









-
Network-based representations have been widely applied in biological systems to model complex interactions, such as protein–protein interactions (PPIs)[1] or neural networks[2]. In immunology, networks have been used to capture the intricate relationships among components of the immune system, particularly T-cell receptor (TCR) repertoires[3]. TCR networks are constructed by defining edges between clonotypes with sequence similarity below a specified threshold[3]. In TCR networks, nodes represent unique clonotypes, and edges represent sequence similarity, where the count of a unique clonotype reflects its abundance. Both global and local network properties are used to quantify the topology, providing insights into the structural and functional dynamics of the immune repertoire[4].
Advances in high-throughput immune repertoire sequencing have enabled the capture of both highly diverse clonotype sequences and clonal expansion, yielding the large-scale construction of TCR networks, especially for bulk TCR sequencing. However, such networks can become extremely large and computationally intensive, particularly when pairwise similarity calculations are required for thousands of sequences. Moreover, clonal expansion during immune activation leads to highly abundant TCR clonotypes, which further increases the computational burden. Previous studies have shown that the size and complexity of these networks pose major challenges for both network construction and downstream analysis[5−7] . TCR repertoires typically exhibit a strongly skewed clonal frequency distribution, with the vast majority of clonotypes occurring at extremely low frequencies and only a small fraction being highly expanded. This long-tailed abundance structure has been observed across various tissues and disease contexts, and can further amplify the computational burden in large-scale repertoire network analyses[3,8,9].
Most research in TCR repertoire analysis mainly focused on optimizing sequence similarity computations during network construction, while few studies have tackled the scalability challenges of analyzing network topology. In clinical immunomonitoring studies, TCR repertoire sequencing is commonly performed on serial peripheral blood samples and, when available, paired tumor biopsies collected before and after therapy, generating large cohorts and longitudinal datasets that motivate scalable network-based analyses[10]. In these settings, network topology is often used to characterize clonal expansion, diversity, and connectivity patterns associated with treatment response, immune activation, or resistance. Effective subsampling strategies that preserve key topological features, therefore, enable comparative analyses across time points, tissues, and patient groups while maintaining clinically relevant immune signatures. For example, in immunotherapy studies, network-level properties such as cluster size, hub clonotypes, or changes in connectivity have been used to compare responders and non-responders over time[11,12]. Subsampling methods that fail to preserve these features may obscure clinically meaningful immune dynamics. These clinical and analytical challenges motivate a systematic evaluation of subsampling strategies that preserve biologically meaningful network topology while enabling scalable analysis. Therefore, in this study, we focus on evaluating various subsampling algorithms and methods (Fig. 1a), such as Random Node Sampling (RNS), Snowball Sampling (SB), PageRank (PR), Metropolis-Hastings (MH), and Simple Random Walk with Fly Back (SRWFB), which have been widely used in social networks, internet topology, and large-scale web graphs to study node importance and network properties. We will also consider different subsampling strategies and examine how well they preserve biologically meaningful network properties, ensuring the immunological relevance of the resulting subsampled networks. To our knowledge, this work represents the first comprehensive evaluation of network subsampling techniques in the context of immune repertoire analysis, providing insights and serving as a reference for future studies.
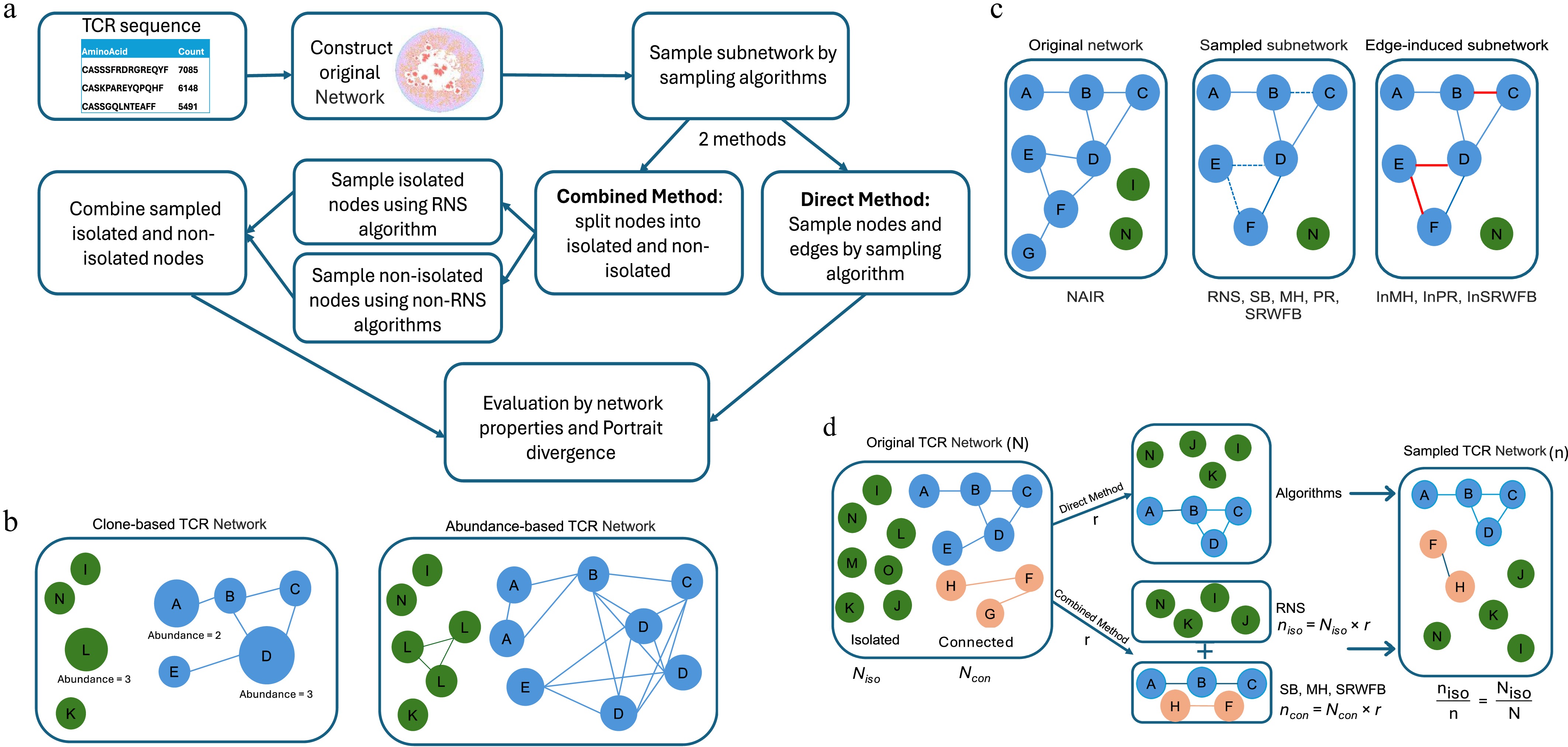
Figure 1.
Schematic chart of subsampling approaches. (a) Analytic flow of network subsampling. (b) Clone-based and abundance-based TCR networks. Both networks are constructed by setting hamming distance between the TCR sequences equals to 1. Nodes in clone-based network correspond individual TCR sequences. In abundance-based network, nodes are expanded based on counts (abundance) of each unique TCR clone. (c) Pseudo examples of original network, and subnetwork by the original algorithm and the induced algorithm. (d) Illustration of the direct and combined strategies. In the direct method, a network is subsampled as a whole using one single algorithm. In the combined method, nodes are partitioned into isolated (Niso) and connected (Ncon) groups. To preserve network sparsity, both groups are subsampled at a consistent rate r, such that niso = Niso × r, and ncon = Ncon × r. This proportional scaling ensures the subnetwork's isolation rate matches the original niso/n = Niso/N, preventing the edge-traversal bias of algorithms from artificially inflating connectivity. The subsampled results are then merged to form the final subnetwork.
