








-
Osteoarthritis (OA) is the most common joint disease, affecting all articular joints (i.e., knee, hip, ankle, temporomandibular joint [TMJ], etc.), resulting in an enormous socioeconomic burden every year[1]. Lifetime prevalence studies estimate that OA will affect over 40% of the global population, with higher rates in female patients[2]. However, the exact mechanism of OA pathogenesis remains unclear, and there are no licensed disease-modifying OA drugs (DMOADs) for OA conditions. Currently, OA patients have few medical options but to take palliatives such as nonsteroidal anti-inflammatory drugs[3], and injections of hyaluronic acid[4].
OA has long been regarded as a joint degenerative disease due to constant wear and tear. Recently, OA has also been defined as a heterogeneous and multifaceted disease with different molecular subtypes and clinical phenotypes. Cartilage degradation-driven OA subtype is characterized by cartilage loss and damage due to mechanical wear-out, and is the most common OA subtype[5]. Another subtype is bone remodeling-driven OA. In this subtype, activated subchondral bone resorption occurs at an early stage, preceding cartilage deterioration. Patients with bone remodeling-driven OA often demonstrate typical MRI signs of subchondral bone edema or bone marrow lesions. With OA development, the subchondral bone plate and trabeculae become thicker, showing sclerosis along with decreased subchondral trabeculae separation[5]. In addition, inflammation-driven subtype is another common OA subtype and is identified by transcriptome atlas of chronic low-grade inflammation[6]. Synovial inflammation gives rise to inflammation symptoms including pain, warmth, and swelling through elevated pro-inflammatory cytokine levels, adipokines, chemokines, and metabolites. Patients with inflammation-driven OA phenotypes have similar manifestations to rheumatoid arthritis, and show positive correlations between high systemic inflammation markers (C-responsive proteins), and OA severity[7]. Apart from the above three subtypes, many other factors also participate in OA pathogenesis and make OA a complex multi-factorial disease beyond the joint. For instance, obesity plays an important role in OA pathogenesis. Obesity-incurred excessive mechanical loading contributes to cartilage damage and malalignment in load-bearing joints[8]. Obesity also increases systematic pro-inflammation cytokines and the resultant synovitis, thus leading to non-load bearing joint OA (i.e., hand and wrist). Besides obesity, other metabolic dysfunctions (i.e., hypertension, dyslipidemia, and glucose intolerance, etc.) also contribute to OA development by inducing the expression of proinflammatory factors, as well as degradative enzymes. Thus, increasing evidence suggests that metabolic OA is another OA subtype[9].
All these joint environmental alterations give rise to the existence of detrimental molecules and pathogenic cellular biology, and the related signaling pathways, thus accelerating OA. OA is therefore a complex pathological process involving interactions between various internal and environmental factors. Mounting evidence indicates that OA is a polygenic inheritance[10]. Epigenetic modification is more versatile and reversible compared with genomic modifications, and varies by cell type and gene[10]. Emerging studies demonstrate that epigenetic dysregulation, such as DNA hyper-/hypo-methylation, histone post-translational modifications, non-coding RNA networks, and the newly characterized m6A RNA methylation plays a pivotal role in driving osteoarthritis pathogenesis[11]. Currently, some interventions such as physical exercise and muscle activity-related myokine, show therapeutic effects on OA pathogenesis via epigenetic mechanisms[12]. However, the key coupling factors between the environmentally epigenetic stimuli (i.e., mechanical overloading, environmental inflammation, and metabolite stimulation, etc.) and OA initiation and progression, have not been fully identified. Thus, this study aims to summarize the recently identified epigenetic studies and the relevant therapeutic targets in OA. Based on this, the future epigenetic OA research directions and underlying epigenetic DMOADs are further discussed.
-
In mammals, DNA methylation compromises three steps: DNA methyl group writing, reading, and erasing[13]. DNA methyl writers (enzymes) catalyze the additional methyl groups onto cytosine residues, and initiate DNA methylation (Fig. 1a). In OA joints, DNA hypermethylation can trigger silencing of specific genes. DNA methyl readers recognize and subsequently bind to methyl groups to regulate the target gene expression. Erasers finally modify and remove the methyl group[13]. Physiologically, these processes are precisely coordinated and maintain homeostasis within the joint.
