara, Dalian, China). Amplification was performed on a T100 Thermal Cycler (Bio-Rad) using standard amplification protocol anealing at 55 °C. Following amplification, 5 μL of each PCR product was separated by electrophoresis in a 1% agarose gel.
-
The 'Benihoppe' strawberry is a key cultivar widely used in research and breeding, but its existing genome annotation (v1.0.a1) contained numerous mislabeled or incomplete gene features. To systematically address these issues, a large collection of RNA-seq data from the present research group, and publicly available datasets[10,11,27,28], were integrated to update the genome annotation. These datasets ensure the capture of a broad range of transcripts under diverse conditions. Long-read RNA-seq data were also used for improving annotation quality[10,11,28]. An initial de novo gene prediction was performed using Helixer (v0.3.4), a tool demonstrated to offer high accuracy and sensitivity[36]. This approach predicted 101,937 candidate gene features. All RNA-seq reads were aligned to the reference genome using splice-aware mapping, achieving mapping rates of 90%–98.4%. Genome-guided transcriptome assembly using these alignments yielded 607,695 transcripts, which served as input for iterative refinement of Helixer gene models via the PASA pipeline. This process generated 233,672 gene models with 309,276 associated untranslated region (UTR) annotations. To retain only the most reliable gene models, isoform expression levels were quantified using Kallisto (v0.46.1), and only the structures of dominantly expressed isoforms were retained. The resulting annotations were manually curated using IGV-GSAman, referencing BAM alignment files and the original genome GFF3 file. Over 6,000 genes were manually corrected, primarily to address inaccurate intron boundaries, overly long UTRs lacking read support, short intron (< 10 bp) retentions, and gene fusions. The BHv1.0.a2 annotation has been deposited in the Genome Database for Rosaceae (GDR,
www.rosaceae.org ), under Accession No. tfGDR1089.Evaluation of the updated FxaBHv1.0.a2 annotation
-
FxaBHv1.0.a2 was compared with the accompanying original annotation, as well as the phased 'Benihoppe' assembly (Table 1). The updated annotation comprises 103,316 genes, fewer than the original v1.0.a1 annotation, and the haplotype one, but with a longer average gene length (3,211 bp vs 2,462/2,932 bp). This increase is primarily attributed to two factors: (1) the merging of originally adjacent gene pairs, as illustrated in Fig. 2a; and (2) the extension of gene models supported by confident read-mapping evidence (Fig. 2d). Additionally, some exons were expanded to correct errors in the original annotation (Fig. 2c). In rare instances, original genes were split into two independent genes, each supported by distinct read coverage and lacking bridging reads to connect them (Fig. 2b). The curated genes encode 113,259 mRNAs, indicating that approximately 10% of genes have multiple high-confidence transcripts in the updated annotation. This number was fewer than that of the haplotypic version. Nearly half of the original single-exon genes were revised to include multiple exons, which was similar to the haplotypic gene definition. The most significant improvements were observed in UTR annotations, with 89% of transcripts now annotated with UTR features at one or both ends, largely due to the incorporation of long-read RNA-seq data. A total of 33,923 coding sequences (CDS) remained identical between the original and the updated annotations. Consistent with these changes, the average annotation edit distance (AED) was higher for mRNAs (0.434) than for CDS (0.366), reflecting greater modifications in non-coding regions. Detailed feature statistics are provided in Table 1.
Table 1. Annotation feature comparison among the initial release, haplotype release and the updated version in this study.
v1.0.a1 v1.0H
(haplotype)v1.0.a2
(this study)Number of genes 109,320 116,232 103,316 Number of mRNAs 109,320 175,313 113,259 Mean length of genomic loci 2,462 2,823 3,211 Mean exon number 4.5 5.5 5.6 Mean CDS length 1,049 1,232 1,169 Single-exon gene number 31,703 29,344 17,048 Multi-exon gene number 77,617 86,888 86,268 Genes with 5'UTR 37,673 45,601 102,967 Genes with 3'UTR 42,605 46,873 101,449 Genes with both 5'UTR and 3'UTR 31,198 40,542 101,404 Mean 5'UTR length 173 371 248 Mean 3'UTR length 350 631 414 Shortest gene length 228 155 104 Longest gene length 151,412 808,995 67,370 Shortest intron length 4 20 20 Longest intron length 50,784 483,249 29,051 Complete BUSCOs (%) 96.2 98.7 99.5 Fragmented BUSCOs (%) 1.1 0.3 0 Missing BUSCOs (%) 2.7 1.0 0.5 Genes with GO terms 54,718 NA 64,266 Genes with KEGG assignment 20,318 NA 43,732 
Figure 2.
