








-
The improvement of sugarcane (Saccharum spp.) is severely hindered by complex mixed ploidy, aneuploidy, and interspecific hybridization. Recently, Huang et al. constructed the first multiscale sugarcane pangenome graph integrating nine assemblies[1]. Overcoming traditional linear reference limitations, this framework increased genomic diversity capture from 34% to 82% and identified key agronomic loci (e.g., sugar content, leaf angle) via the novel dosage genome-wide association study (GWAS) method. We highlight this study's technical, statistical, and biological dimensions.
Modern sugarcane cultivars typically possess extremely high chromosome numbers (2n = 100−130) and complex homologous/homoeologous relationships[2], primarily resulting from interspecific hybridization and backcrossing between S. spontaneum and S. officinarum over a century ago. Traditional single linear reference sequences cannot simultaneously accommodate variations arising from different ploidy levels, chromosome numbers, and introgression backgrounds, leading to substantial multimapping and reference bias in multiomics analyses. To overcome these bottlenecks, gap-free assemblies of modern cultivars (e.g., R570, SP80-3280) provide an indispensable baseline for multigenome integration. Concurrently, telomere-to-telomere (T2T) assemblies of wild relatives (e.g., Erianthus rockii) have enriched the allele pool for tracing introgression and stress-resilient traits[3]. Furthermore, massive resequencing of global germplasm has accumulated critical data for large-scale population studies.
-
The research team utilized the PGGB (PanGenome Graph Builder) pipeline (Fig. 1) to integrate nine chromosome-level assemblies covering four species, including modern cultivars, progenitor species, and outgroups. Statistically, this super-pangenome graph contains approximately 425.9 million nodes with a cumulative length of 14.7 Gb, achieving a compression ratio of 34.03%. The graph structure retains 47–57 haplotypes and captures approximately 82% of sugarcane's genomic diversity, significantly higher than the 34% captured by a single linear reference genome. The study achieved a multiscale span from genome to gene and protein levels, defining complexity metrics such as "graph Link" (gL) and "graph Bubble" (gB) to characterize the degree of variation within homologous gene clusters.
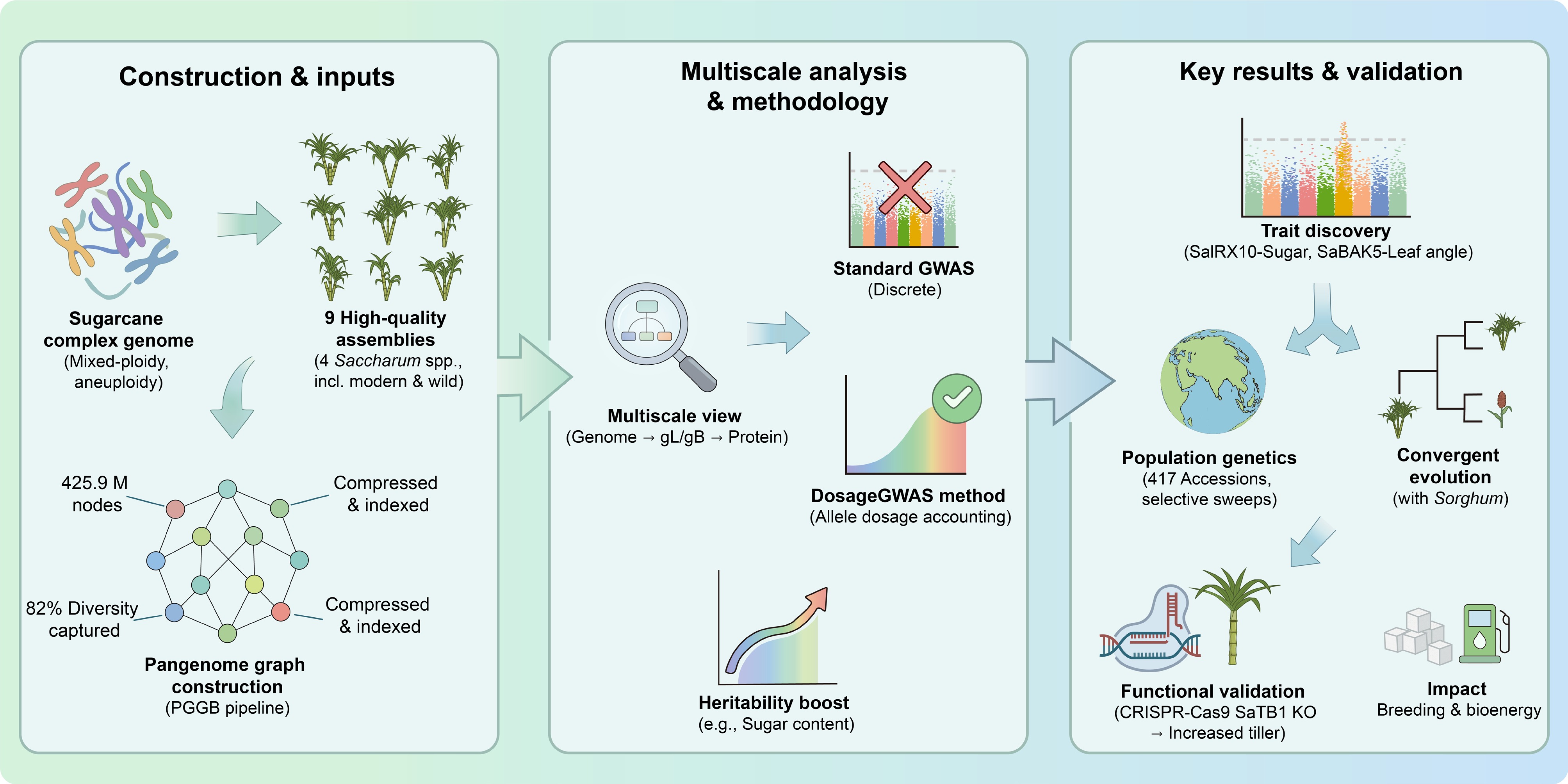
Figure 1.