-
This study included 11 metastatic pancreatic adenocarcinoma patients enrolled in Phase 1b clinical trial NCT02562898, who received ibrutinib at the University of California, San Francisco, and Oregon Health and Science University in Portland, USA. Baseline blood samples were collected prior to treatment, and post-timepoint samples were obtained after at least one administration of the drug. Cryopreserved peripheral blood mononuclear cells (PBMCs) were isolated from blood samples, and genomic DNA was extracted for sequencing of the TCR beta CDR3 region using the immunoSEQ platform (Adaptive Biotechnologies). Detailed clinical and demographic information for the cohort is available in the original publication[10].
TCR repertoires network
-
TCR networks were constructed for each repertoire based on amino acid sequence similarity using the Network Analysis of Immune Repertoire (NAIR) package[3]. We set the Hamming distance less than or equal to 1 to allow at most one residue at the same location between two unique TCR clones. Two types of TCR networks were analyzed in this study (Fig. 1b): (1) clone-based networks, where each unique TCR amino acid sequence was represented as a single node regardless of its abundance; and (2) abundance-based networks, where nodes were replicated according to clonal frequency, allowing high-abundance TCRs to appear multiple times. The distribution of TCR clone abundance is typically left-skewed, with the top 1% of expanded clones accounting for up to 20% of total reads, reflecting immune response specificity[9,13]. We observed a similar abundance distribution in our samples, with most clones showing fewer than 500 reads. Based on these patterns, we classified the samples into high-, medium-, and low-abundance groups according to the proportion of highly expanded clones (HECs, abundance > 500 reads) > 2%, 1%–2%, and < 1%, respectively (Table 1). In our datasets, the abundance distribution is extremely heavy-tailed, and a handful of extreme counts can dominate weight magnitudes and lead to prohibitive memory/runtime during network construction; therefore, nodes with an abundance greater than 500 were truncated to 500 before subsampling. This threshold was specifically selected to prevent extremely expanded clones from disproportionately dominating the network topology as super-hubs[14]. By mitigating the influence of these outliers, we ensure that subsampling algorithms can effectively capture the underlying structural diversity and connectivity patterns of the broader repertoire while maintaining computational stability during systematic evaluation. Such strategies are consistent with established practices in immune repertoire analysis for controlling clonal dominance and stabilizing diversity- and network-based metrics[15,16].
Table 1. Distribution of TCR node abundance.
Patient Proportion of nodes Abundance level ≥ 100 ≥ 200 ≥ 500 P1 6.50% 2.90% 1.40% Medium P2 17.70% 10.50% 6.20% High P3 3.40% 1.10% 0.50% Low P4 8.20% 4.10% 1.80% Medium P5 0.90% 0.30% 0.00% Low P6 8.60% 6.10% 3.00% High* P7 4.70% 2.80% 1.50% Medium P8 5.60% 3.30% 1.60% Medium* P9 2.80% 0.90% 0.50% Low P10 6.70% 5.10% 2.40% High P11 0.20% 0.10% 0.00% Low * * Indicates representative patients selected from each group for evaluation of sampling methods. Review of network sampling algorithms
-
The following will review five commonly used network sampling algorithms (Table 2). RandomNode Sampling (RNS) is a node-based method that selects nodes uniformly at random, independent of edge connectivity[17]. It is especially effective for sampling isolated or sparsely connected nodes. Metropolis-Hastings (MH) is an edge-informed sampler based on Markov Chain Monte Carlo (MCMC), where acceptance of a candidate node depends on the properties of the current node, including its degree[18]. Both RNS and MH have no tunable hyperparameters in our implementation. SnowBall (SB) is a hybrid approach that begins with a randomly selected set of seed nodes of size k and iteratively expands by adding neighboring nodes through connected edges[19]. The parameter k represents the maximum number of neighbors that can be added in each sampling cycle. In the context of TCR networks with many small, disjoint clusters, k balances local expansion within a similarity neighborhood against the need to reach the target sample size without over-sampling a few large components. Smaller values of k favor highly local neighborhood preservation but may require more iterations to reach the target sample size, whereas larger values of k accelerate sampling at the risk of over-representing highly connected components. Page Rank (PR)[20] is an edge-based technique that follows an iterative process, where nodes are subsampled based on their PageRank scores. The PageRank score measures the relative importance of a node by recursively considering both the number and significance of incoming edges, while incorporating a damping factor[20]. The damping factor (α) represents the probability of the nodes being randomly selected instead of walking along the edge continuously. For disjoint TCR landscapes, α is critical for balancing edge-based importance with random jumping to ensure navigation across isolated components. Lower values α emphasize connectivity-driven importance within components, while higher values increase random jumps, improving coverage across disjoint clusters but reducing sensitivity to local topology. Simple Random Walk Sampling with Fly Back (SRWFB) starts with a random node and performs a random walk along its edges, choosing adjacent nodes sequentially with a predefined Fly Back probability (p)[20]. The Fly Back probability (p) is the probability of returning to the starting point to explore other neighbors. Higher fly-back probabilities promote broader exploration across components, whereas lower values favor deeper traversal within individual clusters. This prevents the walker from staying within a single component, facilitating exploration across the many disjoint clusters in TCR networks. An iteration time parameter determines the total number of steps in the walk, ensuring that both densely and sparsely connected regions of the graph are covered.
Table 2. Summary of sampling algorithms.
Algorithm type Description Key parameters Random Node Sampling (RNS) Selects nodes uniformly at random from the network. − SnowBall (SB) Starts from a set of seed nodes and expands by connecting edges. k − Max number of neighbors added per cycle Page Rank (PR) Nodes are sampled based on their PageRank score in an iterative process. α (damping factor) − 0.85 Metropolis-Hastings (MH) Relies on edge connections and follows a Markov Chain Monte Carlo (MCMC) process. Acceptance depends on node's degree Simple Random Walk with Fly Back (SRWFB) Starts with a random node and performs a random walk with a predefined probability of returning to the starting node. p (fly-back probability) − 0.15,
iteration time − 100Induced-Page Rank (InPR) Retains all original edges between selected nodes. Inherits from PR Induced-Metropolis-Hastings (InMH) Retains all original edges between selected nodes. Inherits from MH Induced-Simple Random Walk with Fly Back (InSRWFB) Retains all original edges between selected nodes. Inherits from SRWFB In addition, we also evaluated induced alternatives of some algorithms, such as Induced Metropolis-Hastings (InMH), Induced Page Rank (InPR), and Induced Simple Random Walk Sampling with Fly Back (InSRWFB). After obtaining a sampled node set, we construct the final network as the node-induced subgraph by recovering all edges from the original graph whose endpoints are both sampled[17]. Those induced algorithms retain all original edges among the selected nodes from the full network, ensuring that local connectivity and topological measures are evaluated on a structurally faithful subgraph rather than being distorted by edge loss during sampling (Fig. 1c)[21].