Examples of modified gene structures. (a) IGV view of genes Fxa6B02488 and Fxa6B02489 merged into one gene, FxaBH_6B_003577. (b) IGV view of the gene structure for Fxa1Bg01984, split into two neighboring independent genes, each with markedly distinct read coverage. (c) IGV view of the modified FxaBH_7D_000580 gene, updated by extension of the second exon of the original Fxa7Dg01200 gene. (d) Correction of defects associated with incomplete 3' ends of the Fxa6Cg02195 gene. (e) Schematic representation of primer binding sites and expected product sizes. (f) Agarose gel electrophoresis of locus-specific PCR products.
The completeness of the updated FxaBHv1.0.a2 genome annotation was evaluated using BUSCO (v5.4.3) with the embryophyta_odb10 dataset. The analysis revealed significant improvements over the original v1.0.a1 and the haplotypic annotations, with complete BUSCOs increasing from 96.2%(98.7%) to 99.5%, and fragmented BUSCOs decreasing from 1.1%(0.3%) to 0%. Additionally, missing BUSCOs were reduced from 2.7%(1.0%) to 0.5%, indicating a more comprehensive and accurate annotation set. Both amplification size and Sanger-sequenced PCR products from green fruit cDNA consistently matched the sizes and sequences predicted by the present improved annotation (Fig. 2e, f). These results confirm the presence of previously missing exons, corrected splice junctions, and large-scale structural rearrangements in the updated gene models, thereby verifying the enhanced accuracy of FxaBHv1.0.a2.
Functional annotation of the FxaBHv1.0.a2 genes
-
Functional annotation of the proteins encoded by the updated 'Benihoppe' genome (FxaBHv1.0.a2) was performed using a multi-step homology and domain search strategy, adapting the Annocript pipeline (v2.0). TransDecoder (v5.7.0) was used to predict protein-coding regions, generating peptide sequences, coding sequences (CDS), GFF3 files, and non-coding RNA (ncRNA) candidates by systematically excluding coding regions from the input transcripts. Protein sequences underwent a hierarchical homology search in series databases, with unmatched sequences progressively filtered to ensure high-confidence annotations. Given Swiss-Prot's manually curated, experimentally validated protein annotations, hits to this database were prioritized for their superior accuracy over the automated TrEMBL and UniRef90 databases. Consequently, 72,659 genes (70.3% of the 103,316 total genes) were assigned homologs in Swiss-Prot, reflecting reliable protein function inferences. Sequences unmatched in Swiss-Prot were further searched against eggNOG and other databases. InterProScan identified known protein domains or features for 64,266 genes, which were assigned GO terms. Additionally, 43,732 genes were assigned KEGG Orthology identifiers. Non-coding candidates were scanned against the Rfam database using Infernal (v1.1.4) to identify ncRNAs, resulting in 6,167 ncRNA-encoding genes, each exhibiting high identity to at least one Rfam database entry. By integrating all results, 93,041 genes (90.0%) in the FxaBHv1.0.a2 annotation received at least one functional annotation. These results were compiled into a portable SQLite database, publicly available at figshare:
https://doi.org/10.6084/m9.figshare.30271885.v1 , facilitating reproducible and scalable downstream analyses.Identification of key regulators orchestrating the development and maturation of strawberry fruits
-
Interactions between achenes and receptacles are essential for initiating fruit development in F. × ananassa, although their roles in later fruit maturation remain unclear[37]. To elucidate the genetic regulators underlying fruit growth and maturation, RNA-seq data from achenes and receptacles across four sequential developmental stages (green, white, turning, and red) were analyzed. It was hypothesized that genes encoding key regulatory factors would exhibit significant expression changes in at least one stage compared to others in either tissue. Genes with an expression range (maximum–minimum) of less than two after variance-stabilizing transformation were filtered out, leaving 33,585 genes. These genes were clustered based on similar expression patterns using Mfuzz, one for receptacle samples and one for achene samples. A total of 18 clusters were generated in both tissues, with the receptacle one shown in Fig. 3a. The clustering pattern of genes in achenes can be found in Supplementary Fig. S1. As a general principle, it was assumed that receptacle genes with increasing expression trends are positively associated with fruit development and maturation, while those peaking at early stages and declining thereafter are related to early fruit development or negatively associated with receptacle maturation. GO and KEGG pathway enrichment analyses were performed for each cluster. Notably, cluster 5 showed significant over-representation of maturation-related pathways, including a large number of genes involved in flavonoid biosynthesis and metabolism (Fig. 3b). In contrast, cluster 14 was enriched for genes associated with cell wall component synthesis and metabolism, which are primarily linked to early-stage fruit growth (Fig. 3c). These genes were pooled to serve as a candidate list for identifying key regulators in subsequent analyses.
