








-
In a recent issue of Cancer Cell, Zhang et al.[1] identify that Paneth-like phenotypic transition plays a pivotal role in mediating resistance to combined KRAS and epidermal growth factor receptor (EGFR) inhibition in colorectal cancer (CRC). Recent breakthroughs in KRAS-targeted therapeutics have reshaped the therapeutic paradigm for KRAS-mutant cancers. In CRC, selective inhibitors against oncogenic variants (e.g., G12C and G12D mutations) are being combined with anti-EGFR monoclonal antibodies[2,3]. These combinatorial regimens have exciting clinical efficacy in early-phase trials. Despite these advances, the emergence of acquired resistance inevitably limits their long-term utility[4,5]. Previously documented resistance mechanisms included the acquisition of secondary mutations and adaptive feedback activation of parallel oncogenic signaling cascades, such as hyperactivated extracellular signal-regulated kinase (ERK) or AKT pathways[6−8]. Beyond these canonical mechanisms, lineage plasticity, including processes such as epithelial-mesenchymal transition and neuroendocrine-like transitions[9], which enable cancer cells to switch cellular identities under therapeutic stress, could drive tumor progression and therapeutic responsiveness[10]. However, the role of lineage plasticity in resistance to dual KRAS-EGFR blockade remains unclear, highlighting the importance of illustrating these mechanisms to prolong the durability of targeted therapy responses in KRAS-mutant CRC.
Zhang and colleagues employ a range of experimental approaches, including genetically engineered mice, patient-derived organoids (PDO), and clinical specimens, to investigate resistance mechanisms in CRC. They identify an evolutionarily conserved resistance pathway linking the preclinical models to the human condition. Multiple sequencing analyses show that CRC cells undergo a Paneth-like cell state after dual inhibition. Paneth-like cells are a secretory lineage normally restricted to the bases of small intestinal crypts, which sustain tissue homeostasis by secreting antimicrobial peptides[11]. Previous research has shown that Paneth-like cells can arise directly from olfactomedin 4+ (OLFM4+) stem cells[12] and that Regenerating islet-derived 4+ (Reg4+) deep crypt secretory cells serve as Paneth-like cell equivalents in the colon crypt niche[13]. CRISPR-Cas9-edited reporter lineage-tracing assays confirm that this shift results from the transdifferentiation of CRC cells rather than the expansion of existing Paneth-like cells. Notably, the proportion of Paneth-like cells increases after treatment, but this phenotypic change is completely reversible upon withdrawal of therapy, highlighting its role as a stress adaptation. Single-cell RNA sequencing (scRNA-seq) analysis reveals a stepwise transition in CRC cells. Initially, CRC cells enter a diapause-like, drug-tolerant persister state, which has been linked to cell adaptation under therapeutic stress. Over time, these drug-tolerant persisters acquire the Paneth-like phenotype. This sequential adaptive program provides the mechanistic insights into how cancer cells survive initial therapy and develop acquired resistance. These findings have broad implications for targeting minimal residual disease (MRD), a central objective in delaying tumor relapse.
-
To identify drivers of Paneth-like lineage plasticity, the authors perform an unbiased CRISPR knockout screen focused on transcription factors. The approach reveals SMAD family member 1 (SMAD1) as a critical regulator. SMAD1 is upregulated after KRAS-EGFR inhibition, co-localized with Paneth-like cell markers, and its genetic ablation abrogates the transition and restores drug sensitivity. Interestingly, SMAD1 functions independently from the canonical bone morphogenetic protein (BMP) signal and SMAD4, acting as a noncanonical role in therapy-induced lineage plasticity. Mechanistic study has shown that SMAD1 directly binds the fibroblast growth factor receptor 3 (FGFR3) promoter and induces transcription of FGFR3 as well as its ligands. Activation of the SMAD1-FGFR3 axis promotes both Paneth-like plasticity and reactivation of the mitogen-activated protein kinase (MAPK) pathway, a process sustained in Paneth-like cells to mediate resistance (Fig.1). Ultimately, Paneth-like cells exhibit higher basal KRAS-GTP levels and a better MAPK rebound after treatment, explaining the lower sensitivity of these cells to combined KRAS-EGFR inhibition compared to non-Paneth-like cells.
