






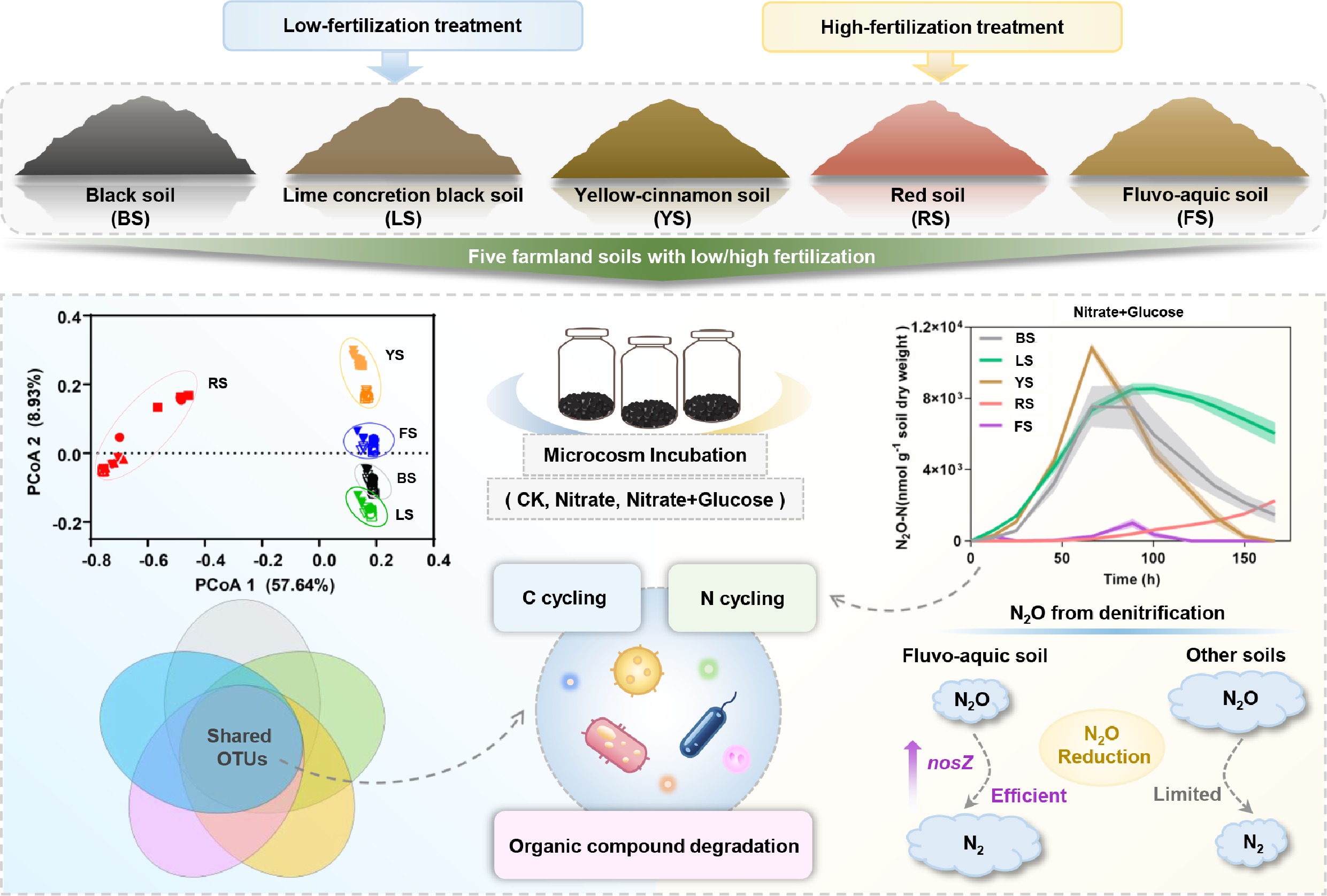



-
Soils harbor some of the most diverse microbial communities on Earth and play a fundamental role in regulating biogeochemical cycles essential to ecosystem functioning[1]. Soil physicochemical properties—particularly pH, organic carbon availability, and redox conditions—strongly shape microbial community structure and function expression, thereby constraining soil biogeochemical processes[2]. In China, farmland soils exhibit pronounced heterogeneity, with major soil types such as black soil, fluvo-aquic soil, and red soil differing substantially in texture, nutrient status, and acidity, thereby creating distinct microbial niches and driving divergent biogeochemical and denitrification phenotypes[3,4]. Core microbiomes—taxa consistently present across sites within a habitat—are increasingly recognized for sustaining ecosystem stability and multifunctionality[5,6], with recent studies showing their disproportionate contribution to nutrient cycling and resilience in both upland and paddy soils[7,8]. However, how intrinsic soil physicochemical properties shape the composition and functional potential of core microbial communities under varying carbon and nitrogen inputs remains largely unexplored.
Given the critical role of soil microbial communities in mediating biogeochemical cycles, their specific contributions to nitrogen cycling are of particular interest due to their environmental implications. Nitrogen cycling mediated by soil microorganisms is of particular environmental concern because it directly governs nitrous oxide (N2O) emissions. Nitrous oxide is a potent greenhouse gas with a global warming potential approximately 273 times that of CO2 over a 100-year horizon and is also a major contributor to stratospheric ozone depletion[9,10]. Agricultural soils account for nearly 60% of global anthropogenic N2O emissions, largely driven by microbial processes associated with fertilization practices[11,12]. Among these processes, denitrification—the stepwise microbial reduction of nitrate (NO3−) to dinitrogen (N2)—represents a major (and often dominant) pathway for N2O production in agroecosystems[13]. This process is catalyzed by a sequential suite of enzymes encoded by narG, nirK/nirS, norB, and nosZ, which encode nitrate reductase, nitrite reductase, nitric oxide reductase, and nitrous oxide reductase, respectively[13,14]. Previous studies have examined the effects of individual management practices—such as fertilization regimes, irrigation, nitrification inhibitors, and biochar amendments—on N2O emissions[15−18]. However, comparative investigations across contrasting soil types under controlled carbon and nitrogen input conditions remain scarce. As a result, a fundamental question remains unresolved: why does the same carbon and nitrogen input lead to systematically different N2O emissions across soils? Addressing this question requires disentangling the relative roles of soil physicochemical constraints and microbial community functional potential in regulating denitrification outcomes.
In this study, these knowledge gaps are investigated by five typical Chinese farmland soils spanning a wide range of physicochemical properties and fertilization histories. High-throughput 16S rRNA gene sequencing is combined with functional prediction (Functional Annotation of Prokaryotic Taxa, FAPROTAX), quantification of key denitrification genes (nirK, nirS, and nosZ), and controlled anaerobic microcosm incubations using a robotic system for continuous measurement of N2O and N2 fluxes. By examining microbial community composition, shared and soil-specific taxa, and denitrification gas partitioning under different carbon and nitrogen addition regimes, we aimed to elucidate how soil physicochemical backgrounds constrain denitrification phenotypes and how microbial functional heterogeneity mediates soil-specific responses. Overall, this study seeks to advance a mechanistic understanding of how interactions between soil physicochemical properties and microbial functional potential regulate the partitioning between N2O and N2 during denitrification. Such insights are essential for developing soil-specific strategies to mitigate agricultural N2O emissions and improve nitrogen management across diverse agroecosystem contexts.
-
Five distinct soil types were collected for incubation experiments in this study (Fig. 1a): (1) black soil from a long-term experimental field in Lishu County, Jilin Province; (2) lime concretion black soil from the North Anhui Experimental Station of Anhui Agricultural University; (3) yellow-cinnamon soil from the Nongcuiyuan Experimental Base of Anhui Agricultural University; (4) red soil from Dongting Village, Rongjiawan Town, Yueyang County, Hunan Province; and (5) fluvo-aquic soil from the long-term experimental field at Shangzhuang Experimental Station of China Agricultural University. Each of the five in situ farmland soils included two fertilization histories: low fertilization (LF) and high fertilization (HF). The low-fertilization soils had received no fertilizer input for at least two years before sampling, whereas the high-fertilization soils had received fertilizer inputs ranging from 100 to 240 kg N ha−1. In total, ten soil samples were collected and designated as follows: BFL and BFH represent black soil under LF and HF conditions, respectively; LFL and LFH represent lime concretion black soil under LF and HF conditions, respectively; YFL and YFH represent yellow-cinnamon soil under LF and HF conditions, respectively; RFL and RFH represent red soil under LF and HF conditions, respectively; and FFL and FFH represent fluvo-aquic soil under LF and HF conditions, respectively. For each soil type, subsamples were collected from five randomly selected points at 0–20 cm depth and thoroughly mixed to form a composite sample. All soil samples were placed in black plastic bags and stored at 4 °C before analysis. Before the incubation experiments, soils were passed through a 2 mm sieve to remove stones, plant residues, and coarse roots.