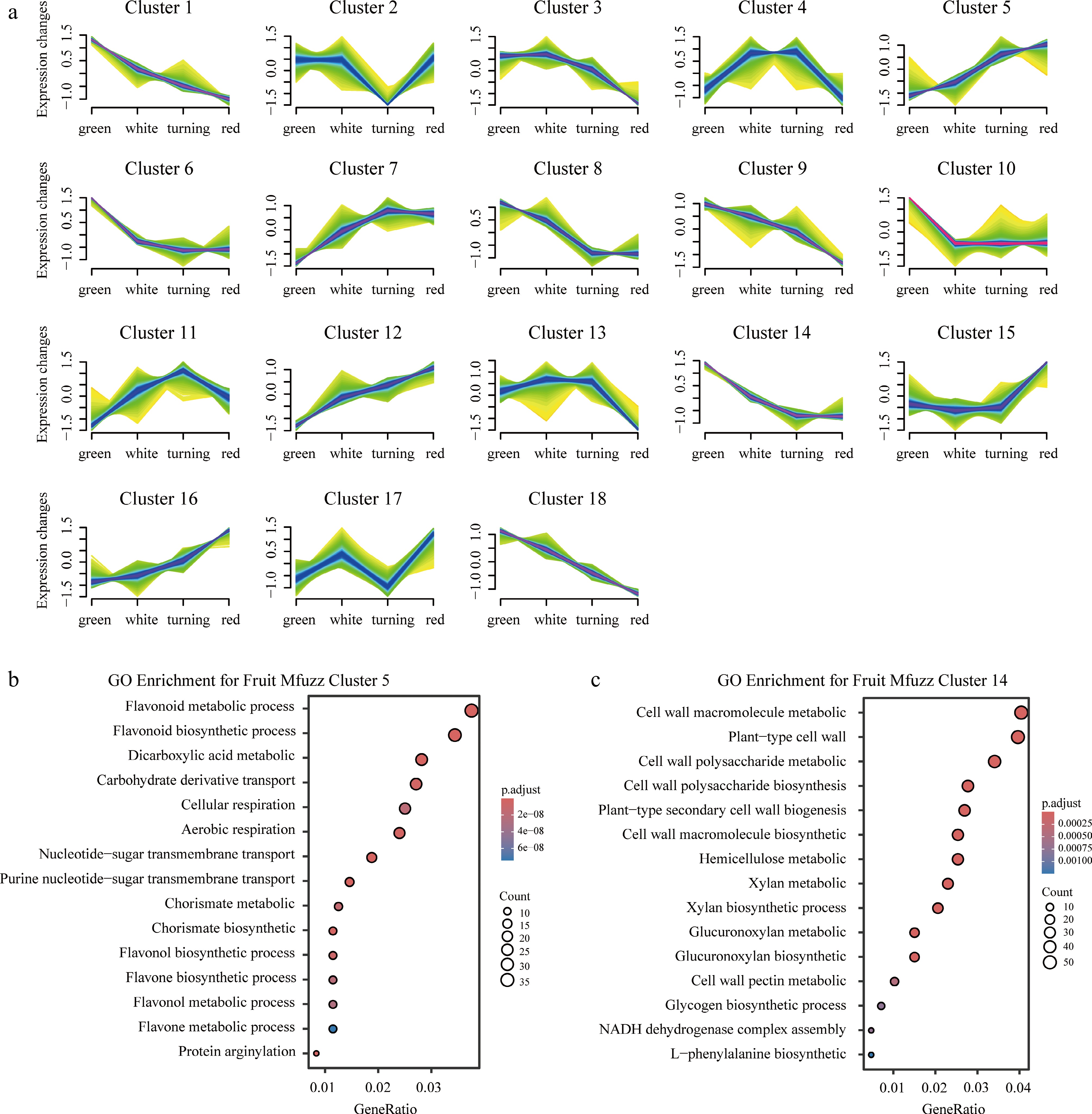
Figure 3.