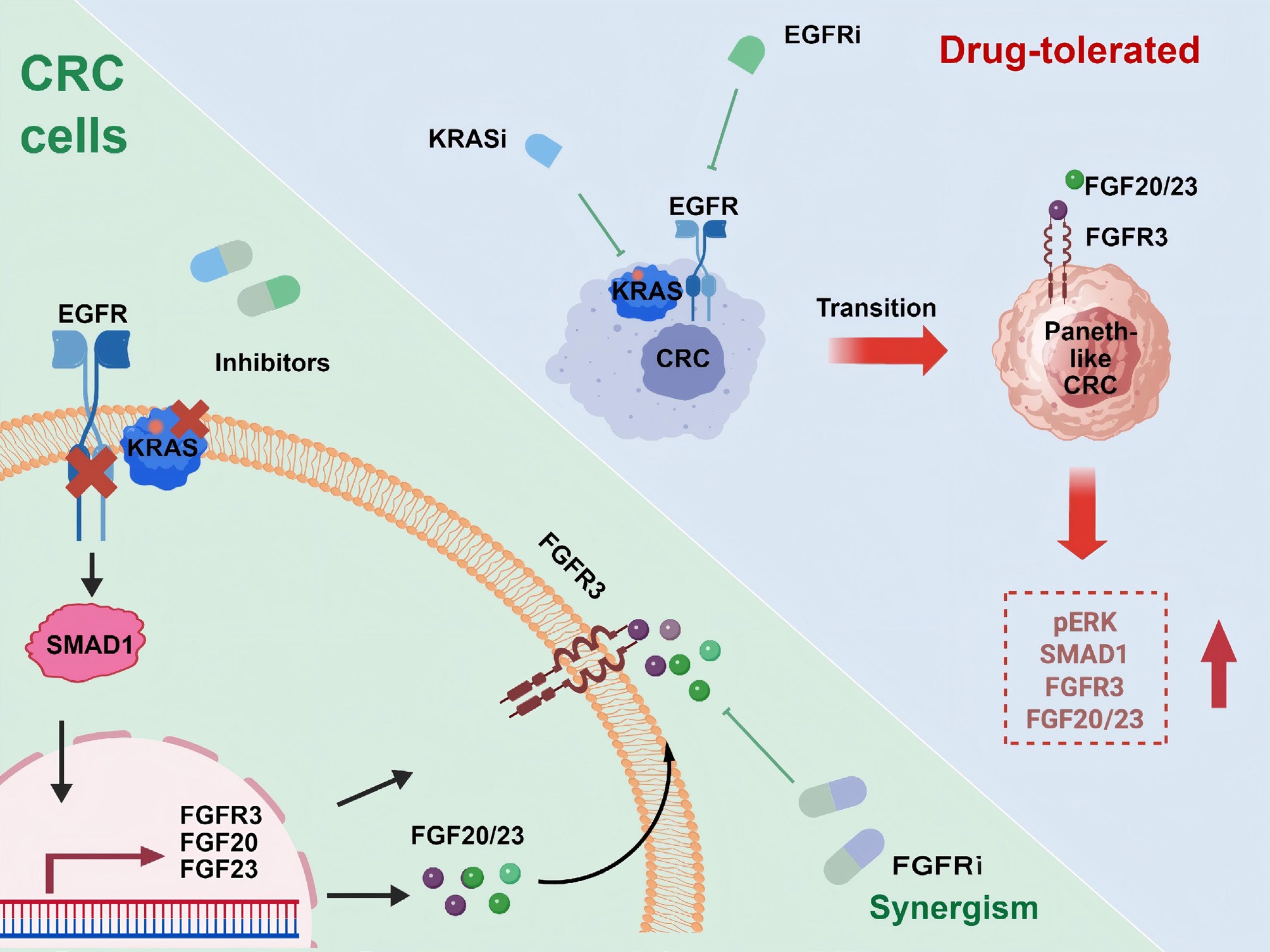
Figure 1.
Schematic representation of CRC cell plasticity in resistance to dual KRAS and EGFR inhibition. Dual inhibition of KRAS and EGFR induces resistance in CRC via Paneth-like transdifferentiation of CRC cells (top right). Non-classic SMAD1 signaling activation drives Paneth-like lineage plasticity through upregulation of FGFR3 and its ligands, and FGFR3 blockade reverses resistance to KRAS and EGFR dual inhibition in CRC (bottom-left).