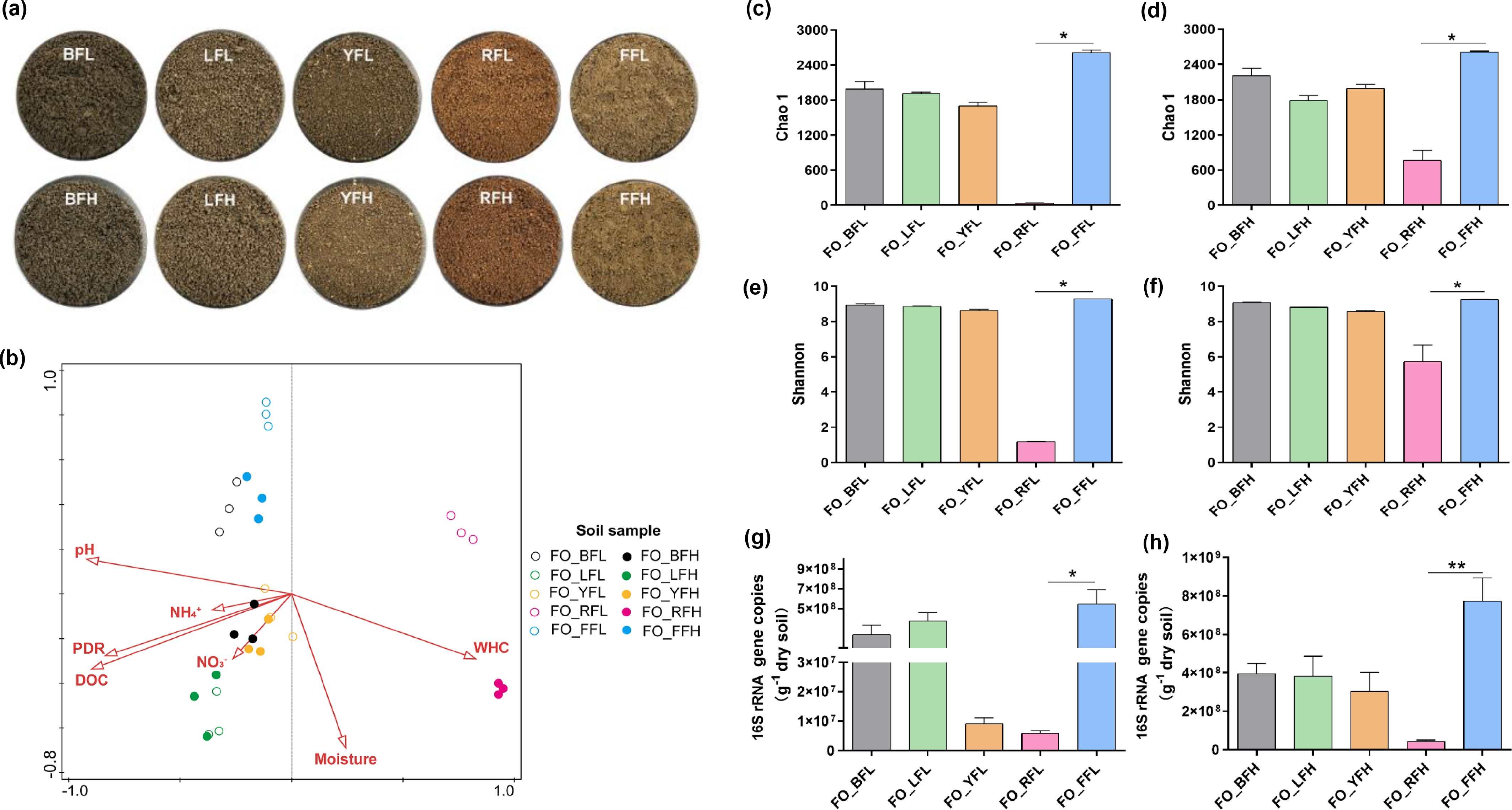
Figure 1.
Characterization of bacterial communities in five soils. (a) Physical appearance of the five soils after air-drying and sieving to 2 mm. (b) Relationships between environmental factors and bacterial community in the five soils of the FO group. (c)–(f) Alpha diversity indices of bacterial communities in the five soils of the FO group. (g), (h) qPCR quantification of total bacteria in the five soils of the FO group. * p < 0.05, ** p < 0.01.
Experimental design and microcosm incubation
-
This study aimed to compare bacterial community composition and functional metabolic potential across five soils under varying carbon and nitrogen conditions. For each soil, 20 g (dry weight equivalent) of soil was placed into 120 mL serum vials and aerobically pre-incubated at 25 °C for 7 d to stabilize microbial communities and minimize initial pulse respiration. Following the 7-d aerobic pre-incubation, five soils were assigned to the FO group to determine baseline nitrate and dissolved organic carbon (DOC) concentrations before anaerobic incubation. Based on the initial nitrate levels measured in the FO group, three anaerobic treatments were subsequently established: (1) anaerobic incubation for 7 d without nitrate addition (CK); (2) addition of nitrate (KNO3) to 250 mg N kg−1 followed by anaerobic incubation (SN); and (3) the SN treatment coupled with a uniform glucose addition at a final concentration of 1,000 mg kg−1, regardless of initial differences in DOC (GN). Each treatment was performed in triplicate. Soil moisture content was determined before microcosm establishment, and the volumes of sterile water, KNO3 solution, and/or glucose solution added to each vial were adjusted to maintain soil moisture at 70% of the water-holding capacity (WHC) across all treatments. The required volumes were sprayed evenly onto the soil surface by moving the syringe needle around the vial opening. The vials were then immediately sealed with sterile butyl rubber septa and aluminum crimp caps. The vial headspace was subsequently evacuated and refilled with high-purity helium (99.999%) four times to establish strictly anaerobic conditions. All microcosms were incubated at 25 °C for 7 d in the dark. Due to the airtight sealing with butyl rubber septa and aluminum crimp caps, soil moisture was assumed to remain stable during the 7-d anaerobic incubation at 25 °C.
Measurement of denitrification gases and soil parameters
-
During the 7-d anaerobic incubation, headspace gas samples were collected from the serum vials every 4 h and analyzed for N2O and N2 concentrations using a robotic incubation system as described by Molstad et al.[19]. To characterize the proportion of N2O in the total denitrification gases (N2O + N2), an N2O production index was calculated as N2O/(N2O + N2) during anaerobic incubation, as described in previous studies[20]. To characterize the proportion of N2 in the total denitrification gases (N2O + N2), an N2 production index was calculated as 1 minus the N2O production index. Soil pH was measured in a 1:5 (w/v) soil-to-water suspension using a pH meter (Mettler-Toledo, Switzerland). DOC was determined using an Elab-TOC analyzer (Suzhou Elab Analytical Instrument Co., Ltd, China). Soil water content was measured by oven drying at 105 °C. Inorganic nitrogen species, including ammonium-N (NH4+-N), nitrite-N (NO2−-N), and nitrate-N (NO3−-N), were extracted from soil samples with 1 M KCl[21]. Nitrite concentrations in the extracts were quantified spectrophotometrically using the N-(1-naphthyl) ethylenediamine dihydrochloride method[22] before and after cadmium reduction, and the difference between the two measurements was taken as the nitrate concentration[23]. Soil ammonium-N was determined using the indophenol blue method[24]. Emission rates of soil N2O and N2 were calculated as the slopes of linear regressions of their respective concentration changes over time. The potential denitrification rate (PDR) was calculated as the sum of the N2O and N2 emission rates[25].