(a) Mfuzz soft-cluster heat map, and (b), (c) GO/KEGG enrichment bubble plots for clusters 5 and 14 of the receptacle. Color intensity indicates relative expression; bubble size represents gene ratio; color scale denotes adjusted p-value.
For a second round of gene cluster/module identification, WGCNA analysis was also carried out with the variance stabilizing transformation expression values for the same 33585 gene set. To enhance the robustness and interpretability of the co-expression results, a combined expression matrix of genes in both the receptacle and achenes was used. Twenty-seven co-expression modules were generated, with at least 47 genes, or at most 4,880 genes in the modules. To prioritize modules relevant to genes critical for both fruit development and maturation, hypergeometric enrichment tests were performed to identify modules enriched for the candidate genes picked from the above Mfuzz clusters. The pink module, highly correlated with both the red stage (r = 0.43, p = 0.02), and the receptacle tissue (r = 0.73, p = 4e-9), was enriched for cluster-5 and cluster-14 genes. Genes in the module were further analyzed to extract high-confidence transcription factors (TFs) with strong module membership (kME > 0.8), yielding 45 high-confidence TFs (Supplementary Table S3). Among these, predominant TF families included six ethylene-responsive transcription factors (ERFs), six zinc finger CCCH domain-containing factors, four MYB transcription factors, and four NAC transcription factors, which are likely critical for regulating fruit development processes due to their known roles in ethylene signaling, stress response, and developmental regulation. These TFs provide a prioritized list of candidates for further experimental validation.
To elucidate the regulatory network governing strawberry fruit maturation, GENIE3 was employed to infer causal regulatory networks, focusing on transcription factors (TFs) that regulate genes associated with fruit maturation. The green module, which showed a near-linearly increasing kME value along the fruit maturation process (Supplementary Fig. S2) and had the highest correlation to the red stage, was selected. This module included 197 TFs. GENIE3 was applied using these TFs as regulators against the rest of the fruit maturation gene set, producing the top 500 regulatory links (Supplementary Table S4). The top TF, FxaBH_1D_001688, encoding a zinc finger protein Sensitive to Proton Rhizotoxicity 1 (STOP1), which is critical for proton tolerance in Arabidopsis[38], showed an accumulated regulatory weight of 19.76, suggesting its key role in driving late-stage maturation. It was recently reported to directly control strawberry fruit ripening[39]. This excellent agreement strongly supports the predictive power of our approach and underscores the high priority of functionally characterizing the remaining hub TFs that currently lack experimental validation. The top 100 regulator-target interactions were plotted, as shown in Fig. 4a. This network highlights late receptacle TFs (dark red nodes, e.g., SCL8, XBAT31, MYB20, JA2, AL5, NBCL) regulating fruit target genes (gray nodes, e.g., GH3, ABI5, RPL30, BAM2, RNP1, CAC3, among others), emphasizing interactions in hormonal signaling (e.g., auxin via GH3, ABA via ABI5), ribosomal processes (e.g., RPL30), and cell wall modification (e.g., BAM2) during strawberry fruit maturation. Most of the TF genes have been experimentally validated to be involved in regulating fruit maturation (Table 2), but some have not yet been investigated.