Implementation strategies
-
Two subsampling strategies were employed in this study: the direct method and the combined method (Fig. 1d). In the direct method, each subsampling algorithm described above was applied directly to the entire TCR network without distinguishing between isolated and connected nodes. However, clone-based networks and some abundance-based networks exhibited high sparsity, with over 50% of isolated nodes (Supplementary Fig. S1). In such graphs, directly applying topology-aware samplers to the full network may under-sample isolated nodes (which have no incident edges), thereby altering the proportion of isolated nodes and artificially increasing apparent connectivity in the sampled subgraph. To address this limitation, we designed a combined subsampling strategy motivated by the observation that isolated and connected nodes represent fundamentally different structural entities in sparse TCR networks. Because topology-aware samplers rely on edge traversal, they are inherently biased against isolated nodes, motivating a hybrid design that treats these two node types separately when sparsity is substantial and downstream analysis is sensitive to component-level structure. In the combined method, nodes are first partitioned into isolated (Niso) and connected (Ncon) sets, and each set is subsampled at a fixed rate r (Fig. 1d). This proportional scaling preserves network sparsity by construction, and prevents topology-aware algorithms from artificially inflating connectivity through systematic undersampling of isolated nodes. The combined strategy is therefore most advantageous for highly sparse TCR networks with a large fraction of isolated clonotypes, particularly when downstream analyses are sensitive to sparsity, component structure, or the relative prevalence of expanded vs unexpanded clonotypes.
By defining the sample sizes as
$ {n}_{\text{iso}}={N}_{\text{iso}}\times r $ $ {n}_{\text{con}}={N}_{\text{con}}\times r $ $ \dfrac{{{n}}_{\text{iso}}}{{{n}}_{\text{iso}}+{{n}}_{\text{con}}}=\dfrac{{{N}}_{\text{iso}}\times {r}}{\left({{N}}_{\text{iso}}+{{N}}_{\text{con}}\right)\times {r}}=\dfrac{{{N}}_{\text{iso}}}{{N}} $ This proportional scaling preserves network sparsity by construction and prevents topology-aware algorithms from artificially inflating connectivity through systematic under-sampling of isolated nodes. The combined strategy is therefore most advantageous for highly sparse TCR networks with a large fraction of isolated clonotypes, particularly when downstream analyses are sensitive to sparsity, component structure, or the relative prevalence of expanded vs unexpanded clonotypes.
Aligning each algorithm with the part of the graph where its preference was beneficial, together with separating isolates from the connected part, provided the methodological justification for the combined strategy and explained the lower distortion observed for PDiv and other component-sensitive metrics. A range of subsampling percentages (i.e., 5%, 10%, 15%, 20%, 25%, and 30%) was used to evaluate the performance under different combinations of algorithms and strategies. For each setting, twenty independent replicates (with distinct random seeds) were generated. Results were summarized as the mean across replicates, with standard error (SE) indicated.
Performance evaluation metrics
-
To evaluate the effectiveness of different sampling methods and strategies in preserving the original characteristics of TCR networks, we used a topological metric, PDiv. PDiv first calculates each network's 'portrait', a matrix representation that captures the network's structure, and then computes the Jensen-Shannon divergence between these portrait matrices to quantify the structural similarity[22]. Compared to traditional comparison metrics, such as Graph Edit Distance (GED), which is computationally NP-hard, or DeltaCon, which requires strict node correspondence, PDiv is a permutation-invariant and parameter-free measure that is ideal for subsampling evaluation where node identities are not preserved[22,23]. Furthermore, unlike individual network properties that capture local or fragmented features, PDiv provides a holistic, multi-scale comparison of the system-level topology[23]. Taken together, these properties make PDiv particularly well-suited for evaluating network subsampling in TCR repertoire analyses, where networks are large, node identities are not preserved, and preservation of global topological structure is essential. PDiv values range from 0 to 1, with values close to 0 indicating higher structural similarity and minimal distortion.
Network properties have been widely used to quantify the networks at the cluster and node levels, which have been shown to associate with clinical outcomes and to provide causal inference in simulation studies[24,25]. We employed four cluster-level network properties, i.e., degree, assortativity, transitivity, and density (Table 3), to evaluate the robustness of the subsampling methods on a real-world dataset. Degree and transitivity capture local clustering and centrality[26,27], while assortativity and transitivity are global metrics. Density measures the proportion of possible edges present and indicates the overall level of network connectivity[14]. The maximum degree is more sensitive than mean or median values for capturing rare but critical topological features relevant to immunological interpretation in left-skewed TCR network structures[25]. In addition, we quantified the assortativity coefficient to capture how similarly connected TCR clones tend to link in repertoire networks using standard definitions[28]. This metric is informative in repertoires as a positive value indicates that similarly connected clones preferentially link (assortative mixing), whereas a negative value indicates disassortative mixing (high-degree clones link to low-degree ones)[5]. Besides direct comparisons of network metrics, we further examined the preservation of dynamic network behavior by calculating Cohen's d effect sizes to quantify changes in these metrics between the baseline and post-treatment periods[29]. Shifts in assortativity reflect centralization of sequence similarity under antigen-driven selection and clonal expansion, and temporally specific changes provide a concise readout of repertoire centralization vs dispersion.
Table 3. Evaluation metrics.
Metric Description Network Portrait Divergence (PDiv) Assesses similarity of 2 networks by analyzing 'Network Portrait'. Values range from 0 to 1. Network properties Max degree The maximum number of edges connected to a single node. Density The ratio of the number of actual edges to the possible number of edges. Assortativity Measures how strongly nodes with similar properties preferentially connect. Transitivity Measures the tendency of similar nodes to connect to each other. -
We first constructed original TCR networks at both clone and abundance levels for each of the 22 samples from 11 patients at both baseline and post-treatment time points. In clone-based networks, the total number of nodes ranged from 27,274 to 102,899, with a median of 73,758 at baseline, and from 20,317 to 80,751, with a median of 57,524 post-treatment. In abundance-based networks, the median node counts are 109,838 (ranging from 41,935 to 148,895) and 76,278 (ranging from 34,533 to 131,753) at baseline and post-treatment, respectively (Supplementary Table S1). Based on the proportion of HECs (abundance > 500), patients were categorized into three groups: three patients (with HECs proportion 6.2%, 3.0%, and 2.4%) were classified as the high-abundance group; four (with HECs proportion 1.8%, 1.6%, 1.5% and 1.4%) as the medium-abundance group, and the remaining four (with HECs proportion < 1%) as the low-abundance group (Table 1). As expected, the proportion of isolated nodes exceeded 50% in all samples in clone-based networks, indicating relatively sparse network structures (Supplementary Fig. S1a) while most abundance-based networks exhibited lower isolation rates, suggesting a denser and more compact structure, likely due to the impact of the presence of high-abundance clones (Supplementary Fig. S1b). The distribution of isolation rates across samples remained consistent within each network type regardless of the clinical time point.
Parameter sensitivity analysis and optimization using PDiv
-
To determine the optimal settings for each algorithm, we conducted a sensitivity analysis using PDiv as the objective function (Supplementary Figs S2a–S2c). In our sensitivity analysis for SB, k values of 1, 10, 100, and 1,000 were evaluated. PDiv decreased from k = 1 to 10 across all subsampling percentages; however, it began increasing at k = 100 for subsampling percentages of 5%, 10%, and 15% of the nodes (Supplementary Fig. S2a). Hence, we considered k = 100. For PR, we evaluated a = 0.5, 0.65, 0.75, 0.85, 0.90, and selected a = 0.85 because it minimized the replicate-mean, dataset-averaged PDiv (Supplementary Fig. S2b). For SRWFB, we considered p = 0.10, 0.15, 0.20 in the sensitivity analysis, and chose p = 0.15 as it minimized replicate-mean PDiv (Supplementary Fig. S2c). The iteration time was set to 100 to ensure full subgraph exploration. We also performed parameter sensitivity for InPR (Supplementary Fig. S2d) and InSRWFB (Supplementary Fig. S2e). The sensitivity analysis across all algorithms showed that PDiv values were optimized within the tested parameter ranges, supporting our selection of hyperparameters.