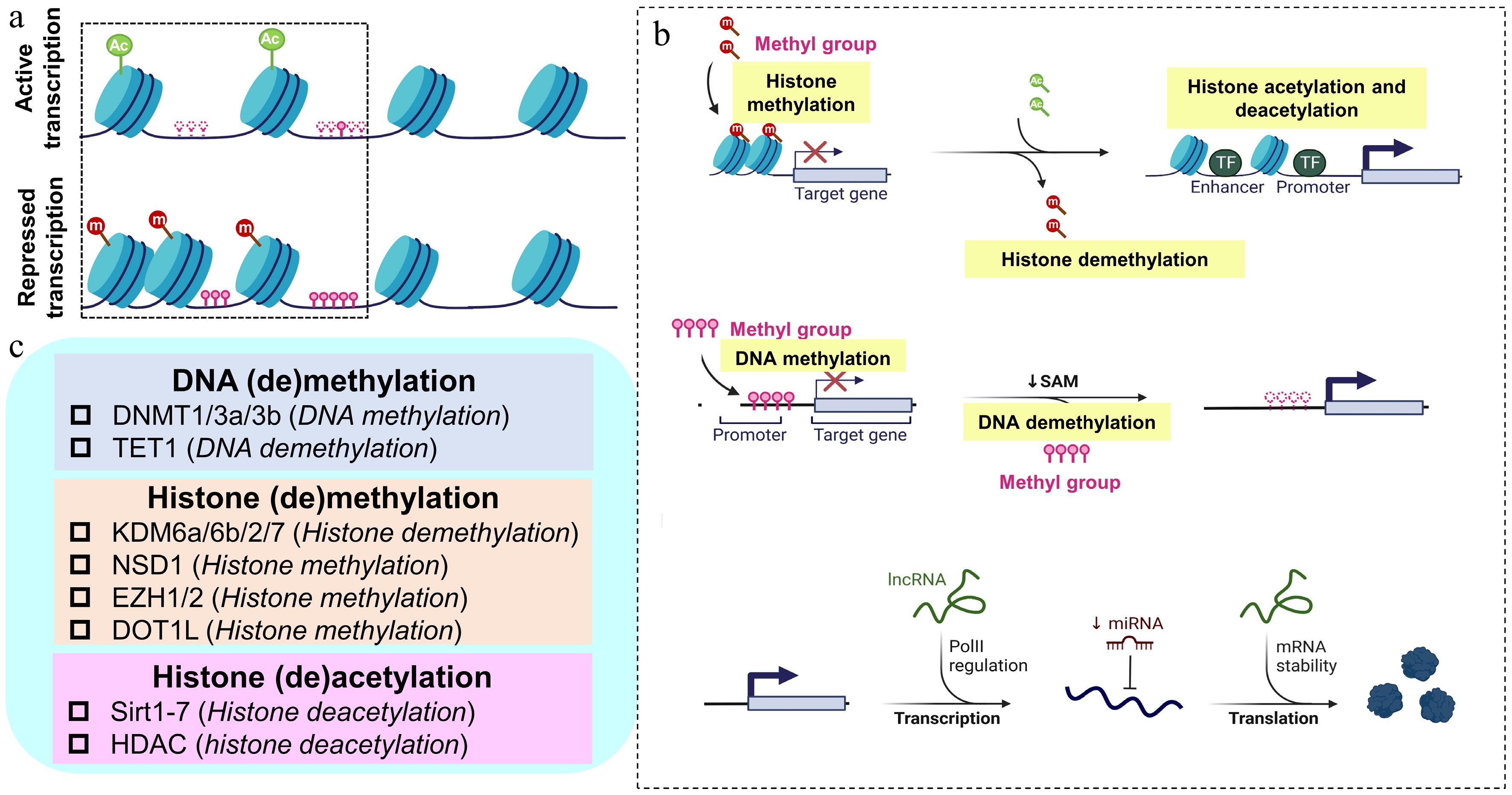
Figure 1.