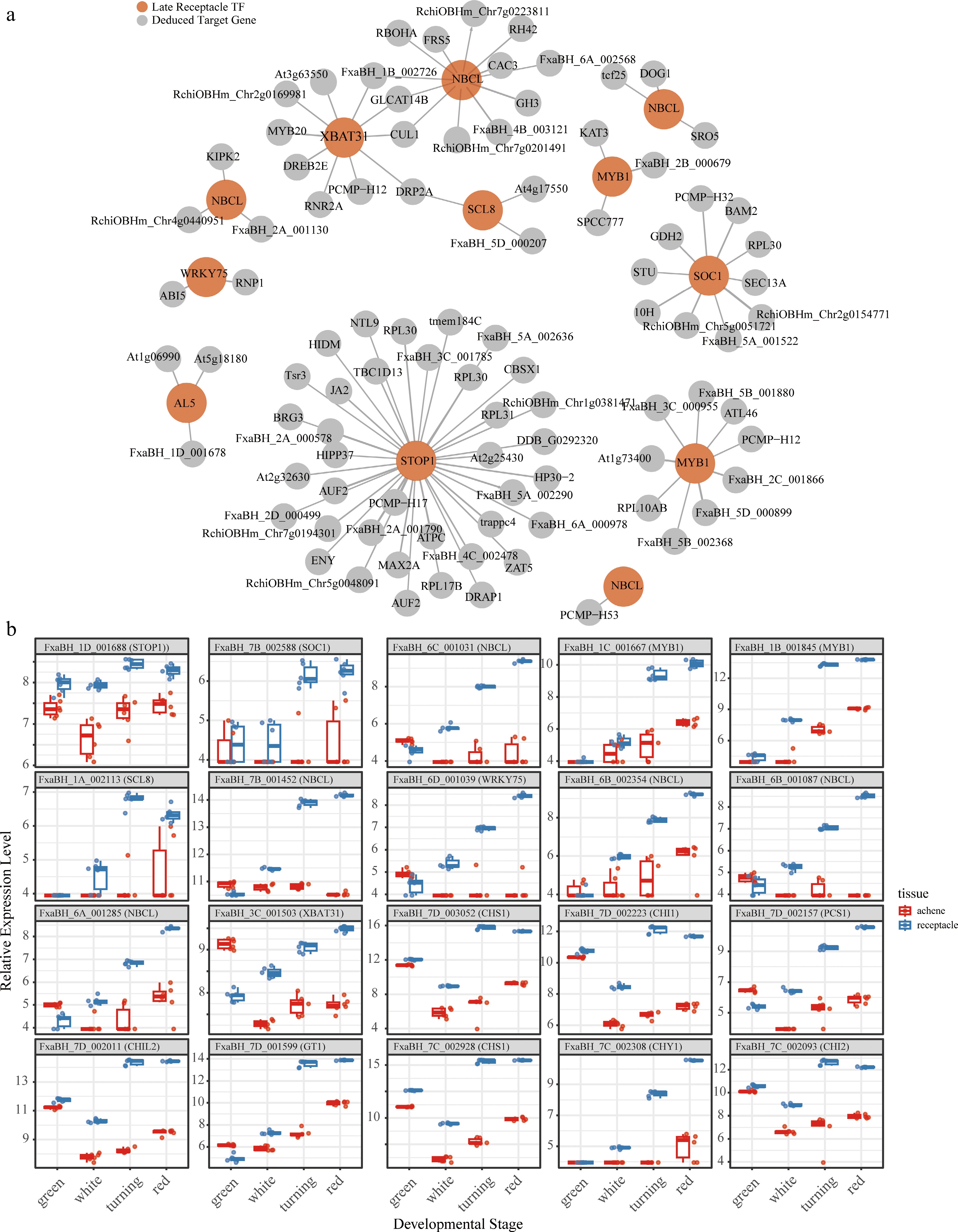
Figure 4.
(a) The top 100 regulator-target interactions unveiled in the preent study, and (b) relative expression patterns of the top 20 in receptacle and achene tissues from different developmental stages.
Table 2. TOP candidate genes in regulating strawberry fruit maturation.