Performance evaluation for subsampling clone-based TCR networks
-
To evaluate the performance of various subsampling algorithms and strategies, we selected one baseline sample from each abundance group, i.e., Patient 6 (P6, high-abundance), Patient 8 (P8, medium-abundance), and Patient 11 (P11, low-abundance). For each sample, 20 subsampling replicates were performed using the implemented subsampling algorithms under both the direct and combined strategies. PDiv value and network properties were computed for each subnetwork and averaged across the 20 replicates.
As expected, PDiv values generally decreased with increasing subsampling percentages from 5% to 30% for all algorithms under both direct (Fig. 2a−c) and combined strategies (Figs 2d−f), except for MH. This plateau in MH performance likely results from slow mixing on highly disjoint TCR networks. Due to its rejection-based mechanism for correcting degree bias, MH can become trapped within isolated clusters, limiting global structural coverage even as the sampling proportion increases. Networks with higher abundance levels had lower PDiv values at the same sampling percentage and algorithm (Fig. 2c, f). Under the direct strategy (Fig. 2a−c), subsampling algorithms exhibited different patterns in performance, more specifically, SRWFB and InSRWFB consistently have the lowest PDiv, SB performed moderately; and MH, InMH, RNS, PR, and InPR show the poorest performance. This separation was observed across different subsampling percentages for different abundance levels for the direct strategy. However, the combined strategy (Fig. 2d–f) displayed a more scattered pattern of PDiv. Among all algorithms, InSRWFB consistently achieved the lowest PDiv across all subsampling percentages and strategies, demonstrating superior preservation of the original network structure. This pattern was observed consistently across low (Fig. 2a), medium (Fig. 2b), and high (Fig. 2c) abundance networks, demonstrating the robustness of InSRWFB in maintaining global structural similarity. However, under the combined strategy, SB outperformed InSRWFB in the high-abundance clone-based network (Fig. 2f).
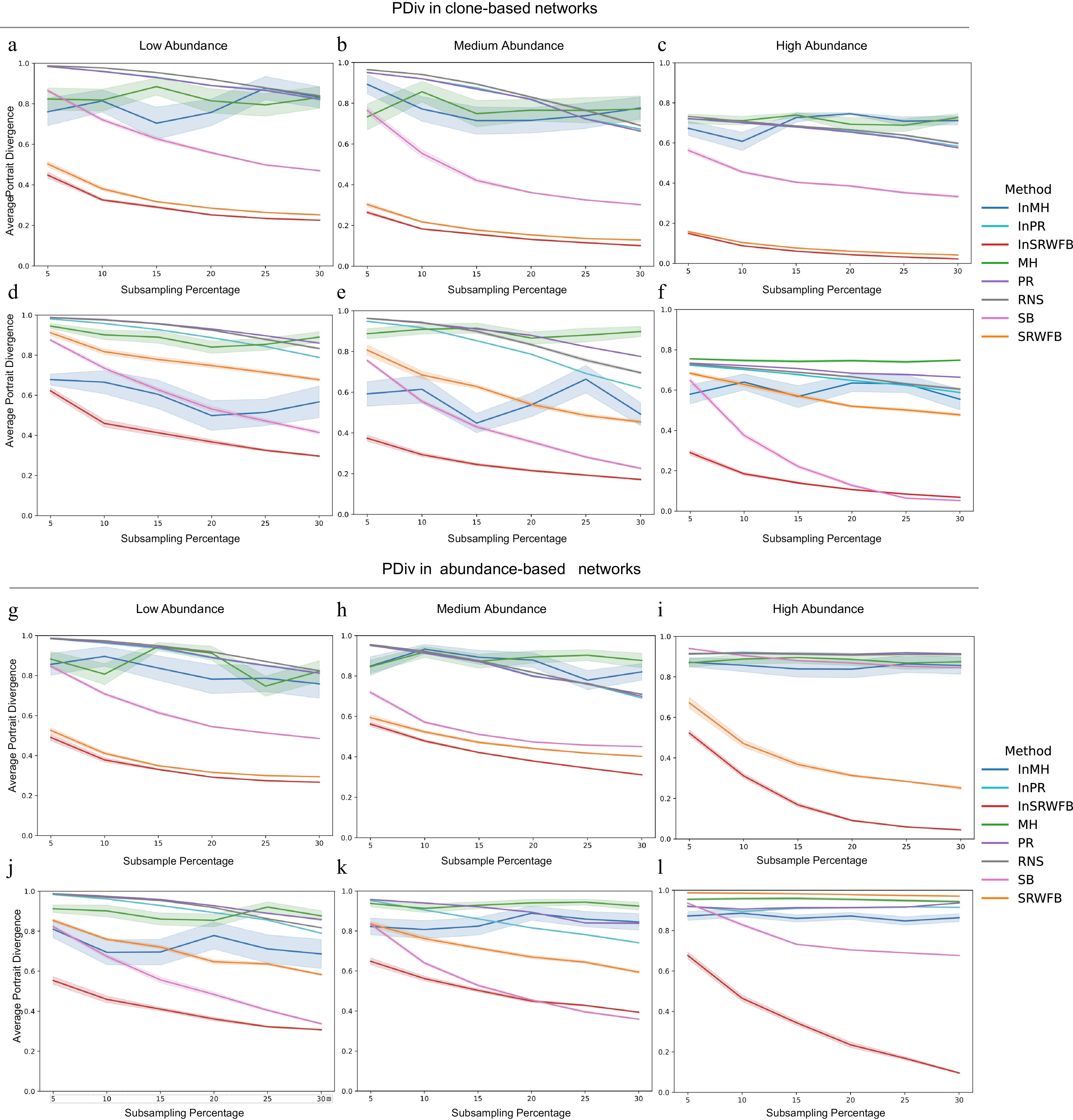
Figure 2.
Average Portrait Divergence (PDiv) between the original network and the subnetwork. PDiv under the direct strategy for clone-based networks with (a) low, (b) medium, and (c) high abundance level. PDiv under the combined strategy for clone-based networks with (d) low, (e) medium, and (f) high (f) abundance level. PDiv under the direct strategy for abundance-based networks with (g) low, (h) medium, and (i) high abundance level. PDiv under the combined strategy for abundance-based networks with (j) low, (k) medium, and (l) high abundance level. Each curve represents PDiv change across different subsampling percentages (5% to 30%) for one of the subsampling algorithms, including Metropolis-Hastings (MH), PageRank (PR), Random Node Sampling (RNS), Snowball Sampling (SB), and SRWFB, and Induced Metropolis-Hastings (InMH), Induced PageRank (InPR), Induced Simple Random Walk with Fly Back (InSRWFB). For each subsampling percentage, 20 replicates were performed per method, and the lines represent the mean PD across replicates. Shaded areas indicate mean+/- standard error. Lower PD values indicate greater structural similarity between the subnetwork and original networks.
Next, we examined how each subsampling algorithm preserved network structure and characteristics. As shown in the heatmaps (Supplementary Fig. S3), changes in maximum degree decreased with higher subsampling percentages. The smallest changes were found in InSRWFB under the direct strategy, indicating its ability to maintain the largest clusters. For assortativity, while SRWFB and InSRWFB were found consistently decreased with increased subsampling percentage, we also noticed that SB maintained relative low difference under both strategies (Supplementary Fig. S4). For transitivity, which measures local compactness, InSRWFB consistently preserved original values across abundance levels, especially at higher subsampling percentages. In contrast, InMH, InPR, and PR were more influenced by abundance level (Supplementary Fig. S5), while SB demonstrated stability across abundance levels.