Quantification of denitrifying genes and 16S rRNA gene sequencing analysis
-
Genomic DNA was extracted from 0.3 g of frozen soil samples collected from the four treatment groups (FO, CK, SN, and GN) following previously described protocols[26,27]. The extracted DNA was used as the template for quantitative PCR (qPCR) amplification of bacterial 16S rRNA genes and denitrification functional genes, including nirK, nirS, and nosZ. All qPCR assays were performed using a Light Cycler 96 system (Roche, Basel, Switzerland). Detailed primer information and amplification conditions have been described previously[28].
Amplicon libraries targeting the V3-V4 regions of the bacterial 16S rRNA gene were constructed and sequenced on the MiSeq platform (Illumina Inc., USA). Sequence quality control and downstream processing were performed as described previously[29]. After quality filtering, 2,358,830 high-quality sequences were obtained, with sequence counts per sample ranging from 12,144 to 25,964. Representative operational taxonomic units (OTUs) were clustered using the UPARSE pipeline with default parameters[30]. Reference-based chimera detection was additionally performed using UCHIME[31] against the RDP classifier training database (version 9)[32]. An OTU table was generated by mapping quality-filtered reads to the representative OTU sequences using the USEARCH global alignment algorithm with a 97% similarity threshold[33]. Representative sequences of each OTU were taxonomically assigned using the RDP classifier (database version 2.10) with a confidence threshold of 80%. To ensure uniformity, sequences were normalized to 12,000 per sample. The bacterial community composition of the five farmland soil types was subsequently analyzed. Alpha and beta diversity analyses were performed using QIIME (version 1.8)[34]. Canonical correspondence analysis (CCA) was conducted to assess the relationships between physicochemical properties and bacterial community structures across the five soils.
Shared OTUs were defined as those detected simultaneously under the same treatment conditions across all five soils (or across four soils when red soil was excluded, due to its markedly distinct microbial community composition compared to the other four soils). Unique OTUs were defined as those detected exclusively in a single soil and absent from all other soils under the same treatment conditions. Functional profiles of shared OTUs across the five farmland soils were predicted using FAPROTAX.
Statistical analysis
-
Principal coordinate analysis (PCoA) of microbial communities across the five farmland soils was performed using MATLAB 2018a (MathWorks Inc., USA) and GraphPad Prism 8.0. Multivariate analysis of variance (MANOVA) was conducted in MATLAB 2018a to test for differences in bacterial community composition among soil types, fertilization histories, and incubation treatments. Significant differences among treatments for all measured soil variables were assessed using two-way analysis of variance (ANOVA).
-
The physicochemical properties of the five soils collected in situ are summarized in Supplementary Table S1. The pH values of soils subjected to high fertilization (BFH, LFH, YFH, RFH, and FFH) are consistently lower than those of their corresponding low-fertilization counterparts (BFL, LFL, YFL, RFL, and FFL), indicating that long-term fertilization led to soil acidification. Among the soil types, lime concretion black soil exhibited the highest pH and the highest potential denitrification rate, whereas red soil showed the lowest pH and the lowest potential denitrification rate (Supplementary Table S1). Nitrate content varied markedly among soils, with the highest in YFH (240.27 mg N kg−1), followed by LFH (107.29 mg N kg−1), and the remaining soils ranged from 2.51–26.11 mg N kg−1. Nitrite was not detected in any of the five soils at the time of sampling. Ammonium concentrations were higher in black and yellow-cinnamon soils than in red and fluvo-aquic soils, which exhibited comparatively lower levels. In addition, the DOC content of red soil was significantly lower than that of the other four soils (Supplementary Table S1).
Bacterial community compositions in soil samples
Relationships between community compositions and edaphic factors
-
Bacterial community structure differed markedly among the five farmland soils under both low- and high-fertilization treatments within the FO group (Supplementary Fig. S1). Notably, the bacterial community structure of red soil was significantly distinct from that of the other four soils (Supplementary Fig. S1a–S1d). Among the measured edaphic factors, soil pH and nitrate availability exerted significant influences on bacterial community composition. Soil pH explained 46.1% of the total variation in community structure, representing the primary driving factor, whereas nitrate accounted for an additional 9.6% of the variation (Fig. 1b).
In situ microbial community diversity in farmland soils
-
Microbial community richness (Chao1 index) and diversity (Shannon index) in red soil were consistently the lowest among the five soils under both low-fertilization (Fig. 1c, e) and high-fertilization (Fig. 1d, f) conditions, and were significantly lower than those observed in fluvo-aquic soil. Although microbial richness and diversity also varied among black soil, lime concretion black soil, yellow-cinnamon soil, and fluvo-aquic soil, these differences were not statistically significant (Fig. 1c–f). In addition, bacterial abundance differed among soils, with red soil exhibiting the lowest abundance and fluvo-aquic soil exhibiting the highest abundance across both fertilization regimes (Fig. 1g, h).
Effects of soil properties on the dominant bacterial communities
-
Bacterial community diversity showed similar patterns under low- and high-fertilization treatments (Fig. 2a), suggesting that fertilization had a limited influence on bacterial community diversity within each farmland soil. In contrast, PCoA based on Bray-Curtis distances, together with multivariate analysis of variance (MANOVA), revealed significant differences in bacterial community structure among the five farmland soils subjected to different carbon and nitrogen addition treatments (Fig. 2b). Notably, the bacterial community structure of red soil was distinctly separated from those of black soil, lime concretion black soil, yellow-cinnamon soil, and fluvo-aquic soil (Fig. 2b, c). Except for red soil, the bacterial community structure in each of the other soil types under the GN treatment differed from that under the FO, CK, and SN treatments (Supplementary Fig. S2).
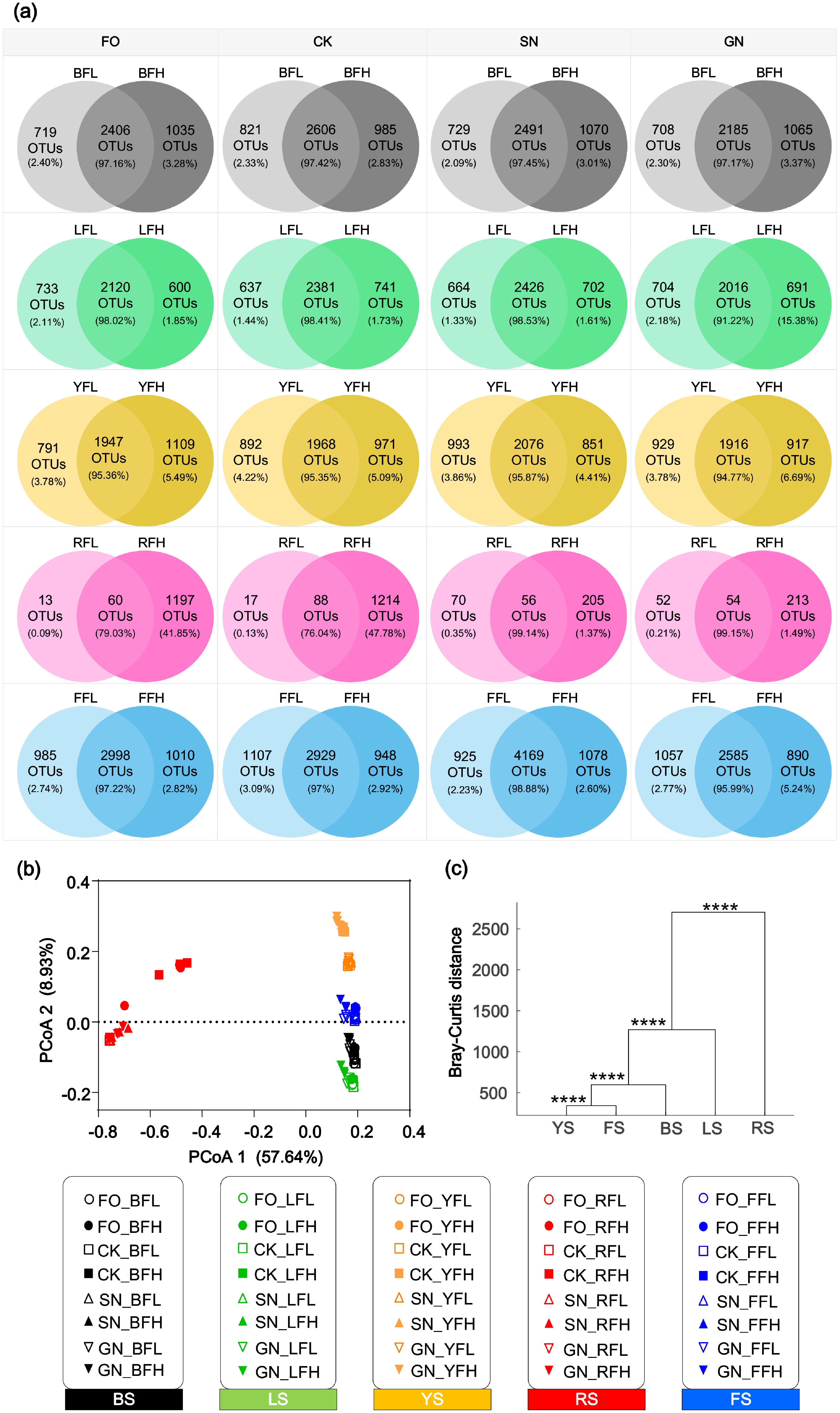
Figure 2.