Summary of DNA methylation, histone modifications, and non-coding RNA in OA. (a) Dynamic changes of chromatin and the resultant gene expression in OA. Upper panel: regions of chromatin with activate transcription; lower panel: regions of chromatin with repressed transcription. (b) The epigenetic modulatory process of histone modifications, DNA methylation, and non-coding RNA; upper panel: histone modifications (histone modifications included methylation and acetylation); middle panel: DNA methylation (DNA methylation is dynamically maintaining the balance by adding methyl groups and deleting methyl groups in normal chondrocytes, and abnormal changes in DNA methylation occur in the promoter regions of related genes and signaling pathways in OA chondrocytes); lower panel: non-coding RNAs including lncRNA and miRNA modulate the gene expression during and post transcription. (c) Different classes of epigenetic enzymes responsible for the structural modification of chromosomes (DNA and histone) and its regulation effects. Ac: acetylation; DNMT: DNA methyltransferase; DOT1L: Disruptor of telomeric silencing 1-like; m: methylation; EZH: enhancer of zeste homolog; HDAC: histone deacetylase; KDM: histone lysine demethylase; NSD: H3K36 methyltransferase; Sirtuins: Sirt; TET: ten–eleven translocation enzyme.
-
DNA methylation is catalyzed by DNA methyltransferases (Dnmts, DNA methyl writer) that transfer a methyl group from C to the fifth carbon of a cytosine residue to form 5mC. Dnmt3a and Dnmt3b are de novo DNA methyl writers, and are responsible for transferring methyl groups onto naked DNA strands; meanwhile, Dnmt1 is responsible for maintaining the DNA methylation pattern during replication.
Dnmt1/3a are elevated in both human and mice OA chondrocytes, meanwhile, the hypermethylation and resultant reduction of chondroprotective PPARγ accelerates OA progression[14]. Besides knee cartilage, PPARγ and its promoter hypermethylation also contribute to TMJ OA progression in a partial discectomy (PDE)-induced rat TMJOA model[15]. Knee cartilage is covered with hyaline cartilage; meanwhile, the TMJ surface is coated with fibrocartilage instead[16]. These results imply that epigenetically aberrant hypermethylation of the PPARγ promoter is involved in both hyaline cartilage and fibrocartilage degeneration. Therefore, targeting aberrant epigenetic modifications shows bright therapeutic promise for both hyaline cartilage and fibrocartilage degeneration and OA. Inhibition of Dnmt1/3a by 5aza[14,15], or diacerein[17] effectively restores the suppression of PPARγ and alleviates surgically induced OA in mice. In addition to restoring chondro-protective genes, 5Aza also boosts the stemness of BMSCs and enhances cartilage regeneration by upregulating the heterochromatin-regulatory gene SPI1[18].
In contrast to Dnmt1/3a, Dnmt3b is downregulated in OA chondrocytes[19]. Dnmt3b gain-of-function mice demonstrate a lower mitochondrial activity in knee chondrocytes and exhibit a joint protection against cartilage deterioration after meniscal ligament injury[19]. Interestingly, Dnmt3b−/− mice exhibit a remarkable delay in cartilage formation, maturation, and removal in the murine tibial fracture models[19]. These Dnmt3b−/− mice displayed less bone formation and impaired bony callus remodeling during fracture repair[19]. These results indicate that the epigenetic aberration in chondrocytes not only impacts cartilage homeostasis, but also affects endochondral ossification in fracture healing. In addition to PPARγ, in OA chondrocytes the Sirt1 promoter is also hypermethylated, which impairs the binding of C/EBPα on the SIRT1 promoter in OA chondrocytes[20]. Inhibition of DNMTs by 5aza (5μM) suppresses the OA-like phenotype (i.e., elevation of MMPs) in chondrocytes[20]. Interestingly, the hypomethylation of some genes also contributes to OA. For instance, a Ser/Thr phosphatase, Phlpp1, is found to undergo a promoter hypomethylation in OA chondrocytes, and the resultant upregulation of Phlpp1 is correlated with OA severity[21]. This phenomenon might probably be attributed to the accumulation of inflammatory transcription factor binding sites within the Phlpp1 promoter. Thus, the hypomethylation of Phlpp1 breaks the normally condensed and blocked binding sites of inflammation transcription factors such as the NF-κB (−1,532 bp from the transcription starting site), THAP1 (−1,462 bp transcription starting site), and HIF1 (−1,504 to −1,491 bp transcription starting site), thus creating an inflammation environment within the knee joint.