Given the inherently low density of both the original and subsampled TCR networks, we normalized the density change by the original values. InSRWFB, PR, and RNS exhibited low-density distortion across subsampling percentages (Supplementary Fig. S6). RNS generally performed the best, likely due to the network sparsity, but was less robust than InSRWFB and PR, especially in high-abundance networks with more uneven connectivity. Based on the above findings, we conclude that InSRWFB is the most effective and consistent subsampling algorithm for preserving both the overall network structure and consistent subsampling algorithm for preserving both the overall structure and key topological properties of TCR networks across different abundance levels.
Performance evaluation for subsampling abundance-based TCR networks
-
Following the same approach for evaluating the subsampling performance in clone-based TCR networks, we selected one baseline sample from each abundance group and performed 20 subsampling replicates for each sample. The performance patterns of the subsampling algorithms in abundance-based TCR networks were similar to those observed in clone-based networks in terms of PDiv. A similar separation in low-abundance networks under the direct strategy was observed (Fig. 2g), which became less distinguishable in other scenarios (Fig. 2h−l). SRWFB and InSRWFB consistently having lower PDiv values, indicating their robust performance across different network settings (Fig. 2g−i). InSRWFB consistently achieved the lowest PDiv across all subsampling percentages and abundance levels. The advantage of InSRWFB was most evident in high-abundance TCR networks (Fig. 2i and l). Additionally, SB showed lower PDiv values under the combined strategy than the direct strategy, suggesting a compensatory effect, particularly at higher sampling percentages.
In terms of network properties, InSRWFB, SRWFB, and SB maintained a consistent advantage in preserving maximum degree and assortativity, while also showing competitive performance in transitivity (Supplementary Figs S7–S9). For transitivity, as observed in clone-based networks, performance differences among algorithms diminished in medium- and high-abundance networks (Supplementary Figs S5 and S9). Regarding density, InSRWFB and PR again showed higher stability than RNS, while some extreme distortions were observed in MH, SB, and SRWFB, particularly at lower subsampling percentages (Supplementary Fig. S10).
Evaluation based on clinical interpretation
-
In addition to quantifying network structures, researchers have also considered network properties as potential biomarkers for correlating with clinical outcomes and evaluating the treatment effects. Therefore, we used Cohen's d to measure the effect size of changes in network properties from baseline to post-treatment. Given the superior performance of InSRWFB in preserving network characteristics, we focused on evaluating the effect size derived from original and InSRWFB-subsampled networks.
Overall, InSRWFB demonstrated a stronger ability to reflect the magnitude of change, with the direct strategy showing more stable performance than the combined strategy in clone-based networks (Fig. 3a−d). As the subsampling percentage increased, Cohen's d values from the direct InSRWFB approach closely approximated those from the original networks, whereas the combined InSRWFB showed deviation and fluctuation. Specifically, for assortativity, maximum degree, transitivity, and density, the direct InSRWFB closely matched the original effect size at subsampling percentages of 15% or higher (Fig. 3a−d). The change of maximum degree gradually converges toward those of the original networks as the subsampling percentage increases (Fig. 3b).
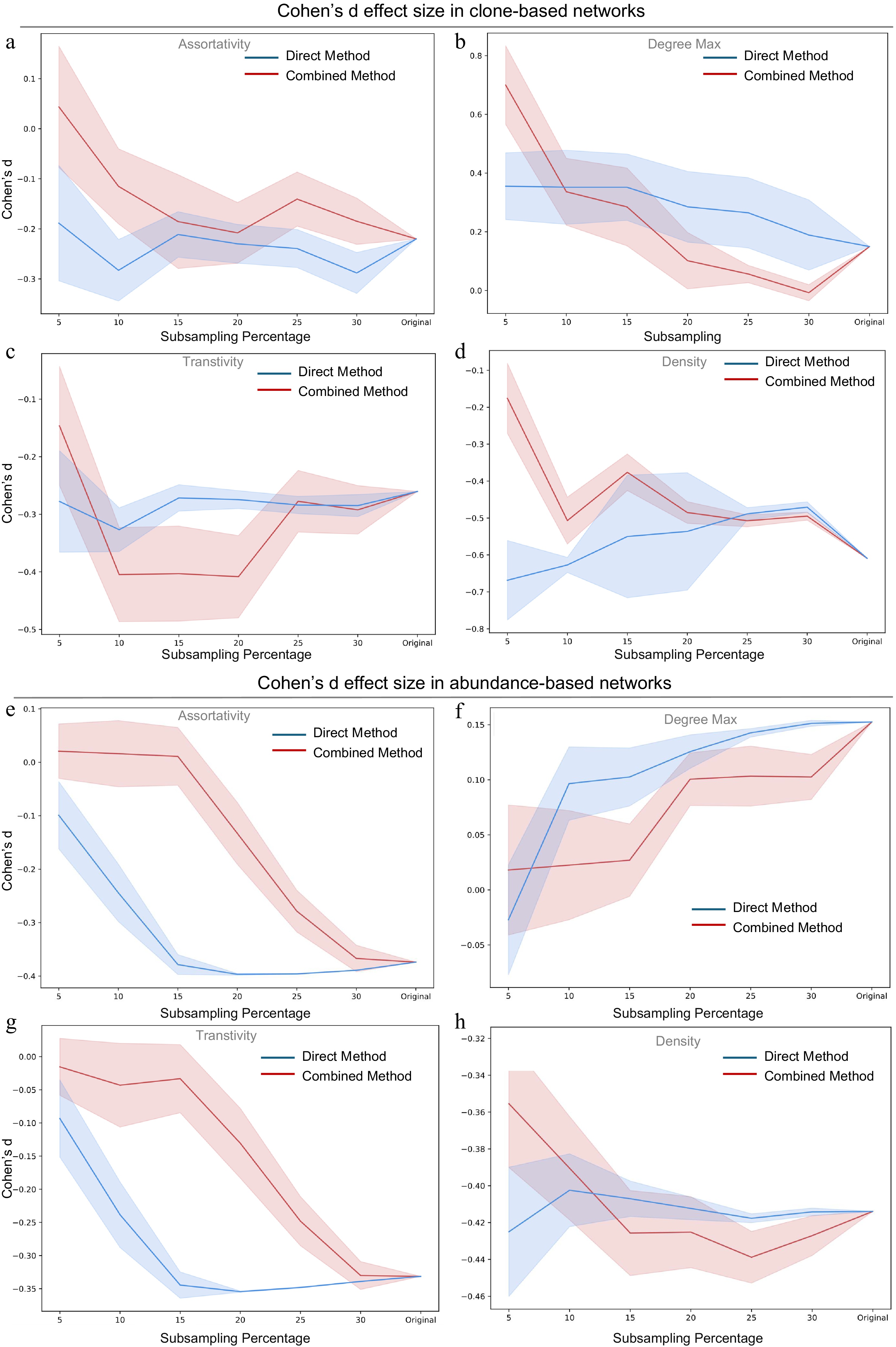
Figure 3.
Cohen's d effect size of original network and subnetworks using Induced Simple Random Walk with Fly Back (InSRWFB) at different subsampling percentages (5% to 30%). Cohen's d effect size of (a) assortativity, (b) maximum degree, (c) transitivity, and (d) density by InSRWFB for clone-based networks. Cohen's d effect size of (e) assortativity, (f) maximum degree, (g) transitivity, and (h) density by InSRWFB for abundance-based networks. Cohen's d values were computed based on 11 patients at two time points to assess the magnitude and direction of change in the four network properties. For each patient and time point, 20 independent subsampling replicates were generated, and the resulting d values were averaged across replicates. Blue and red lines represent the direct and combined strategies, respectively. Shaded areas indicate mean +/− standard error.