Comparison of OTU composition and community structure of bacterial communities under different carbon and nitrogen addition treatments. (a) Venn diagram showing the number and relative abundance of shared and unique OTUs in each soil type under low- and high-fertilization treatments. (b) Bacterial community structure in five farmland soils under different carbon and nitrogen additions. (c) PCoA of bacterial communities in the five farmland soils based on Bray-Curtis distance. **** p < 0.0001 (MANOVA test).
Across all soil types, the dominant bacterial phyla consistently included Acidobacteria, Actinobacteria, Firmicutes, Gemmatimonadetes, and Proteobacteria (Fig. 3a, c, e, g, i). Red soil exhibited a distinct community structure, characterized by significant enrichment of Proteobacteria, particularly the genus Pseudomonas (Fig. 3g, h). In addition, red soil uniquely contained several phyla, including Candidatus Saccharibacteria and Cyanobacteria/Chloroplast (Fig. 3g). At the genus level, Ammoniphilus, Arthrobacter, Gaiella, and Gemmatimonas were dominant across most soils. In contrast, several genera displayed clear soil-specific distributions (Fig. 3b, d, f, h, j). For instance, Bacillus and Ensifer were prevalent in black soil, lime concretion black soil, and fluvo-aquic soil, whereas Streptomyces was more abundant in lime concretion black soil, yellow-cinnamon soil, and fluvo-aquic soil. Consistent with these patterns, red soil exhibited a markedly different bacterial composition compared with the other four soils. Under the SN and GN treatments, red soil under high fertilization further showed significant enrichment of Firmicutes and Alicyclobacillus (Fig. 3g, h). Detailed information on the dominant bacterial taxa at both the phylum and genus levels is provided in Supplementary Table S2. Furthermore, the GN treatment resulted in consistent enrichment of Firmicutes and a concomitant decrease in Actinobacteria across all five soils compared with the other three treatments. At the genus level, Ammoniphilus was significantly enriched under the GN treatment in most soils (Fig. 3).
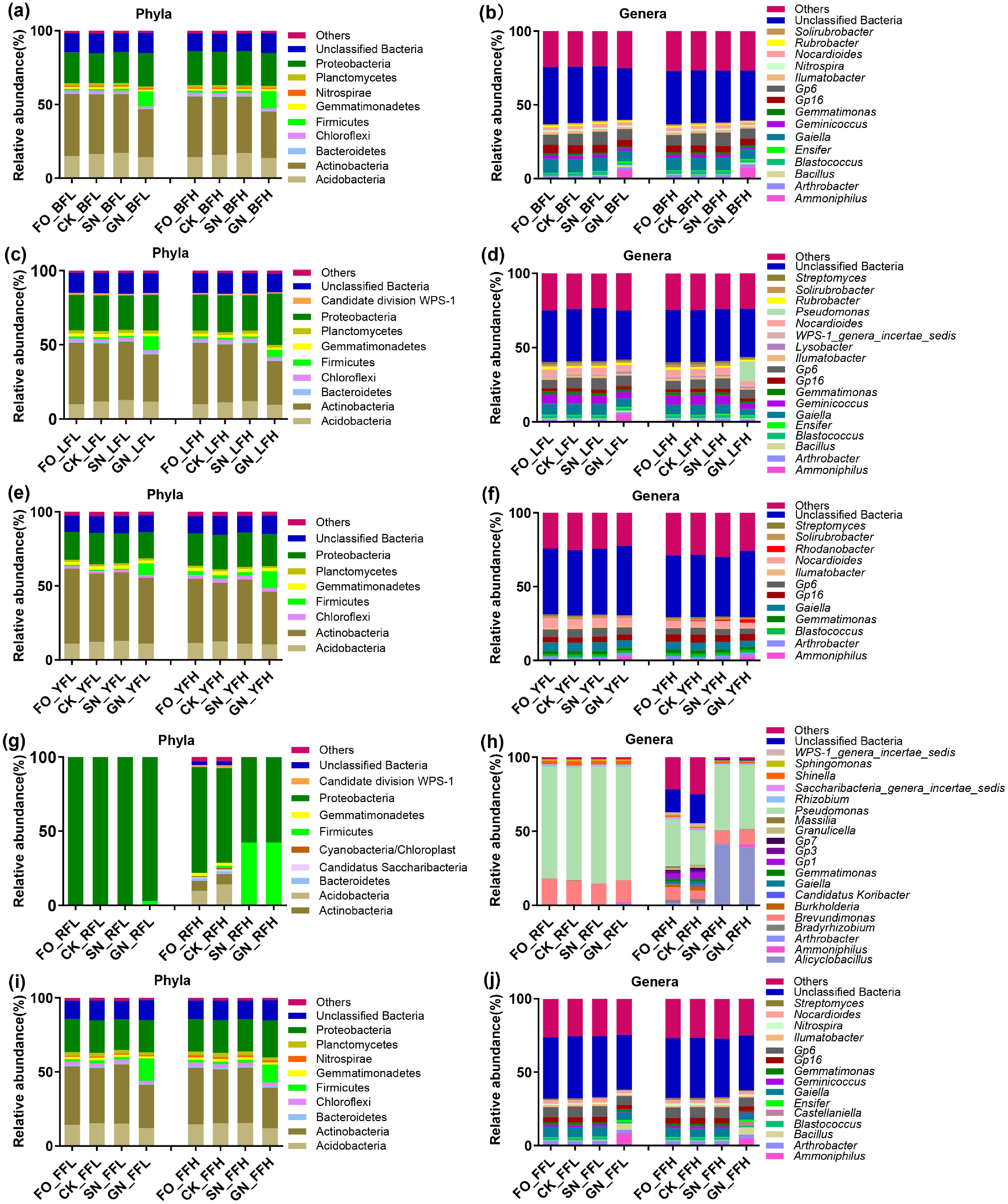
Figure 3.
Taxonomic profiles of the dominant microbial communities in five Chinese farmland soils. (a), (b) Black soil. (c), (d) Lime concretion black soil. (e), (f) Yellow-cinnamon soil. (g), (h) Red soil. (i), (j) Fluvo-aquic soil. Panels (a), (c), (e), (g), and (i) show phylum-level composition; panels (b), (d), (f), (h), and (j) show genus-level composition.
Community composition and shared OTUs among the soils
-
Venn diagram analysis was performed to assess the distribution of OTUs among the five soils under FO, CK, SN, and GN treatments (Fig. 4). The results showed that, across all treatments, the number of shared OTUs among the five soils was consistently higher under high fertilization than under low fertilization. Under low fertilization, the total number and average relative abundance of shared OTUs in the five soils were 30 (22.80%), 36 (22.72%), 71 (33.42%), and 58 (37%) for the FO, CK, SN, and GN groups, respectively. In contrast, under high fertilization, the corresponding values increased to 293 (43.10%), 307 (40.99%), 109 (32.81%), and 91 (35.99%). The relative abundance of soil-specific OTUs for black soil (BFL, BFH), lime concretion black soil (LFL, LFH), yellow-cinnamon soil (YFL, YFH), red soil under low fertilization (RFL), and fluvo-aquic soil (FFL, FFH) ranged from 0.25% to 3.53% across the four treatments (Fig. 4). Notably, red soil under high fertilization (RFH) consistently exhibited a much higher number and relative abundance of soil-specific OTUs, with values of 572 (15.20%), 593 (24.23%), 76 (41.98%), and 92 (40.05%) under the FO, CK, SN, and GN treatments, respectively (Fig. 4b, d, f, h).