GeneID TotalScore Functional description ShortName FxaBH_1D_001688 7.5 Protein SENSITIVE TO PROTON RHIZOTOXICITY 1 STOP1 FxaBH_7B_002588 7.5 MADS-box protein SOC1 SOC1 FxaBH_6C_001031 7.5 BTB/POZ domain and ankyrin repeat-containing protein NBCL NBCL FxaBH_1C_001667 7.5 Transcription factor MYB1 MYB1 FxaBH_1B_001845 7.5 Transcription factor MYB1 MYB1 FxaBH_1A_002113 7.5 Transcription factor MYB1 MYB1 FxaBH_7B_001452 6.5 Scarecrow-like protein 8 SCL8 FxaBH_6D_001039 6.5 BTB/POZ domain and ankyrin repeat-containing protein NBCL NBCL FxaBH_6B_002354 6.5 Probable WRKY transcription factor 75 WRKY75 FxaBH_6B_001087 6.5 BTB/POZ domain and ankyrin repeat-containing protein NBCL NBCL FxaBH_6A_001285 6.5 BTB/POZ domain and ankyrin repeat-containing protein NBCL NBCL FxaBH_3C_001503 6.5 Putative E3 ubiquitin-protein ligase XBAT31 XBAT31 FxaBH_7D_003052 4.5 Chalcone synthase CHS1 FxaBH_7D_002223 4.5 Chalcone--flavanone isomerase 1 CHI1 FxaBH_7D_002157 4.5 Aspartic proteinase PCS1 PCS1 FxaBH_7D_002011 4.5 Chalcone isomerase-like protein 2 CHIL2 FxaBH_7D_001599 4.5 Anthocyanidin 3-O-glucosyltransferase 1 GT1 FxaBH_7C_002928 4.5 Chalcone synthase CHS1 FxaBH_7C_002308 4.5 3-hydroxyisobutyryl-CoA hydrolase 1 CHY1 FxaBH_7C_002093 4.5 Chalcone--flavanone isomerase 2 CHI2 FxaBH_7C_001900 4.5 Chalcone isomerase-like protein 2 CHIL2 FxaBH_7C_001470 4.5 Anthocyanidin 3-O-glucosyltransferase 1 GT1 FxaBH_7B_003068 4.5 Chalcone synthase CHS1 FxaBH_7B_002249 4.5 Chalcone -flavanone isomerase 2 CHI2 FxaBH_7B_002068 4.5 Chalcone isomerase-like protein 2 CHIL2 FxaBH_7B_000048 4.5 Polygalacturonase FxaBH_7A_003557 4.5 Chalcone synthase CHS1 FxaBH_7A_003556 4.5 Chalcone synthase CHS1 FxaBH_7A_003555 4.5 Chalcone synthase CHS1 FxaBH_7A_002558 4.5 Chalcone--flavanone isomerase 1 CHI1 To prioritize candidate genes for in-depth functional validation in strawberry fruit maturation, an integrative approach was employed by assigning different weight to the above described analytical layers. This system evaluates all genes, extending beyond transcription factors to include structural, enzymatic, and signaling genes, thereby identifying a comprehensive set of high-priority targets for experimental studies, such as CRISPR editing or overexpression assays in the future. The top 30 candidate genes are listed in Table 2. Several well-known anthocyanidin biosynthesis pathway structural genes, including CHS, CHI, CHIL2, and GT1 paralogs, were consistently recovered. Three MYB1-encoding genes (FxaBH_1C_001667, FxaBH_1B_001845, FxaBH_1A_002113), which share very high sequence identity with the extensively reported MYB10 protein, a master regulator of color formation[40,41], were ranked at the top of this list.
The expression levels of the top 20 candidate genes were investigated across different developmental stages of strawberry fruit (Fig. 4b). As expected, most of these genes peaked in expression during the receptacle ripening stages, indicating their specific roles in regulating maturation. Interestingly, several of them also exhibited high abundance in early achene tissue. For example, FxaBH_3C_001503, which encodes an E3 ubiquitin-protein ligase XBAT31 reached its highest abundance in the achenes, surpassing levels observed in the receptacle tissues (Fig. 4b). This gene is a known positive factor mediating thermotolerance in the reproductive organs of Arabidopsis under heat stress conditions[42]. The underlying mechanism of its involvement in the fruit maturation process warrants further investigation.