In abundance-based networks, InSRWFB also closely approximated the original network's effect sizes as the subsampling percentage increased (Fig. 3e−h). The Cohen's d of maximum degree and density gradually converged towards the original network's value (Fig. 3f, h), while the effect sizes for assortativity and transitivity aligned with the original effect size at subsampling percentages of 15% (Fig. 3e, g). The direct method exhibited smoother trajectories and more closely matched the original network properties even at low subsampling percentages, whereas the combined method only showed comparable performance at higher sampling percentages (i.e., 30%).
By definition, positive and negative values of Cohen's d indicate an increase and a decrease in the network properties after the treatment, respectively. For example, a positive assortativity effect size reflects greater dispersion of sequence similarity, consistent with broader polyclonal activity and adequate diversification. In contrast, a negative value indicates a shift toward disassortativity, where similarity concentrates around a few expanded clones, consistent with antigen-driven activation and centralized clonal expansion[5]. In our data, both clone-based and abundance-based, the assortativity effect size was negative, implying that post-treatment networks became more disassortative. An increase in degree post-treatment indicates that, on average, clones acquire more sequence-similar neighbors, consistent with greater convergence around shared motifs/epitopes or expansion of similarity neighborhoods. A decrease in transitivity indicates looser local structure, consistent with dispersed diversification. A negative density effect size indicates sparser connectivity, consistent with increased repertoire breadth and novel/unique clones.
Comparison of direct vs combined strategies
-
While we initially expected improved performance from the combined strategy, it was generally less effective than the direct strategy for most algorithms based on both PDiv and network properties. InSRWFB under the direct strategy consistently achieved comparable or lower PDiv across conditions. SRWFB also performed well under the direct strategy for clone-based and abundance-based networks at different abundance levels (Fig. 2a−c, g, h), except for the abundance-based networks with high abundance, where its effectiveness declined. Under the combined strategy, SRWFB was consistently less effective than InSRWFB (Fig. 2d−f, j−l). While PR and InPR yielded similar performance under the direct strategy, their results diverged under the combined strategy as the subsampling percentage increased (Fig. 2). InMH displayed promising performance in low- and middle-abundant networks under the combined strategy; however, this advantage diminished under the direct strategy in abundance-based networks (Fig. 2i, l).
Nevertheless, SB consistently yielded lower PDiv under the combined strategy than under the direct strategy, especially at high subsampling percentages (Fig. 2d−f, j−l). At the same time, a similar pattern was observed for maximum degree, suggesting a substantial compensatory effect of SB under the combined strategy. Further investigation on Cohen's d effect size in two types of networks supported this improvement as well (Supplementary Fig. S11). Unlike InSRWFB, SB under the combined strategies outperformed the direct strategy from 10% subsampling onward across all four network properties in clone-based networks, indicating that the combined strategy partially corrected the deviations in both direction and magnitude (Supplementary Fig. S11a−S11d). From 15% subsampling onward in abundance-based networks, the combined approach consistently shows smaller absolute deviation from the original effect size than the direct approach at every subsampling percentage, indicating better fidelity in assortativity, transitivity, and density, while both strategies produced similar trends (Supplementary Fig. S11e, S11g, S11h).
To facilitate a clear, results-oriented comparison across scenarios, we include a summary figure (Supplementary Fig. S13) that identifies the method achieving the minimum PDiv for each dataset–abundance stratum–sampling condition. The only scenarios not dominated by InSRWFB are the Abundance-based, Medium-abundance networks and the Clone-based, High-abundance networks at 25% and 30% sampling, where SB attains the PDiv. Across all remaining dataset–abundance–sampling combinations, InSRWFB is consistently selected as the best performer, with no other methods emerging as optimal.
Computation time and memory consumption
-
Time and memory requirements have long presented challenges in the analysis of large biological networks such as TCR networks. To evaluate how effectively subsampling methods alleviate this computational burden, we recorded runtime and peak memory usage during the construction of both the original networks and their corresponding subnetworks. All networks were built on a local Mac personal computer with an Apple M3 Max chip and 64 GB of memory. We investigated all algorithms by calculating the ratios of time or memory usage for subsampled networks relative to their original counterparts across all 22 TCR samples at different subsampling percentages (5%, 10%, 15%, 20%, 25%, and 30%). Results are summarized across samples using the median with the range (Min, Max) (Table 4). Given the inherent difference in network complexity, clone-based and abundance-based networks were investigated separately. As expected, both relative runtime and peak memory usage increased with higher subsampling percentages (Fig. 4). Across both network types, the relative runtime of all algorithms showed a consistent linear relationship with the subsampling percentage (Fig. 4a, b). In contrast, memory consumption displayed a distinct and stable pattern for both clone-based and abundance-based networks. Peak memory consumption reached a plateau at a 10% subsampling percentage across all evaluated algorithms, indicating that the dominant memory cost arises during the initial stage of loading the full repertoire network into memory. Increasing the subsampling fraction has a negligible impact on additional memory demand. This stability demonstrates that higher subsampling percentages can be scaled without substantially increasing hardware requirements.
Table 4. Relative time and memory consumption of InSRWFB across subsampling percentages.