Femoroacetabular impingement (FAI) is a condition that occurs when the femoral head pinches up against the acetabulum[22]. The constant mechanical overloading makes FAI a pre-status of OA[22,23]. Recently, clinical specimens from FAI patients' results indicate that Dnmt3b is downregulated in FAI and advanced hip OA cartilage. Further, due to the reduction of Dnmt3b in OA chondrocytes, the 4-aminobutyrate aminotransferase promoter hypomethylates and subsequently accumulates in cartilage, leading to a series of catabolic changes in hip cartilage[24]. These results strengthen the importance of DNA methylation in early OA initiation. In another study, Evseenko et al. showed Dnmt3b was regulated by the upstream STAT3[25]. Deleting STAT3 in fetal chondrocytes could induce global hypermethylation in chondrocytes and accelerated OA progression in the mouse OA (DMM) model. In contrast, activating STAT3 by a small molecule could induce hypomethylation in primary human chondrocytes and relive the OA-like phenotype[25]. In the future, the mechanism of epigenetic balance maintained by Dnmts (Dnmt1/3a and Dnmt3b) still needs to be illustrated, and mechanistic investigation of different responses of Dnmt1/3a (increase), and Dnmt3b (decrease) in OA conditions is also worthy of attention.
Apart from directly modulating gene expression in chondrocytes, Dnmts can also regulate chondrocyte-derived factors in the ECM. The increased stiffness of cartilage is a hallmark of cartilage aging and OA, which is also a crucial external environmental mechanical factor. Stiff cartilage-incurred mechanical changes drive epigenetic repression of the anti-senescence factor α-Klotho in aged mice[26]. During cartilage aging, the downregulation of α-Klotho is modulated by high stiffness—triggered Dnmt1 upregulation[26]. Blockage of Dnmt1 by 5Aza (10 μM) could ameliorate high stiffness-incurred aging phenotype in chondrocytes[26]. In addition to cartilage ECM aging, environmental chemicals may also contribute to OA by epigenetic modification. Triclocarban (TCC, C13H9Cl3N2O) is a widely used bacteriostatic compound and has been identified as an environmental endocrine-disrupting chemical[27]. Soaked within a TCC (3 μM) contained environment, zebrafish anal fin joints exhibit an OA-like change, including a narrow joint space and loss of alcian blue staining. The mechanistic exploration in human chondrocytes reveals that type II collagen is suppressed in TCC (3 μM) treatment due to Dnmt1 activation and subsequent hypermethylation[27].
DNA methyl reader and eraser: reading and erasing methylation information
-
In mammal cells, DNA methylation and demethylation are precisely coordinated by the DNA methylation writer, reader, and eraser. In contrast to DNA methyl writers, DNA methyl readers can selectively recognize methylated DNA sites and direct the recruitment of chromatin remodelers such as methyltransferases to establish target-specific transcriptional repression. The methyl-CpG-binding domain (MBD) family is the primary DNA methylation reader, and the MBD family is implicated in many diseases (i.e., pulmonary fibrosis, acute kidney injury, etc.), currently, little attention has been attached to the roles of DNA methyl readers in OA, which might be a potential target in OA research. The erasing mechanism is mediated by erasing-related enzymes i.e., the ten-eleven translocation (TET) enzymes, including TET1, TET2, and TET3. The TET family facilitates DNA demethylation by oxidizing 5mC through iterative hydroxylation reactions, converting it into 5hmC (5-methylcytosine), 5fC (5-formylcytosine), and 5caC (5-carboxylcytosine) intermediates that are subsequently excised by base excision repair machinery[28].