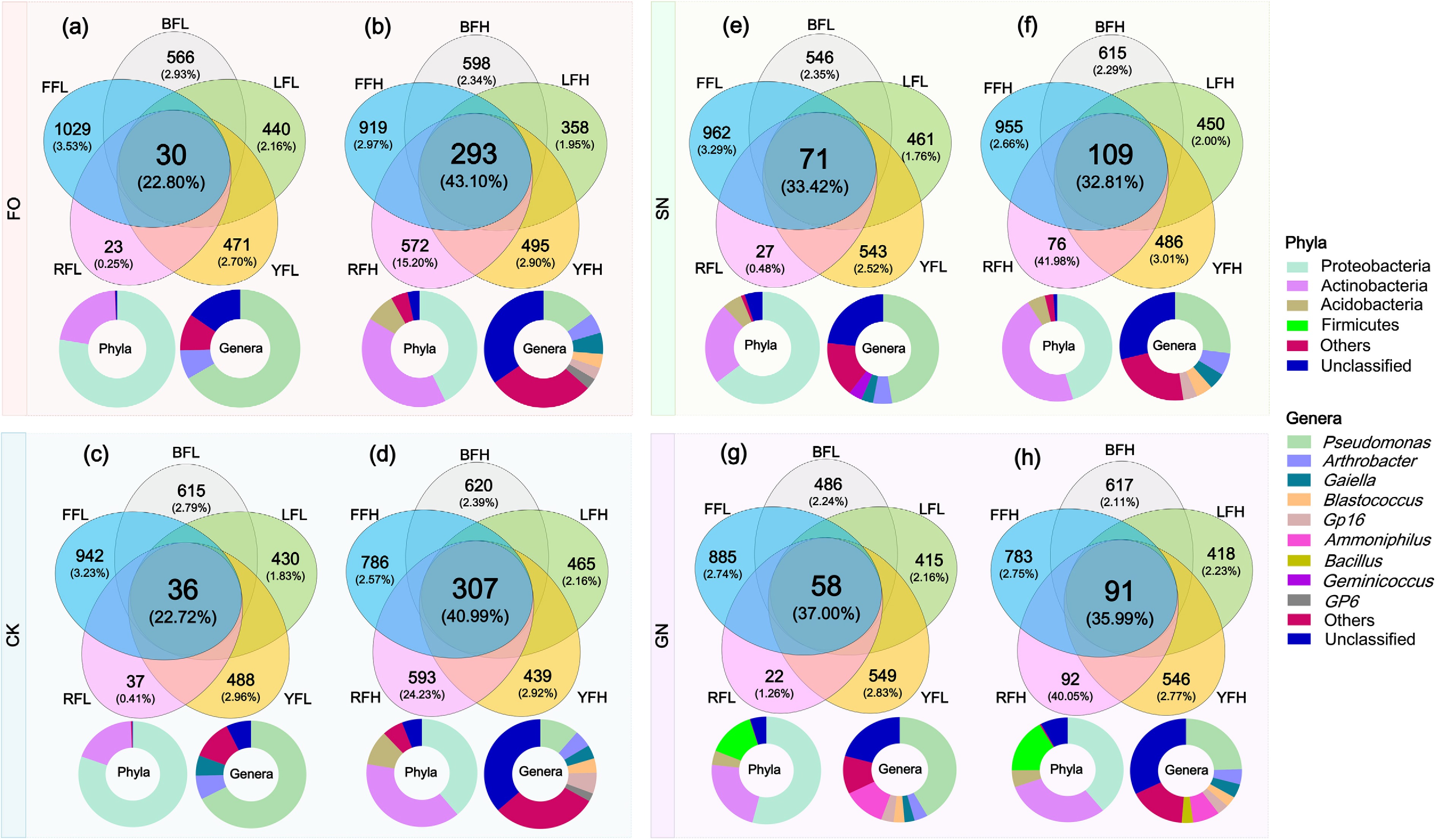
Figure 4.
Shared and unique OTUs in five farmland soils under different fertilization histories and treatments. Venn diagrams illustrating the counts and relative abundances of shared OTUs (common to all five soils) and unique OTUs (specific to each soil), along with the taxonomic classification of dominant taxa among the shared OTUs. (a), (b) FO treatment. (c), (d) CK treatment. (e), (f) SN treatment. (g), (h) GN treatment. Within each pair of panels, the left panels (a), (c), (e), (g) represent soils with low fertilization history, and the right panels (b), (d), (f), (h) represent soils with high fertilization history.
Across all treatments and fertilization regimes, a subset of shared OTUs was consistently detected across all five soils, indicating a core bacterial assemblage resilient to soil type and carbon-nitrogen perturbations. These shared OTUs were primarily affiliated with phyla Proteobacteria and Actinobacteria, and dominant shared genera included Pseudomonas and Arthrobacter across all treatments. Detailed taxonomic composition of the shared microbiome across five farmland soils is provided in the Supplementary materials. A total of 457 shared OTUs among five soils were defined as the union of OTUs detected under either low- or high-fertilization histories across all incubation treatments. Correlation analyses revealed no significant relationships between the relative abundances of these shared OTUs (n = 457) and the N2O/(N2O + N2) ratio after false discovery rate (FDR) correction (all adjusted p > 0.89), indicating that shared taxa alone do not independently explain soil-specific N2O partitioning (Supplementary Fig. S3). The bacterial community structure of red soil was markedly distinct from that of the other four soils. Venn diagram analysis showed that the total number and average relative abundance of shared OTUs among the four farmland soils (black soil, lime concretion black soil, yellow-cinnamon soil, and fluvo-aquic soil) were higher than those shared among all five soils (Supplementary Fig. S4). Detailed taxonomic composition of the shared microbiome across four farmland soils is provided in the Supplementary materials.
Ecological functions of shared microbes in different soils
-
The ecological functions of shared OTUs across the five soils were predicted using FAPROTAX. The results indicated that these shared microorganisms potentially contribute to multiple biogeochemical processes, including carbon, nitrogen, and sulfur cycling, metal ion oxidation, and organic matter degradation (Fig. 5). Specifically, Methylobacterium, which was shared across all five soils, was associated with methanol oxidation. Shared genera, including Methylobacterium, Rhizobacter, Mesorhizobium, Azospirillum, Roseomonas, Massilia, and Singulisphaera, were predicted to participate in urea decomposition (Fig. 5). Nitrosospira and Nitrospira were linked to aerobic ammonia oxidation and nitrite oxidation, respectively. Genera such as Azoarcus, Ensifer, and Cupriavidus were associated with nitrate reduction, while Azospirillum and Bradyrhizobium were linked to nitrogen fixation. Bosea was potentially involved in dark sulfide oxidation (Fig. 5). In addition, numerous shared taxa, including Methylobacterium, Mesorhizobium, Azospirillum, Roseomonas, Singulisphaera, Ensifer, Opitutus, Cupriavidus, Bradyrhizobium, Bosea, Streptomyces, Sphingomonas, Pseudomonas, Nocardioides, Cellulomonas, Skermanella, Rhodococcus, and Lysobacter, were associated with chemoheterotrophy across all soils. Geodermatophilus was implicated in manganese oxidation, Lysobacter in chitin degradation, and Rhodococcus and Nocardioides in the degradation of aromatic compounds (Fig. 5).