Subsampling percentage Relative time: Median (Min, Max) Relative memory: Median (Min, Max) Clone-based network Abundance-based network Clone-based network Abundance-based network 5 0.96% (0.6%, 3.1%) 2.4% (1.0%, 7.2%) 3.1% (0.1%, 7.2%) 10.8% (0.8%, 29.9%) 10 2.01% (1.2%, 4.6%) 4.5% (2.5%, 14.6%) 2.8% (1.6%, 10.6%) 13.3% (1.1%, 38.4%) 15 3.62% (2.1%, 8.5%) 7.3% (4.1%, 15.2%) 4.4% (1.5%, 19.2%) 17.2% (1.1%, 37.1%) 20 5.10% (3.4%, 9.8%) 9.2% (5.7%, 18.1%) 6.8% (1.2%, 19.7%) 16.8% (1.2%, 30.7%) 25 7.43% (4.8%, 12.9%) 11.8% (7.9%, 20.9%) 10.9% (1.6%, 38.7%) 20.3% (3.9%, 31.3%) 30 10.17% (6.9%, 16.3%) 13.8% (10.5%, 23.2%) 13.7% (1.2%, 48.0%) 20.7% (4.4%, 27.7%) 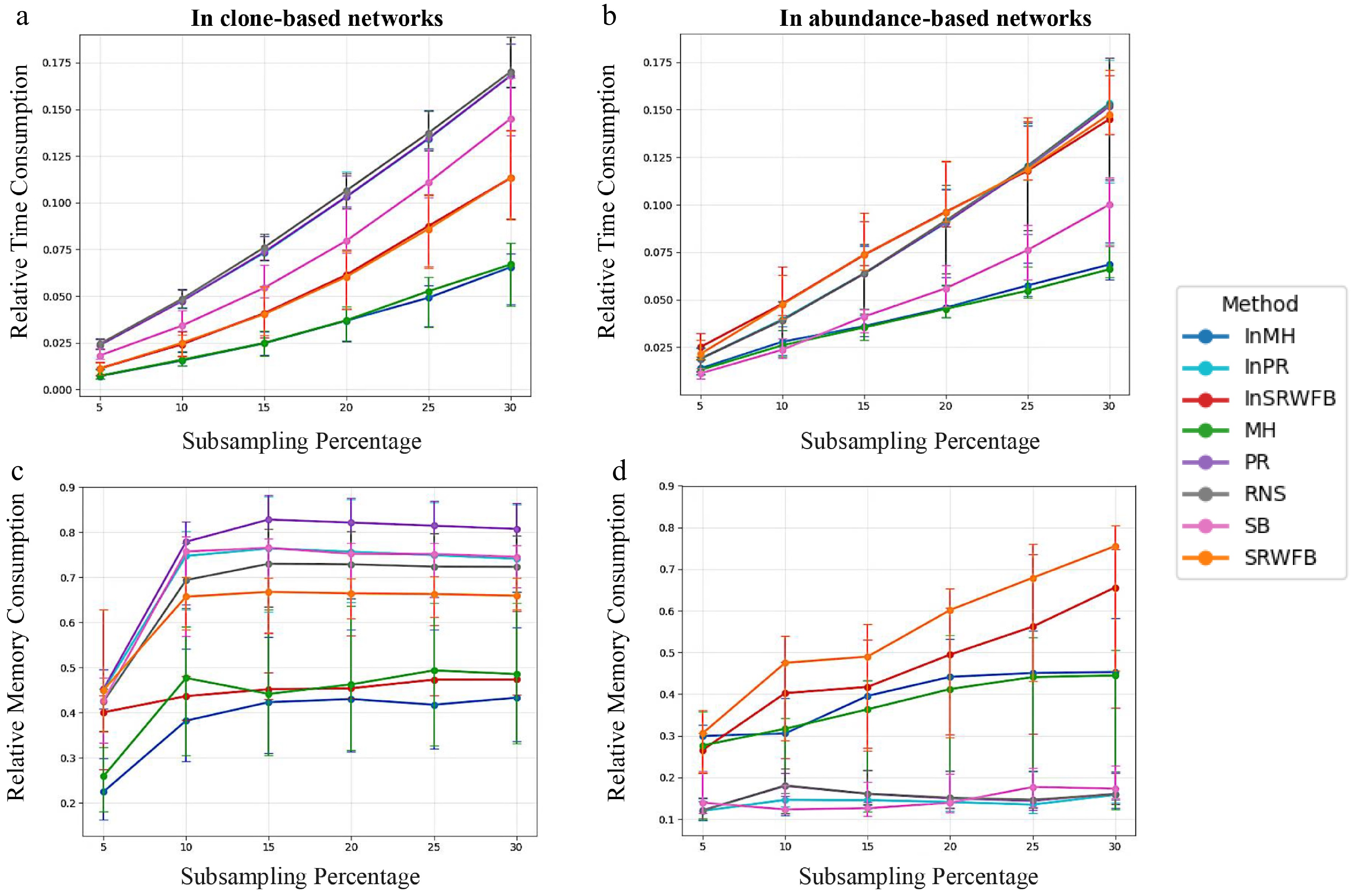
Figure 4.
Computation Time and Memory Consumption Median relative runtime across 22 TCR samples for each subsampling method and percentage (5%–30%) in (a) clone-based, and (b) abundance-based networks. Median relative peak memory across the same samples in (c) clone-based, and (d) abundance-based networks. Error bars indicate the interquartile range (IQR; 25th–75th percentiles).
-
We conducted a systematic benchmarking of established subsampling algorithms and strategies to maintain network structure similarity and support downstream analyses. Our workflow is, in principle, applicable to Adaptive Immune Receptor Repertoire (AIRR) networks beyond TCR, including BCR, but receptor-specific preprocessing (e.g., somatic hypermutation and chain-aware similarity measures) and independent validation are required. Unlike denser biological networks, such as PPI networks or neural networks[23], TCR networks, especially clone-based ones, are extremely sparse. The high sparsity of the TCR network is evidenced by a proportion of isolated nodes (degree = 0) exceeding 50% (Supplementary Fig. S1a) and edge density far below 0.003 (Supplementary Fig. S12). In such extremely sparse TCR networks, structural information is concentrated in a few large, densely connected clusters. Therefore, edge-based sampling approaches that explore and retain these informative regions are better suited for preserving global topology.
Among all the methods tested, InSRWFB consistently yielded the lowest PDiv for both clone-based and abundance-based TCR networks. Although this performance is accompanied by a linear increase in memory demand in abundance-based settings (Fig. 4d), it suggests that InSRWFB's traversal-based strategy effectively prioritizes the preservation of key topological features over mere memory minimization. This makes it particularly suitable for scalable TCR network analysis where structural integrity is paramount. By traversing along edges and incorporating the fly-back mechanism, InSRWFB ensures a thorough exploration of local clusters, preserving the key topological features more effectively than other methods.
InSRWFB performs better in networks with higher abundance (Fig. 2). This may be explained by evidence that antigen-driven clonal expansion often involves multiple TCRs with similar sequences[6,7,30], resulting in higher edge density and more complex clustering. As a result, InSRWFB's traversal-based strategy is better able to capture these structures. Interestingly, the variance of PDiv increased with abundance, suggesting greater structural heterogeneity associated with immune activation.
Given the ability of InSRWFB to capture local structural features, it is expected that it shows superior performance in retaining local (max degree and transitivity) and global (assortativity, density) (Supplementary Figs S3−S6, S7−S10) metrics. This observation suggests that preserving local features contributes to maintaining global structural features indirectly, particularly in denser connected networks. In the context of the TCR repertoire, network density can serve as an indicator of immune activation, where low density suggests limited clonal expansion, implying that the T cell population remains inactive or only weakly stimulated. Therefore, the ability of InSRWFB to effectively preserve network density makes it a promising approach for accurate interpretation of immune response. RNS appeared to perform comparably to InSRWFB, which might be misleading, as uniform node selection in sparse networks can inadvertently mimic structural patterns without capturing biologically relevant features.
Beyond the absolute values of network properties, the direction and magnitude of the change, quantified by Cohen's d effect size, are crucial for interpreting immune dynamics. Cohen's d effect size provides a quantitative summary of whether and to what extent immune activity has changed between time points or conditions, and has been widely used in network studies, including neuroscience[5,31]. InSRWFB consistently preserves the direction and the scale of change across subsampling percentages, aligning closely with the negative value of original networks, ensuring appropriate clinical interpretation. While higher subsampling percentages naturally retain more information, InSRWFB uniquely maintains meaningful effect size estimates even at low subsampling levels, making it a practical choice for large-scale or incomplete datasets. However, we did not perform full downstream validation (e.g., treatment-response prediction) to confirm that subsampled networks reproduce identical clinical conclusions, which requires labeled cohorts and is planned for future work.
Although the combined strategy generally underperformed the direct strategy across most subsampling algorithms, including InSRWFB, SB under the combined strategy demonstrated a compensatory effect in PDiv, assortativity, and effect size. Because the combined strategy separates isolated nodes from connected nodes, SB performs particularly well in capturing the characteristics of connected structures, improving its overall effectiveness in this context. While our study focuses on sparse TCR networks, these findings suggest that SB may demonstrate competitive or superior performance in denser or fully connected biological networks, which requires further investigation in dense networks.
While time and memory consumption increase proportionally with subsampling percentage (Fig. 4), the improvement in portrait divergence for InSRWFB-sampled subnetworks under direct strategy exhibits a marginal effect pattern, beginning to plateau around 15% (Fig. 2a−f). Similarly, the differences in most network properties compared to the original network start to stabilize near this proportion (Supplementary Figs S3−S10). Notably, the effect sizes of key metrics tend to converge toward the original network values at approximately 15% (Fig. 3a−d, g, h), suggesting a marginal gain beyond this point.