Tet1 and Tet3 are upregulated in hip OA cartilage and synovium tissues[29]. Meanwhile, the levels of 5hmC increase in cartilage after OA induction. Nidhi Bhutani et al. demonstrated that Tet1−/− mice lost 98% 5hmC-enriched sites in OA, indicating the important role of Tet1 in regulating 5hmC deposition. Both genetic deletion (TET1−/− mice), or pharmaceutical inhibition (2-HG: I.A. injection, 100 mM twice a week for 5 weeks) of cartilage TET1 attenuates the DMM-induced OA phenotype[30]. Furthermore, they isolated skeletal stem cells (SSCs) from Tet1+/+ and Tet1−/− mice and compared the difference between the two groups of SSCs. Loss of Tet1 alters the lineage tree distribution and niche composition of SSCs, and enhances the chondrogenic differentiation ability of SSCs. The in vivo results indicated that inhibition of Tet1 (I.A. injection of L-2-HG at 100 mM, starting at 4 weeks after tibia-loading) increased the cartilage regeneration after OA induction. More importantly, they discovered that inhibition of TET1 by L-2-HG, primary human SSCs (PDPN+CD146−CD73+CD164+), and CPSC (PDPN+CD146−CD164+CD73−) exhibited significantly higher chondrogenesis effects[31]. Interestingly, they further discovered that melatonin (1 μM) could mimic the effects of Tet inhibition and enhanced GAG accumulation in cartilage pellet[31]. Therefore, melatonin holds the potential to be an available epigenetic DMOAD in the future.
-
Modifications of histone proteins affect cell processes, i.e., the activation/suppression of transcription, chromosome packaging, DNA damage, and DNA repair (Fig. 1a, b). Histone modification is an important post-translational process and is critical in the regulation of gene expression. In addition to directly regulating gene expression at a post-translational level, some histone modifications also remodel the structure of chromatin, or recruit histone modifiers to modulate the target gene[32].
Histone acetylation
-
Histone acetylation breaks the interaction between the histones and DNA, loosens chromatin structure, and usually activates transcription of local genes, thus regulating cell proliferation, differentiation, and development.
One of the most important histone acetylation regulators is the histone deacetylase (HDAC) family. HADCs regulate the transcription process by binding to transcription factors and catalyzing the removal of acetyl groups from both histone and non-histone proteins, particularly targeting lysine residues, thereby influencing transcriptional activity[33]. Generally, HDACs repress transcription by deacetylating histone lysines. The deacetylation increases chromatin density by restoring positive charges, thus hindering transcription factor accessibility. HDACs have also proved to be related with multiple inflammatory factors. Global inhibition of HDACs shows OA protection in mice[34]. Cai et al., evidenced that TSA, an HDAC inhibitor (HDACi), activated the Nrf2 signaling pathway and markedly ameliorated cartilage degeneration in both MIA and DMM-induced OA models (daily subcutaneous injection for 3 weeks; 2.0 mg/kg)[34]. Meanwhile Ohzono et al., found that the administration of another HDACi Panobinostat, could target and activate Foxo1, and alleviate the structural changes and joint pain in OA mice[35]. In addition to global inhibition of HDACs, regional inhibition of HDACs by Panobinostat (weekly I.A. injection of 2 or 10 μg) attenuates nociceptive behaviors in an ACLT-induced rat OA model, including secondary mechanical allodynia and weight-bearing distribution[36]. Specifically, the HDAC family has several subtypes, and different HDAC subtypes have distinctive effects. HDAC6 is well-recognized for its role in tubulin acetylation and microtubule remodeling. HDAC6 is required for both the interleukin (IL)-1 induction of MMP expression and pro-inflammatory interleukin expression in chondrocytes. Mechanistically, Young et al. identified HDAC6 as a critical regulator of IL-1β-triggered NF-κB transduction in OA chondrocytes, controlling downstream expression of MMPs and inflammation-related genes[37]. A small molecule, Tubastatin A, was identified as an HDAC6 inhibitor and can attenuate OA by normalizing mitochondrial function[38].