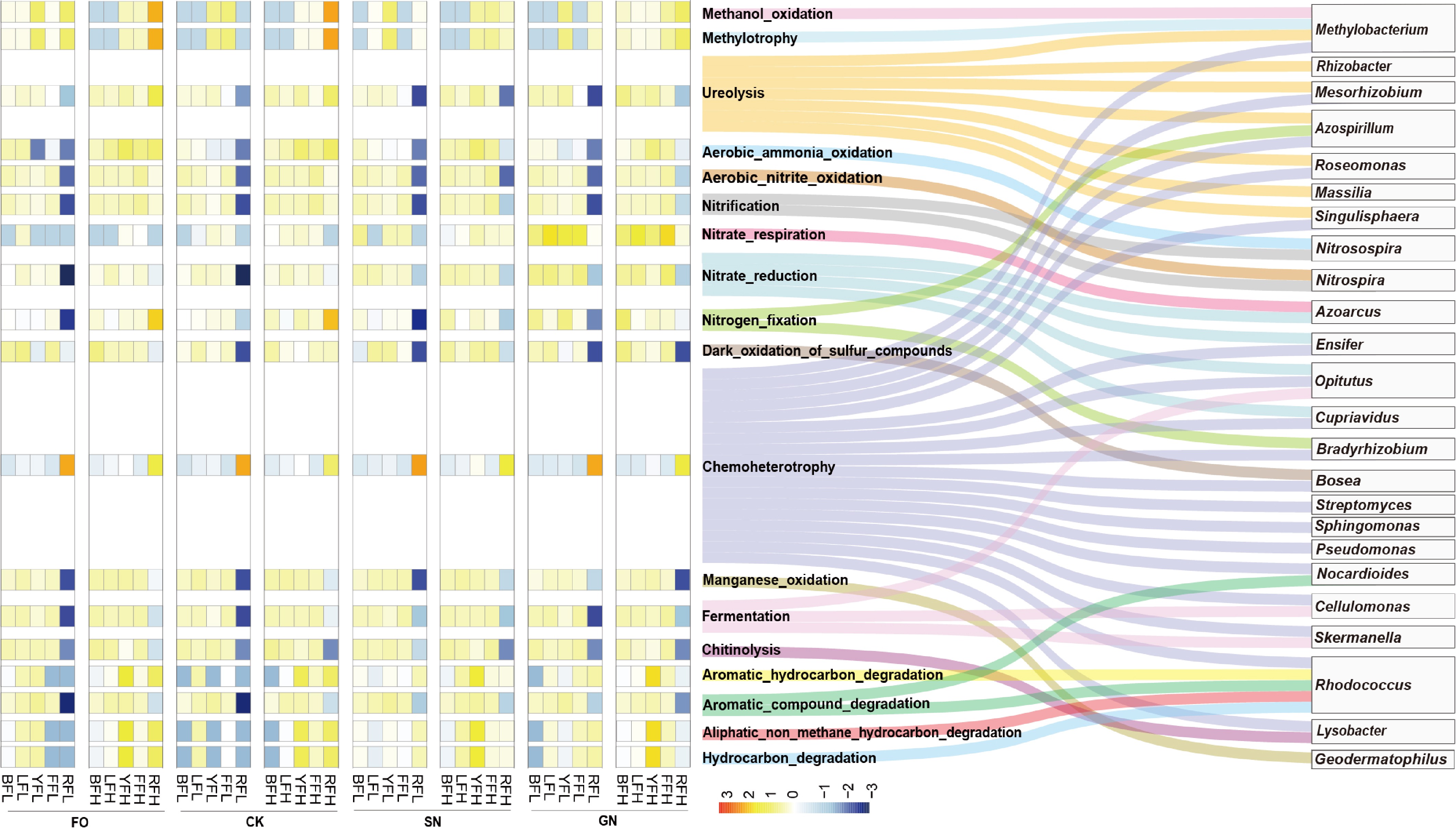
Figure 5.
Functional potential of the core microbial community in five farmland soils under different treatments.
Effects of nitrate and glucose addition on denitrification gases
-
N2O fluxes increased continuously throughout the anaerobic incubation under nitrate-only treatments, resulting in elevated N2O/(N2O + N2) ratios compared with the CK group (Supplementary Fig. S5a, S5b, S5d, S5e and Fig. 6a, b). When nitrate and glucose were added together, N2O fluxes in black soil, lime concretion black soil, yellow-cinnamon soil, and fluvo-aquic soil increased initially and subsequently declined, whereas N2O fluxes in red soil continued to increase throughout the incubation period (Supplementary Fig. S5c, S5f). Compared with nitrate-only treatments, combined carbon and nitrogen addition reduced N2O/(N2O + N2) ratios across all five soils, indicating that increased carbon availability promoted the reduction of N2O to N2 (Fig. 6a, b). Among all treatments, fluvo-aquic soil consistently exhibited lower N2O fluxes, the lowest N2O/(N2O + N2) ratios, and the highest N2/(N2O + N2) ratios, reflecting a greater capacity for complete denitrification (Supplementary Fig. S5a–S5l and Fig. 6a–f). Concurrently, combined carbon and nitrogen addition significantly increased total gaseous nitrogen fluxes (N2O + N2) in all soils. However, denitrification-derived nitrogen losses in red soil remained lower than those in the other four soils (Fig. 6e, f). Overall, variations in N2/(N2O + N2) mirrored inverse trends in N2O/(N2O + N2) (Fig. 6c, d). These results indicate that while carbon and nitrogen inputs promoted a shift toward more complete denitrification, this shift was accompanied by increased total nitrogen losses, revealing a trade-off between reducing N2O accumulation and conserving reactive nitrogen.
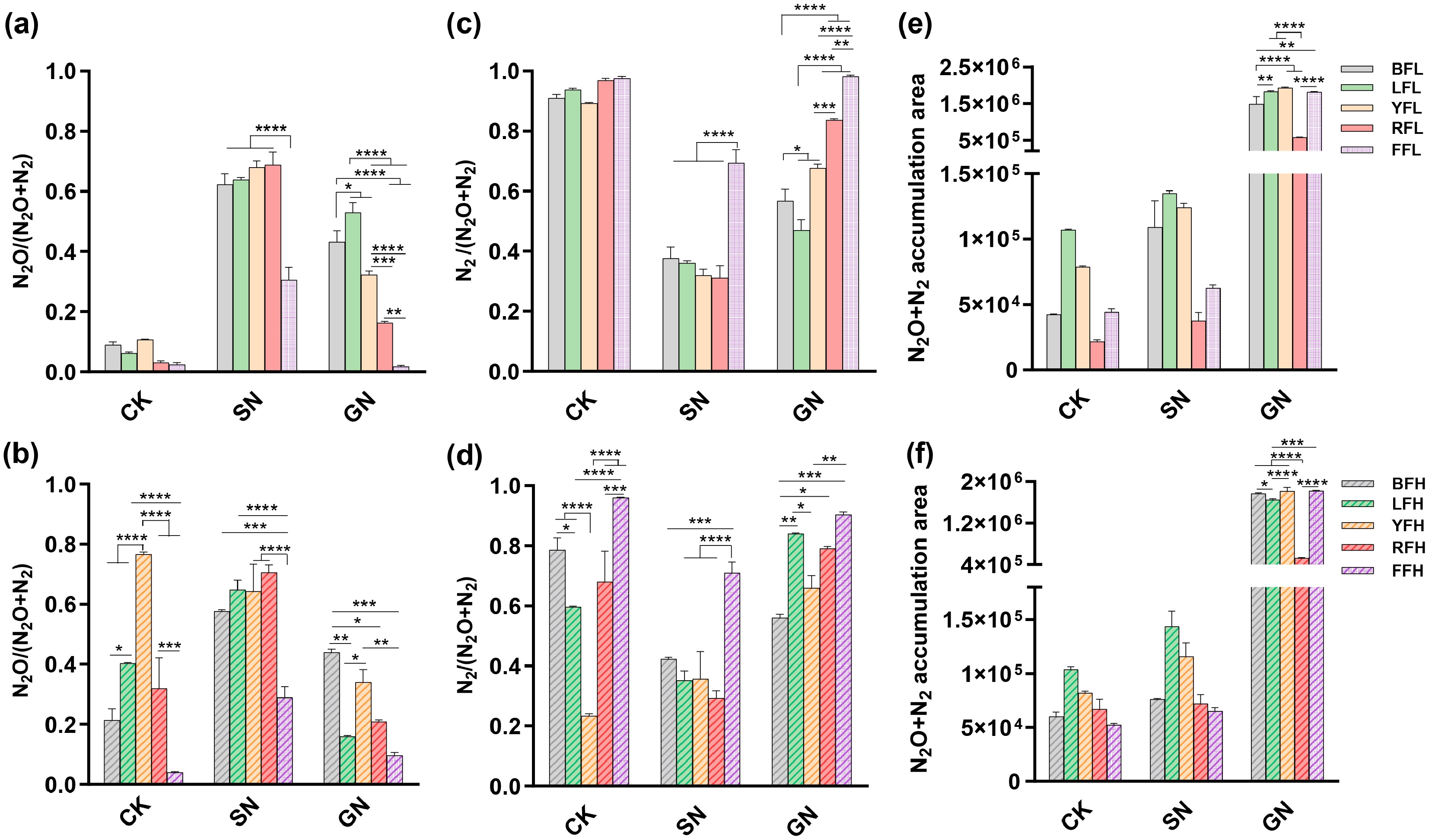
Figure 6.