Our decision to cap clone abundances > 500 prior to constructing abundance-weighted networks was a computational/numerical-stability safeguard to stabilize edge weights, not an attempt to downplay clonal expansion. The cap does not alter node identity or connectivity; it only bounds the influence of extreme counts on weight scaling. Analyses that rely on the magnitude of massive expansions should be performed without a cap or using a monotone transform (e.g., log1p). Our primary claims focus on the comparative behavior of subsampling methods and topology-preserving metrics (density, transitivity, assortativity, PDiv), which depend mainly on connectivity patterns rather than exact weight magnitudes and are qualitatively stable under bounded weight scaling. We document the capping step in the released code and allow users to deactivate it or set a higher threshold as resources permit, enabling re-analysis without the cap when emphasis is on highly expanded clones.
Several limitations of this study should be acknowledged. First, the analysis is based on a relatively small sample size (n = 11), limiting the generalizability of the findings across disease settings, treatment contexts, and cohort characteristics. Although each sample contains a high-complexity TCR repertoire, validation in larger and more diverse clinical cohorts will be necessary to assess the robustness of these conclusions. Second, the number of subsampling replicates was fixed at 20 to balance computational feasibility with empirical stability of network metrics. While this choice was sufficient for the scenarios examined here, different network sizes or evaluation metrics may require alternative replication strategies. Finally, this study evaluates subsampling methods primarily in terms of their ability to preserve network structural fidelity. Although structural preservation is a necessary condition for valid downstream biological and clinical interpretation, it does not guarantee inferential equivalence. The impact of subsampling-induced structural differences on specific downstream tasks, such as differential repertoire analysis or clinical outcome association, was not assessed and remains an important direction for future work.
-
Our analysis highlights InSRWFB as a robust and scalable subsampling approach for preserving critical features of TCR networks while reducing computational burden. InSRWFB consistently outperformed other evaluated algorithms in maintaining both local and global network properties, as well as preserving effect size directionality and magnitude across TCR abundance levels. Our results demonstrate that a subsampling rate of approximately 15% offers an optimal trade-off between computational efficiency and structural fidelity, with marginal gains plateauing beyond this proportion. While InSRWFB was the top-performing algorithm under the conditions examined, the stability patterns observed here arise empirically and may vary across TCR network regimes. Our study also reveals the potential value of alternative strategies such as snowball sampling with the combined strategy in denser or more connected networks, suggesting that algorithm choice should be guided by underlying network topology, motivating future evaluation across a broader range of repertoire contexts.
We would like to thank National Cancer Institute, National Institutes of Health and National Library of Medicine, National Institutes of Health, for their support. Wu H, Le T, Yang H, Le P, and Zhang L are partially supported by the National Cancer Institute, National Institutes of Health (Grant number R21CA264381). Wu H, Le T, Yang H, Le P, and Zhang L are partially supported by the National Library of Medicine, National Institutes of Health (Grant number R01LM013763-01A1). Oh D has received grants from the National Institutes of Health (K08AI139375), the Damon Runyon Cancer Research Foundation (Clinical Investigator Award, CI 110-21), the V Foundation (Translational Adult Cancer Grant), and the Prostate Cancer Foundation (Young Investigator Award). This study incorporates and extends the open-source code from Ashish Aggarwal's Graph_Sampling repository (MIT License). We gratefully acknowledge the original author's contribution.
-
Patients were accrued at the University of California San Francisco and Oregon Health & Science University, Portland, OR, USA. Informed consent was signed by each patient. Institutional Review Boards of both institutions approved the study protocols, including the collection of biospecimens.
-
The authors confirm their contributions to this study as follows: conceptualization: Le T, Yang H, Zhang L; methodology: Wu H, Le T, Yang H, Le P, Zhang L; visualization, validation: Wu H, Le T, Zhang L; writing − original draft: Wu H, Zhang L; formal analysis: Wu H, Le T; writing − review and editing: Le T, Yang H, Le P, Oh D. All authors reviewed the results and approved the final version of the manuscript.
-
The data underlying this article are available upon request. The codes are also available at https://github.com/mlizhangx/Network-Subsampling.
-
DYO has received institutional research support from Merck, PACT Pharma, the Parker Institute for Cancer Immunotherapy, Poseida Therapeutics, TCR2 Therapeutics, Roche/Genentech, Allogene Therapeutics, and Nutcracker Therapeutics; fees for travel and accommodations from Roche/Genentech and Poseida Therapeutics; and consulting fees from Revelation Partners, outside of the submitted work. The other authors declare that they have no conflict of interest.
- Supplementary Table S1 The uumber of nodes in original TCR networks.
- Supplementary Fig. S1 Proportion of isolated nodes of original TCR network.
- Supplementary Fig. S2 Sensitivity analysis: impact of algorithm parameters on portrait divergence across subsampling percentages.
- Supplementary Fig. S3 Difference in maximum degree between subsampled and original clone-based networks across sampling algorithms and abundance levels.
- Supplementary Fig. S4 Difference in Assortativity between subsampled and original clone-based networks across sampling algorithms and abundance levels.
- Supplementary Fig. S5 Difference in transitivity between subsampled and original clone-based networks across sampling algorithms and abundance levels.
- Supplementary Fig. S6 Relative difference in density between subsampled and original clone-based networks across sampling algorithms and abundance levels.
- Supplementary Fig. S7 Difference in maximum degree between subsampled and original abundance-based networks across sampling algorithms and abundance levels.
- Supplementary Fig. S8 Difference in assortativity between subsampled and original abundance-based networks across sampling algorithms and abundance levels.
- Supplementary Fig. S9 Difference in transitivity between subsampled and original abundance-based networks across sampling algorithms and abundance levels.
- Supplementary Fig. S10 Relative difference in density between subsampled and original abundance-based networks across sampling algorithms and abundance levels.
- Supplementary Fig. S11 Cohen's d effect size of original network and subnetworks using Snow Ball (SB) at different subsampling percentages (5% to 30%).
- Supplementary Fig. S12 Edge density across samples in clone-based and abundance-based TCR networks.
- Supplementary Fig. S13 Summary of best subsampling in different scenarios.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wu H, Le T, Yang H, Le P, Oh D, et al. 2026. A systematic evaluation of network subsampling methods for T-cell receptor repertoire network. Statistics Innovation 3: e006 doi: 10.48130/stati-0026-0005 

A systematic evaluation of network subsampling methods for T-cell receptor repertoire network
- Received: 04 June 2025
- Revised: 09 February 2026
- Accepted: 22 February 2026
- Published online: 07 May 2026
Abstract: The T-cell receptor (TCR) is vital in adaptive immune responses. To investigate the structure and function of the TCR repertoire, researchers constructed TCR networks based on TCR sequences. However, the complexity and scale of TCR networks frequently exceed computational capabilities. We aimed to identify effective subsampling approaches to reduce computational burden while preserving essential network properties. We systematically evaluated network subsampling algorithms, including Random Node Sampling (RNS), Page Rank (PR), Metropolis-Hastings (MH), and Simple Random Walk Sampling with Fly Back (SRWFB) and three induced methods: Induced PR (InPR), Induced MH (InMH), and Induced SRWFB (InSRWFB) across varying subsampling percentages (5% to 30%) under direct and combined strategies. We applied these approaches to TCR sequence data from 11 ibrutinib-treated pancreatic cancer patients. For performance evaluation, portrait divergence (PDiv) and several network properties were utilized. We also assessed how well subnetworks preserved the direction and magnitude of changes by Cohen's d effect size. InSRWFB consistently outperformed, yielding the lowest PDiv and best preserving network properties. It also maintained stable effect sizes, becoming more pronounced in higher-abundance repertoires. As the first systematic evaluation of subsampling techniques in the context of immune repertoires, our work provides important insights and a reference for future immunological network analysis.