Another crucial histone acetylation regulator (deacetylases) is the Sirtuin (Sirt) family (Fig. 1c). The Sirt family is composed of seven members (Sirt1-7), and functions as either NAD+-dependent deacetylases, deacylases, or ADP-ribosyltransferases. Interestingly, different Sirt members are located in different subcellular sites, including the nucleus (Sirt6/7), nucleus and cytosol (Sirt1/2), and mitochondria (Sirt3/4/5). The Sirt family maintains chondrocyte function by regulating ECM homeostasis, modulating chondrocyte metabolism, preventing chondrocyte aging, decreasing chondrocyte apoptosis, and enhancing chondrocyte autophagy. For example, the progressive loss of Sirt6 activity in aging chondrocytes disrupts metabolic homeostasis, triggering NF-κB-mediated catabolic responses that promote cartilage matrix breakdown and OA development[39]. Knocking down Sirt6 in cartilage mimics ageing and accelerates OA development in mice models. Several downstream pathways have been identified to be related to Sirt6 in chondrocytes. For instance, Sirt6 deficiency remarkably inhibits IGF-1 signaling and lowers the expression of several ECM-related genes essential for cartilage homeostasis, such as Col2a1. Thus, Sirt6/IGF-1 axis may be necessary to protect cartilage from injury- or age-associated OA[40]. Physiologically, Sirt6 suppresses IL-15/JAK3/STAT5 signaling in cartilage, and the downregulation of Sirt6 in cartilage contributes to the upregulation of IL-15/JAK3/STAT5 signaling, the resultant chondrocytes senescence, and OA progression[41]. OA chondrocyte-specific aptamer (tgg2)-functionalized PEGylated PAMAM nanoparticles are then fabricated to precisely deliver MPL (chemical agonist of Sirt6), in the treatment attenuated OA in mice and human explant samples[41]. These results imply that Sirt6 regulates the histone structural alterations, and impacts the exposure of transcription sites in histone.
In addition to Sirt6, Sirt1 and Sirt3 are also significantly decreased in OA cartilage. As a deacetylase, Sirt1 can directly deacetylate chondrogenesis-related genes and impact cartilage metabolism. In chondrocytes, Sirt1 can deacetylate forkhead box O-4 (FOXO4) and subsequently activate chondrogenesis-related genes (i.e., Sox9) to maintain cartilage homeostasis[42]. Sirt1 can also indirectly modulate cartilage homeostasis by regulating other pathological processes, including autophagy and protein ubiquitination[43,44]. For example, Sirt1 maintains chondrocyte autophagy by interacting and deacetylating of lysine residues on crucial autophagy proteins (Beclin1, ATG5, ATG7, LC3)[44]. Sirt3 deficiency causes COX4I2 loss and leads to mitochondria respiratory chain dysfunction, which accelerate cartilage damage in DMM-induced mice OA models. Interestingly, activating Sirt3 by honokiol (a small molecule derived from Traditional Chinese medicine Houpu) advocates mitochondrial antioxidant defense to preserve cartilage ECM, and ameliorates OA progression[45]. This study indicates that epigenetic modifications can also regulate mitochondria function and are a potential therapeutic target.
In addition to histone regulating enzymes, levels of some circulating metabolites are correlated with chromatin opening/closing-related gene expression changes. A recent study revealed that elevation of free fat acids (FFA) induced acetyl-CoA upregulated H3K27ac level in murine chondrocytes. With the cleavage under targets and tagmentation (CUT Tag) seq technique, the global binding peak spectrums of H3K27ac in FFA-induced OA conditions were clearly demonstrated: increased H3K27 acetylation of cartilage catabolic genes and decreased acetylation of anabolic genes[46].
Histone methylation
-
Histone lysine methylation plays a crucial role in transcriptional regulation through specialized proteins called reader domains that identify methylated histone marks (Fig. 1b). Typically, methylation of histone H3 at lysine 4 (H3K4me), and lysine 36 (H3K36me) is linked to transcriptional activation, while methylation at H3K9, H3K27, and H4K20 correlates with gene silencing and repressed chromatin. These modifications exist in mono-, di-, or tri-methylated states, dynamically regulated by opposing enzymatic activities: histone lysine methyltransferases (KMTs) that add methyl groups (writers); and histone demethylases (KDMs) that remove them (erasers).
H3K36 methylations are found to be decreased in murine ageing chondrocytes and human osteoarthritic cartilage[47]. The upstream H3K36 methyltransferase NSD1 was then identified to be involved in the reduction of H3K36me in OA. Nsd1Prrx1-Cre mice exhibited an OA-like joint change, including cartilage damage and osteophyte formation. Further bioinformatic study showed that a zinc finger-containing transcription factor was regulated by NSD1 in chondrocytes. Osr2 complement (I.A. injection of Osr2 adenovirus) could relieve Nsd1 deletion incurring chondrogenic impairment[47]. Interestingly, deletion of Nsd1 in murine mesenchymal progenitors delayed fracture healing[48], which indicates that histone methylation also plays a crucial role in bone fracture healing.