Pipeline and main results of the pangenome graph of sugarcane genomics.
Multiomics analysis and alignment optimization
-
As reported previously, the graph reference genome significantly mitigates alignment challenges in high-ploidy genomes. In assay for transposase-accessible chromatin (ATAC)-seq analysis, the unique mapping rate using graph alignment exceeds 50%, which is more than double that of a haploid linear reference genome and 10 times that of a homologous allele-aware linear reference genome. This improvement has enabled researchers to identify an additional 6,831–19,202 accessible chromatin regions (ACRs) and resolve allele-specific regulation of sucrose transport loci such as SUT1.
-
The authors conducted deep resequencing (approximately 18 Tb of data) on 417 mixed-ploidy sugarcane accessions. Through selection sweep scans and cross population composite likelihood ratio (XP-CLR) analysis, the study identified 1,664 genes under selection, enriched in sucrose metabolism (e.g., SWEETS, SUS, STP) and plant architecture regulation (e.g., TB1, tin1) pathways. Beyond these established pathways, recent breakthroughs have cloned and functionally verified new members of the sucrose transporter (SUT) gene family, resolving their allele-specific regulation to better dictate the sucrose sink's strength. Moreover, the genomic focus has rapidly broadened to encompass stress resilience; novel networks such as ScDREB and ScbHLH governing drought and cold tolerance, as well as nucleotide-binding site leucine-rich repeat (NBS-LRR) gene clusters for smut resistance, have been isolated, reflecting the urgent diversity of modern breeding targets. Coevolutionary analysis revealed significant genome-wide convergent selection between sugarcane and sorghum (Sorghum bicolor) (232 syntenic orthologous pairs), whereas convergence with maize (Zea mays) was weaker[4].
DosageGWAS: deciphering the enigma of allele dosage
-
To address the challenge of enumerating genotypes in high-ploidy species, the study introduced the DosageGWAS method. It directly models continuous allele dosage rather than relying on traditional discrete genotypes. In an efficacy assessment, DosageGWAS significantly improved the heritability estimates of traits, increasing the average by 0.06 for sugar traits and 0.19 for leaf angle. Through the gene mining method, precise dosage-phenotype gradient associations were discovered near the SaIRX10 (sugar-related) and SaBAK5 (leaf angle-related) loci.