-
Importantly, the authors discover that pharmacological inhibition of FGFR3 synergizes with dual KRAS-EGFR inhibition. This combination more effectively suppresses MAPK signaling and tumor cell proliferation in vitro, while also reducing Paneth-like cell markers and inhibiting CRC progression in both mouse models and PDOs. The clinical relevance of this mechanism is supported by the observation that residual tumors from human CRC treated with dual KRAS-EGFR therapy are enriched for Paneth-like cells. Notably, FGFR inhibitor monotherapy doesn't produce an antitumor response in CRC models, underscoring the specific role of FGFR3 in mediating plasticity-induced resistance. These findings highlight the necessity of combining FGFR inhibition with KRAS-EGFR targeting and have important clinical implications for maximizing the benefit of current targeted therapies.
-
This work opens several promising avenues for future study. First, identifying the upstream signals that drive SMAD1 expression after KRAS-EGFR inhibition could uncover new biomarkers for MRD or reveal therapeutic targets to prevent the Paneth-like transition. Second, employing advanced sequencing technologies, multiplex immunohistochemistry (IHC) staining of biopsy samples, and ctDNA analysis to screen for FGFR pathway mutations would allow the assessment of SMAD/FGF pathway activation and other markers of the Paneth-like transition. These strategies could enable precise patient stratification and help guide combination therapy with FGFR inhibitors. Third, as previously reported, pan-FGFR inhibitors often cause hyperphosphataemia, a side effect that can lead to organ failure, due to inhibition of FGF23 signaling[14]. Developing isoform-selective FGFR3 inhibitors could enhance the therapeutic selectivity and reduce off-target toxicity compared to pan-FGFR inhibitors. Finally, the authors find that FGFR hyperactivation is linked to increased phosphorylated ERK (pERK) activity, a well-established resistance mechanism in KRAS-mutant cancers treated with RAS/MAPK inhibitors. Thus, targeting the SMAD1-FGFR3 axis may broaden the therapeutic options for other KRAS-mutant cancers, such as pancreatic cancer and lung cancers, further enhancing the potential of this approach.
In conclusion, Zhang et al.[1] identify a novel non-genetic resistance mechanism in KRAS-mutant CRC and provide strong preclinical support of combinatorial therapy with FGFR inhibitors. Co-targeting FGFR3 with KRAS and EGFR inhibitors may improve the durability of treatment responses in patients with KRAS-mutant CRC. Ongoing work on Paneth-like lineage plasticity and its regulators will be crucial for overcoming resistance and developing more efficient, long-term therapies against KRAS-mutant malignancies. This study exemplifies how deciphering non-genetic resistance pathways can inform rational combination therapies, offering new hope for improving patient outcomes.
This work is supported by the National Natural Science Foundation of China (Grant Nos 82573166, 82504847) and the Jiangsu Provincial Natural Science Foundation Youth Fund Project (Grant No. BK20251567).
-
Not applicable.
-
The authors confirm contributions to the paper as follows: study conception and design: Yin Z, Guo X, Liu X; literature review: Yin Z, Guo X; draft manuscript preparation: Yin Z, Guo X. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Zeyi Yin, Xingyu Guo
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yin Z, Guo X, Liu X, DeNardo DG. 2026. Reprogramming paneth-like plasticity: FGFR3 as a key to overcoming KRAS-EGFR resistance in CRC. Targetome 2(3): e021 doi: 10.48130/targetome-0026-0020 

Reprogramming paneth-like plasticity: FGFR3 as a key to overcoming KRAS-EGFR resistance in CRC
- Received: 20 February 2026
- Revised: 26 March 2026
- Accepted: 03 April 2026
- Published online: 19 May 2026
Abstract: Dual KRAS-epidermal growth factor receptor (EGFR) inhibition holds promise for KRAS-mutant colorectal cancer (CRC), yet drug resistance remains a key hurdle. This study by Zhang et al. identifies the SMAD family member 1 (SMAD1)-fibroblast growth factor receptor 3 (FGFR3) axis as the driver of Paneth-like lineage plasticity that mediates such resistance and demonstrates that FGFR3 targeting restores drug sensitivity and synergizes with dual pathway inhibition in preclinical models[
-
Key words:
- Colorectal cancer /
- EGFR /
- KRAS /
- FGFR /
- Resistance