Effects of carbon and nitrogen additions on denitrifying nitrogen metabolism in five farmland soils. (a), (b) N2O index, (c), (d) N2 index, and (e), (f) cumulative N2O and N2 fluxes in five farmland soils under (a), (c), (e) low, and (b), (d), (f) high fertilization levels across different treatments. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Abundance patterns of denitrifying genes in five soils
-
The abundances of denitrification functional genes varied moderately among soils under different carbon and nitrogen treatments (Fig. 7a, b). Fluvo-aquic soil consistently exhibited higher abundances of nirK, nirS, and nosZ compared with the other four soils (Fig. 7a, b). Notably, nosZ abundance in fluvo-aquic soil was substantially higher than that of nirK or nirS, which likely contributed to its lower N2O accumulation by enhancing the potential for N2O reduction to N2. Although nosZ abundance in black soil, lime concretion black soil, and yellow-cinnamon soil exceeded that of nirK or nirS (Fig. 7a, b), elevated N2O accumulation could not be explained solely by gene abundance. Red soil exhibited the lowest abundances of nirK, nirS, and nosZ among all soils (Fig. 7a, b), indicating a generally weaker denitrification potential, which was consistent with its lower denitrification-derived nitrogen fluxes.
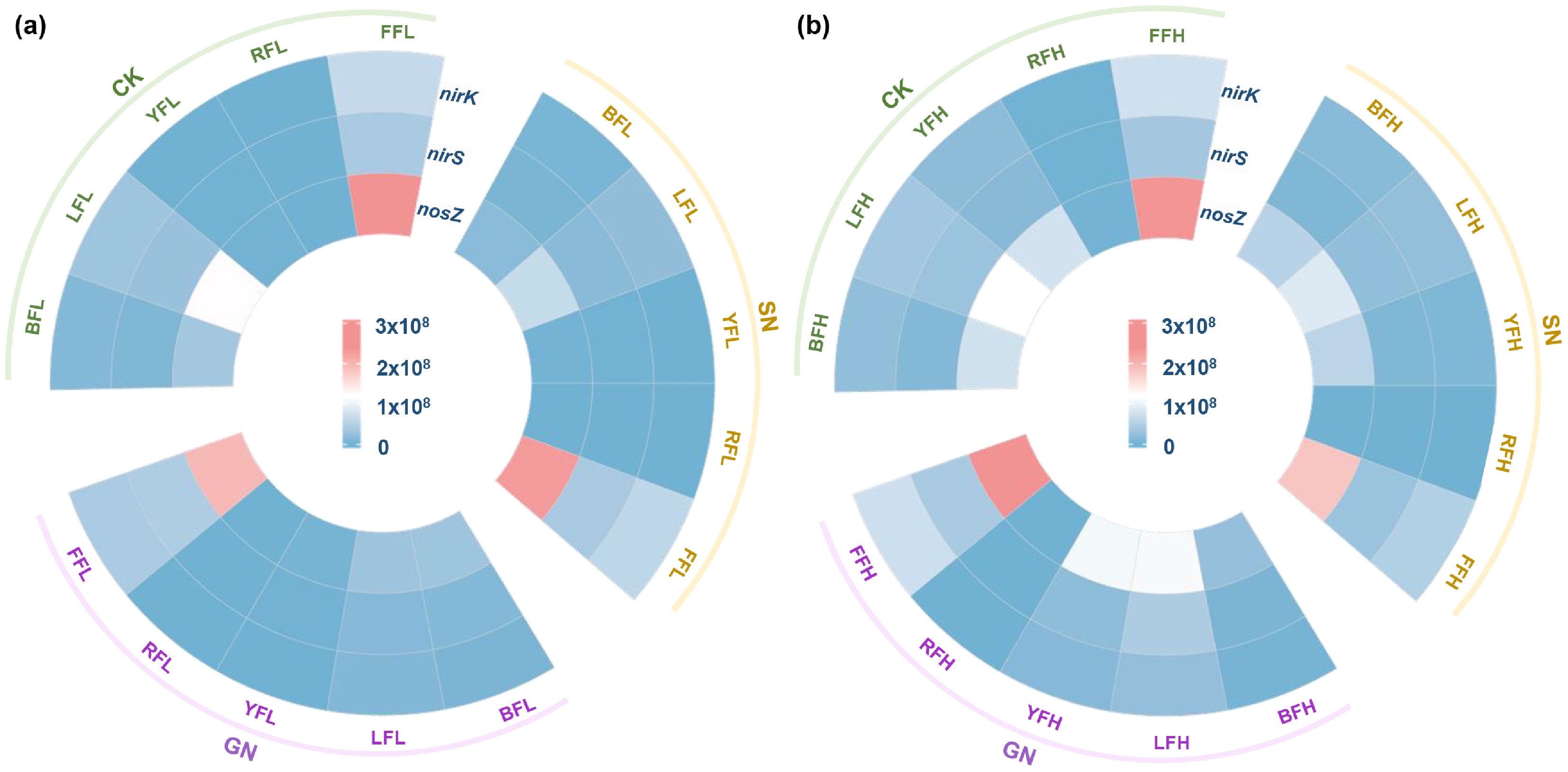
Figure 7.
Abundance of denitrifying genes in five farmland soils under different carbon and nitrogen additions. Gene copy numbers of nirK, nirS, and nosZ under (a) low, and (b) high fertilization levels across treatments.
-
Soil physicochemical properties and microbial communities are tightly coupled, with soil conditions exerting strong selective pressures on microbial community assembly[35,36]. Soil pH is a key driver of bacterial diversity and composition across ecosystems[37−39]. Soil organic carbon availability also strongly influences microbial diversity and functional potential, with nutrient-rich soils generally supporting higher microbial abundance and activity[40]. In this study, variations in bacterial community structures among the five soils were primarily explained by differences in soil pH and nitrate content, with pH emerging as the dominant explanatory factor. Previous studies have shown that most bacteria preferentially thrive at neutral pH, whereas relatively few taxa are adapted to highly acidic or alkaline environments[41]. Consistent with this pattern, red soil exhibited the lowest microbial diversity and abundance, likely due to its acidic conditions and limited nutrient availability[42].
Functional roles of soil microorganisms
-
Soil-driven microbial communities play key roles in regulating climate, nutrient cycling, and pollutant degradation, thereby underpinning soil fertility and ecosystem resilience[43,44]. Through biogeochemical processes, soil microorganisms also modify soil habitats and serve as indicators of soil quality[45,46]. In this study, bacterial community composition at the phylum level remained stable across FO, CK, and SN treatments in all five soils, suggesting resilience to short-term anaerobic conditions or nitrate enrichment[47,48]. Core soil microbes, which are widely distributed across environments, typically occupy broad ecological niches and exhibit high metabolic versatility[49−51].
Shared taxa such as Acidobacteria were consistently detected across all soils. Previous studies have reported that members of Acidobacteria account for approximately 20% of total soil bacterial abundance[52,53], and in some cases can reach up to 50%[54]. They are generally considered oligotrophic microorganisms that typically thrive in environments characterized by recalcitrant carbon substrates or low nutrient availability[55]. Similarly, the ubiquitous Arthrobacter exhibits strong tolerance to environmental stresses and degrades diverse pollutants[56,57]. The plant growth-promoting Bacillus was also shared across soils under sufficient carbon and nitrogen availability[58]. However, the relative abundance of shared OTUs showed no significant correlation with the N2O/(N2O + N2) ratio, indicating that these ubiquitous taxa did not directly explain soil-specific N2O emission patterns.