-
We believe that the core contribution of this work lies in establishing a genomics toolchain that is applicable to complex polyploids. From a technical perspective, the researchers not only solved the issues of compression and representation for the sugarcane genome but, more importantly, utilized the mathematical abstraction of "continuous dosage" to circumvent the difficulties of aneuploidy and precise genotype identification that are notoriously difficult to handle in genetic analyses of polyploid species. This methodological framework demonstrates universality and has been successfully extended to wheat (Triticum aestivum, hexaploid), cotton (Gossypium hirsutum, allotetraploid), and potato (Solanum tuberosum, autotetraploid), exhibiting strong cross-species scalability. This extension aligns seamlessly with recent monumental achievements, such as the gapless assembly of the hexaploid wheat genome, signaling a methodological paradigm shift across all complex Gramineae crops.
Furthermore, the study achieved a closed loop of functional verification; using clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) to knock out the SaTB1 gene successfully increased tiller numbers. As a tillering regulator, SaTB1 can be a direct target for yield improvement in breeding. To further complete the technical closed loop from discovery to field validation, highly efficient ribonucleoprotein (RNP) delivery and multiallelic editing systems in the embryogenic callus of sugarcane are currently being rapidly optimized.
Finally, the graph pangenome supports more precise marker design and allele mining, laying the data foundation for genomic selection (GS) in sugarcane[5]. Although machine learning is currently leveraged to translate genomic data into field performance, there is a critical need for artificial intelligence-based large models that synthesize multiscale omics and field data within agricultural digital twins for dynamic, climate-resilient simulations. Despite this promising trajectory, several foundational limitations remain. Extracting predictive value from multiscale graphs demands massive computational resources, whereas accurate allele dosage estimation still relies on costly deep sequencing. Furthermore, even with accurate models, recalcitrant transformation efficiencies and linkage drag in complex hybrids pose significant hurdles to the physical deployment of these genomic resources.
Altogether, the research by Huang et al. (2026) propels sugarcane genomics from the linear reference era, transitioning from fragmented linear references to comprehensive graph-based panoramic views. It is achieved by multiscale graph pangenome technology. Furthermore, their proposed DosageGWAS method provides a new blueprint for association analysis in all complex polyploid species. This represents a major methodological advancement in plant genomics. Collectively, this study offers a new gene resource map and analytical tools for breeding high-yield and high-quality sugarcane.
This work was funded by the Chinese Academy of Tropical Agricultural Sciences for Science and Technology Innovation Team of National Tropical Agricultural Science Center (CATASCXTD202402), the Project of State Key Laboratory of Tropical Crop Breeding (SKLTCBYWF202503, NKLTCBCXTD24, and NKLTCBCXTD38), and China Agriculture Research System of MOF and MARA (CARS-17). Chen F acknowledges funding from Hainan University (XTC2022NYB04).
-
The authors confirm their contributions to the paper as follows: conceived this opinion article: Que Y, Chen F; analyzed the article and data: Zhang J, Wu Q, Li J, Ye F; drafted the article: Zhang J, Chen F, Que Y. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
-
The authors declare that they have no conflict of interest.
-
Received 12 February 2026; Accepted 30 March 2026; Published online 7 May 2026
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang J, Wu Q, Li J, Ye F, Chen F, et al. 2026. Beyond linearity: a multiscale pangenome map for complex sugarcane. Tropical Plants 5: e014 doi: 10.48130/tp-0026-0015 

Beyond linearity: a multiscale pangenome map for complex sugarcane
- Received: 12 February 2026
- Revised: 12 March 2026
- Accepted: 30 March 2026
- Published online: 07 May 2026
-
Key words:
- Sugarcane (Saccharum spp.) /
- Multiscale pangenome graph /
- DosageGWAS /
- Complex polyploidy