-
Accurate genome annotation is essential for precise gene expression analysis, and supports applications like molecular breeding and functional genomics. In the past five years, the strawberry genome assembly has been significantly improved[3,5,7,9,10,12,15,16], yet annotations remain suboptimal. For example, the phase-resolved 'Benihoppe' strawberry genome defines only one mRNA per gene, with UTRs annotated for just 30% of genes[3], while the Haplotype-resolved 'Benihoppe' genome predicted gene models covering 98.0%–98.4% BUSCO homologes[8]. Conventionally, two main strategies are integratively used for genome annotation. The first employs ab initio tools like AUGUSTUS or GeneMark[43], which predict gene structures using Hidden Markov Models. Recently, AI-driven tools such as DeepAnnotator and Helixer have impressive prediction accuracy and efficiency using machine and deep learning[29,44]. The second strategy, considered more reliable, uses experimental evidence from expressed sequence tags, RNA-seq reads, or homologous gene sequences. Short-read RNA-seq, while widely used, requires complex computational assembly, often producing incomplete or chimeric transcripts. In contrast, long-read RNA-seq captures full mRNA sequences in a single read, also reveals alternative splicing isoforms. Recent strawberry genome reannotations demonstrate the value of integrating these two data types. The diploid F. vesca reannotation, using Illumina and SMRT RNA-seq, increased BUSCO completeness from 91.1% to 98.1%, resolved fragmented models, and added UTRs to 59.1% of genes[45]. The Camarosa reannotation, combining PacBio and Illumina data, annotated 108,447 genes with 97.85% BUSCO completeness, improved UTRs for 79.9% of genes, and identified more transcription factors[21]. The haplotype-resolved 'Yuexin' assembly annotated 110,776 genes with 99.07% BUSCO completeness, revealing structural variants linked to quality traits[46]. In this study, Helixer was used for initial gene prediction, refined with 257 short-read and 33 long-read RNA-seq datasets, achieving 99.5% BUSCO completeness, an average gene length of 3,211 bp. Compared to Camarosa v1.0.a2 (97.85% BUSCO), and Yuexin (99.07%), the present annotation offers superior UTR coverage (89%), and no fragmented BUSCOs, highlighting the benefits of extensive transcriptome sampling. We have to mention that several haplotype-resolved genomes and associated annotations were available[13,14,46]. They provided indispensible data for studying allele-specific expression and structural variants. However, current haplotype-aware alignment and quantification tools remain limited and immature for highly homozygous octoploid genomes. Consequently, haplotype-resolved gene annotation and expression quantification still introduce considerable uncertainty. A conservative subgenome-collapsed strategy was therefore adopted to maximize accuracy in network inference, and anticipate that future advances in haplotype-aware pipelines will unlock the full potential of these high-quality phased assemblies.
The development and ripening of the strawberry accessory fruit is a complex process, orchestrated by an intricate interplay between phytohormones and transcription factors, particularly through signaling between the achenes and the receptacle. It's well-established that achene-derived auxin promotes early receptacle growth while repressing ripening[47]. Previous studies have identified key TFs like RGA1, ARF8, and ARF6 that mediate auxin and gibberellin signaling during these initial stages[47,48]. The present findings provide a deeper mechanistic insight into the transition away from this auxin-dominant phase. The identification of ARF6 within our ripening-associated pink module aligns with its known role in auxin dynamics. More significantly, our GENIE3 regulatory network revealed that top-ranked TFs directly target GH3.6, an auxin-conjugating enzyme. Among these, the novel hub NBCL acts as an adapter of the E3 ubiquitin-protein ligase complex CUL3-RBX1-BTB[49]. This suggests a key ripening strategy involves the active post-translational suppression of auxin signaling to permit maturation. This decline in auxin appears to be a prerequisite for the ascent of abscisic acid, the primary driver of ripening[50]. The interplay between these hormones is critical, as demonstrated by ARF2's repression of the ABA biosynthesis gene FaNCED3[51]. The present network analysis uncovers a complementary regulatory layer: as auxin levels fall, factors like NBCL and STOP1 become active and directly target core ABA signaling components, including ABI5 and the ABA receptor PYL12, respectively. This creates a robust molecular switch that not only removes the auxin 'brake' but also actively 'accelerates' ABA-mediated ripening processes. Once ABA signaling is initiated, a cascade of downstream TFs executes the ripening program.
Recent studies have established the NAC transcription factor FaRIF as a master regulator of strawberry fruit ripening, where it directly activates genes involved in anthocyanin biosynthesis, sugar metabolism, and ABA signaling[52,53]. Additional NAC family members, such as NAC073, antagonistically work with CMB1L to control sucrose accumulation[54], while the ERF3–NAC073 regulatory cascade coordinately modulates both fruit growth and ripening[55]. These findings highlight the extensive crosstalk and hierarchical organization among transcription factor families in orchestrating comprehensive regulatory networks for strawberry fruit maturation. Using an integrative multi-evidence scoring approach, we identified NAC098 as a central hub regulator of receptacle maturation. The same framework confidently prioritized known key players, including a MYB1 homolog of the well-characterized anthocyanin master regulator MYB10, as well as SOC1, which are implicated in flavonoid biosynthesis[40], and developmental timing[56]. In summary, the present study positions TFs like NBCL and STOP1 as pivotal, high-level integrators of the auxin-to-ABA hormonal transition that defines strawberry ripening. These hubs, along with key downstream effectors like NAC, bZIPs, and MYB TFs, form a cohesive transcriptional network controlling fruit quality traits. Their high ranking in the present analysis underscores their potential as prime targets for CRISPR-based functional validation to enhance strawberry agronomic performance.