Limitations of gene abundance in predicting N2O emission potential
-
Net N2O accumulation is the net result of the balance between N2O production and consumption[59]. Fluvo-aquic soil exhibited higher denitrification gene abundances, with nosZ exceeding both nirK and nirS, consistent with its low N2O accumulation and strong reduction potential. However, persistent N2O accumulation in black soil, lime concretion black soil, and yellow-cinnamon soil, despite nosZ abundance being higher than nirK or nirS in these soils, indicates a decoupling between gene abundance and denitrification outcomes. This suggests that N2O reduction depends not only on gene abundance but also on the taxonomic identity and phenotypic traits of denitrifiers[13,60,61]. Microorganisms carrying identical genes may differ in enzyme kinetics and environmental sensitivity, resulting in variable N2O reduction efficiencies[13,59,60]. Keystone denitrifiers may further exert disproportionate effects on N2O emissions through adaptive genomic features and regulatory strategies[61,62]. Thus, high N2O accumulation in black soil, lime concretion black soil, and yellow-cinnamon soil may reflect differences in nosZ-harboring communities or a greater abundance of truncated denitrifiers with limited N2O reduction capacity[13,63]. Although gene abundance data provide mechanistic insights, direct measurements of gene expression or enzyme activity are lacking in this study. Future studies integrating transcriptomics, isotopic tracing, and strain-level characterization are needed to resolve microbial mechanisms underlying soil-specific N2O dynamics[13,64,65].
Soil physicochemical factors influencing N2O emissions
-
Soil physicochemical properties strongly regulate denitrification activity and N2O production[66]. Consistent with previous studies, microbial responses to nitrogen inputs were highly soil-type dependent, highlighting the importance of background soil conditions[67]. Initial nitrate availability was a major determinant of N2O production[68]. Under high fertilization, yellow-cinnamon soil and lime concretion black soil showed higher N2O fluxes, corresponding to their elevated background nitrate levels. However, when nitrate levels were standardized, N2O fluxes increased in all soils, but soil-specific differences persisted, with fluvo-aquic soil consistently exhibiting lower emissions. These results indicate that nitrate availability alone cannot explain soil-specific denitrification phenotypes[68]. Under nitrate and combined carbon-nitrogen additions, the continuous increase in N2O flux in red soil was likely driven by its acidic conditions, as low pH inhibits nitrous oxide reductase activity and limits N2O reduction[20,69]. In contrast, the alkaline pH of fluvo-aquic soil favors efficient N2O reduction, resulting in lower accumulation[20]. These findings highlight soil pH as a key regulator of N2O accumulation. Combined carbon and nitrogen inputs stimulated denitrification across all soils, likely through enhanced electron supply[70]. However, red soil produced less total denitrification gas, probably due to low microbial biomass and acidic constraints[71,72]. This study provides a mechanistic framework linking soil physicochemical constraints with microbial functional heterogeneity to explain divergent denitrification outcomes.
-
Across five Chinese agricultural soils, soil pH and nitrate availability were identified as the primary drivers of bacterial community differentiation, with acidic red soil harboring a distinct microbial assemblage. Despite substantial taxonomic variation, a shared core microbiome consistently participated in key biogeochemical processes, but showed no significant association with soil-specific N2O/(N2O + N2), indicating that core microbial taxa cannot directly predict N2O emission patterns. Carbon-nitrogen co-addition significantly enhanced denitrification completeness by promoting N2O reduction to N2, but induced distinct soil-dependent N2O dynamics. Notably, fluvo-aquic soil exhibited the lowest N2O fluxes and N2O/(N2O + N2) ratios, indicating a superior capacity for complete denitrification. This pattern was associated with higher abundances of denitrification genes, particularly nosZ, suggesting enhanced potential for N2O reduction. In contrast, other soils showed substantial N2O accumulation despite relatively high nosZ abundance, revealing a decoupling between functional gene abundance and actual denitrification outcomes. Red soil exhibited the lowest abundances of nirK, nirS, and nosZ, consistent with its limited denitrification capacity, likely constrained by low organic carbon availability and acidic conditions.
Collectively, our results demonstrate that soil-specific N2O emissions arise from interactions between physicochemical constraints and microbial functional heterogeneity. Carbon and nitrogen inputs do not uniformly mitigate N2O emissions but instead regulate denitrification product partitioning in a soil-dependent manner, highlighting the need for soil-specific strategies in agricultural N2O mitigation and nitrogen management. The observed gene-function decoupling provides new insight into the regulatory complexity of soil denitrification. It highlights the need for future studies that integrate gene expression, enzyme activity, and strain-level functional traits to resolve the microbial mechanisms underlying divergent N2O emissions across soils.
The authors gratefully acknowledge Wei Zhang for her assistance in collecting black soil, Song He for his contributions to collecting lime concretion black soil and yellow-cinnamon soil, Jiong Wen for his role in collecting red soil, and Liuqing Yang for her support in collecting fluvo-aquic soil.
-
It accompanies this paper at: https://doi.org/10.48130/nc-0026-0006.
-
Not applicable.
-
The authors confirm their contributions to the paper as follows: Qiaoyu Wu: contribution data collection, methodology and software, figure visualization, analysis and interpretation of results, draft manuscript preparation; Siyu Yu: contribution methodology and software; Zhen Xie: contribution methodology and software; Xianchao Qin: contribution methodology and software; Ji Li: contribution methodology and software; Xiaojun Zhang: contribution study conception and design, supervision and funding acquisition, writing-review and editing. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
-
This work was supported by the National Natural Science Foundation of China (Grant Nos NSFC 42577128 and 31971526), the Key R&D project of Ministry of Science and Technology (Grant No. 2017YFD0200102).
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
Full list of author information is available at the end of the article.
- The supplementary files can be downloaded from here.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wu Q, Yu S, Xie Z, Qin X, Li J, et al. 2026. Comparative study of microbial communities and denitrification gas emissions in typical Chinese farmland soils under varying C/N conditions. Nitrogen Cycling 2: e019 doi: 10.48130/nc-0026-0006 

Comparative study of microbial communities and denitrification gas emissions in typical Chinese farmland soils under varying C/N conditions
- Received: 18 December 2025
- Revised: 30 March 2026
- Accepted: 06 April 2026
- Published online: 21 April 2026
Abstract: Soils play a fundamental role in regulating biogeochemical cycles, yet how soil physicochemical properties regulate microbial communities and denitrification outcomes under varying carbon and nitrogen inputs remains poorly understood in Chinese farmland soils. Here, bacterial community composition and denitrification responses in five Chinese agricultural soils (black soil, lime concretion black soil, yellow-cinnamon soil, red soil, and fluvo-aquic soil) with contrasting fertilization histories under controlled carbon and nitrogen addition regimes are investigated. Across soils, pH and nitrate availability were identified as the primary drivers of bacterial community structure. A core set of shared dominant taxa was consistently associated with ecological functions related to carbon and nitrogen cycling and organic matter degradation. However, these shared taxa showed no significant correlation with soil-specific N2O/(N2O + N2) ratios, indicating that core microbial composition alone cannot reliably predict N2O emission patterns. Microcosm incubations revealed strong soil-dependent N2O dynamics. Fluvo-aquic soil consistently exhibited the lowest N2O accumulation and N2O/(N2O + N2) ratios, associated with higher abundances of denitrification genes, particularly nosZ, suggesting greater potential for N2O reduction. In contrast, substantial N2O accumulation in black soil, lime concretion black soil, and yellow-cinnamon soil, despite relatively high nosZ abundance, revealed a decoupling between functional gene abundance and actual N2O reduction capacity. Red soil showed the lowest denitrification gene abundances, consistent with its limited denitrification potential under acidic, low-carbon conditions. Overall, soil physicochemical properties exert hierarchical control over denitrification phenotypes, while microbial functional heterogeneity drives soil-specific N2O production and reduction. Carbon and nitrogen inputs regulate denitrification product partitioning in a soil-dependent manner rather than uniformly mitigating N2O emissions, highlighting the need for soil-specific N2O mitigation strategies in agriculture.