-
With the implementation of 257 short-read, and 33 long-read RNA-sequencing data, the genome annotation of the cultivated strawberry 'Benihoppe' was significantly improved. In this new v1.0.a2 annotation, approximately 10% of genes have multiple high-confidence transcripts, and 89% of transcripts are now annotated with UTR features at one or both ends. BUSCO evaluation further validated the completeness of this new annotation. Based on this new genomic information, gene regulatory network analysis revealed several key transcription factors like STOP1 and NBCL, integrating hormone signalling crosstalk between auxin and ABA in orchestrating strawberry fruit development and maturation.
-
The authors confirm contributions to the paper as follows: study conception and design, funding acquisition, resources: Chen Q; methodology: Ma W, Wang Y; software: Ma W, Wang Y, Lin Y; validation, investigation, data curation: Zhang Y, Luo Y; writing − original draft preparation: Ma W, Chen Q; review and editing: Tang H, Chen Q; formal analysis, visualization, supervision: Ma W, Chen Q. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
This work was financially supported by grants from the National Natural Science Foundation of China (Grant No. 32572957).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/frures-0026-0001.
- Supplementary Table S1 RNAseq data used in this study.
- Supplementary Table S2 Primers used in this study.
- Supplementary Table S3 Key bridging transcription factors that colosely related to red stage and receptacle tissue.
- Supplementary Table S4 Top 500 regulatory links of the key TFs and its deduced target from GENIE3 analysis.
- Supplementary Fig. S1 Mfuzz soft-clustering pattern of genes in achenes.
- Supplementary Fig. S2 Eigengene expression pattern in the green module during strawberry fruit development.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Ma W, Wang Y, Lin Y, Zhang Y, Luo Y, et al. 2026. Update of the octoploid strawberry genome annotation and gene regulatory network analysis revealed key factors in strawberry fruit maturation. Fruit Research 6: e010 doi: 10.48130/frures-0026-0001 

Update of the octoploid strawberry genome annotation and gene regulatory network analysis revealed key factors in strawberry fruit maturation
- Received: 07 October 2025
- Revised: 03 December 2025
- Accepted: 29 December 2025
- Published online: 25 March 2026
Abstract: The existing genome annotations for the strawberry cultivar 'Benihoppe' contain incomplete or inaccurate gene models, particularly lacking alternative isoforms and complete untranslated regions, which impedes precise transcriptomic analysis. Here FxaBHv1.0.a2 is presented, a substantially improved genome annotation generated by integrating 257 short-read, and 33 full-length PacBio/Nanopore RNA-seq libraries that encompass diverse tissues, developmental stages, and abiotic stress treatments. Benchmarking Universal Single-Copy Orthologs (BUSCO) completeness increased from 96.2% to 99.5%, fragmented models were eliminated, and 89% of genes now possess complete 5′ and 3′ untranslated regions. Comprehensive functional information was assigned to 90% of the 103,316 protein-coding loci, and 6,167 ncRNAs. Leveraging this high-quality annotation, a high-confidence transcriptional network controlling fruit development and ripening was reconstructed. Using an integrative approach that combined Mfuzz clustering, Weighted Gene Co-expression Network Analysis (WGCNA), and Gene Network Inference with Ensemble of Trees (GENIE3) causal network inference, key regulators were identified and prioritised. The present results pinpoint several high-confidence candidate master regulators, including the known factor MYB1 (a MYB10 homolog), the recently validated SENSITIVE TO PROTON RHIZOTOXICITY 1 (STOP1), and a novel hub, a BTB/POZ domain and ankyrin repeat-containing protein (NBCL). These transcription factors are predicted to orchestrate the critical hormonal transition from auxin repression to abscisic acid (ABA)-driven maturation by targeting core components of their respective signaling pathways. This work not only provides a foundational genomic tool for the strawberry research community, but also delivers novel insights into the regulatory architecture of fruit ripening, identifying high-priority targets for future functional validation and crop improvement.
-
Key words:
- Strawberry /
- Genome annotation /
- Fruit maturation /
- Regulation network