The enzyme H3K27 demethylase facilitates the removal of methyl groups from H3K27me2/3. H3K27me3 is generally linked to transcriptional repression, whereas H3K27me1 tends to correlate with gene activation. Kdm6b is an H3K27me3/me1 demethylase that mediates the stepwise removal of methyl groups from histone H3 lysine 27 residues[49]. In OA, Kdm6b was firstly identified to be implicated in chondrogenesis and cartilage development. Kdm6b knockout in chondrocytes (Col2a1CreERT2; Kdm6bf/f) leads to more severe cartilage damage in DMM-induced OA model, due to the inhibition of the anabolic metabolism of chondrocytes[50]. Interestingly, in ACLT-induced mice OA model, ablation of Kdm6b with a small molecule (GSK-J4) attenuated cartilage deterioration[51]. Another H3K27me regulator, H3K27me3 demethylase Kdm6a (also named as UTX), can also regulate joint homeostasis and deterioration. When OA develops, the UTX is upregulated in cartilage, and contributes to the downstream signaling, including Wnt10a and Fzd10 transcription, thus leading to cartilage degeneration and osteophyte formation[52]. In mice, deletion of UTX in chondrocytes (Col2a1Cre UTXfl/fl) led to fewer signs of OA and delayed OA development[52]. The small molecule, GSK-J4 (previously mentioned) could inhibit UTX activity and prevent cartilage damage in OA mice[53]. Further studies are to be conducted to identify the exact drug target of GSK-J4, as current data could not identify the exact working target of GSK-J4 in OA. Another group of histone lysine methylases are enhancers of the zeste homolog (EZH) family: EZH1/2, which function as catalytic components of the Polycomb Repressive Complex 2 (PRC2), responsible for establishing H3K27 mono-, di-, and tri-methylation (H3K27me1/2/3). In medial meniscectomy (MMx) induced OA models, conditional knockout of Ezh2 deteriorated OA pathological conditions. Ezh2 is further evidenced to boost the healing of injured cartilage via increasing the phosphorylation of Akt by activating TNFSF13B in OA chondrocytes[54]. In addition, methylation of lysine 79 on histone H3 (H3K79me), a chondro-protective epigenetic mechanism, is also reduced in OA cartilage. Kdm7A/B histone demethylases are capable of reducing H3K79 methylation status and enhancing the expression of H3K79 in chondrocytes. Inhibition of Kdm2/7 histone demethylases by daminozide increases the expression of H3K79me and glycosaminoglycans in human chondrocytes[55]. H3K79 methylation can also be regulated by Disruptor of telomeric silencing 1-like (DOT1L), a histone methyltransferase. Inhibition of DOT1L by I.A. injecting EPZ-5676 disrupts chondrocyte homeostasis and triggers OA-like changes in mice even without OA-induction[56]. Mechanistically, DOT1L maintains cartilage homeostasis and protects against OA progression via preventing Wnt signaling activation in chondrocytes[56]. Thus, restoring DOT1L function is critical to preserve joint health and prevent OA, and might be a therapeutic target for OA. The same group then evidenced that HIF1α-mediated hypoxia-signaling promotes DOT1L expression, driving H3K79 hypermethylation to remodel chromatin landscapes. The hypoxia mimetic IOX2 (I.A. injection, 0.5 mg/kg, every 10 d for 7 weeks) stabilized HIF1α and increased DOT1L protein and H3K79 methylation in primary human chondrocytes and DMM-induced murine OA chondrocytes[57]. Physiologically, the microenvironment of cartilage is lacking oxygen, and IOX2 mimics the physiological status of chondrocytes by histone methylation[57]. These results imply that appropriate epigenetic intervention helps restore the physiological status in OA conditions. Although mounting studies have been conducted on the mechanisms of histone acetylation and methylation, few studies have researched other protein modifications, such as lactylation, ubiquitination, and SUMO modification. Further studies are required on the alterations of these modifications in OA.
-
Mature miRNAs direct RNA-induced silencing complexes to post-transcriptionally silence genes via mRNA degradation (fully complementarity), or translational inhibition (partial complementarity) (Fig. 1b). Numbers of miRNAs have been proven to participate in OA pathogenesis. Prof. Kapoor's team, evidenced that miRNA-34a-5p[58], and miRNA-181-5p[59] contributed to OA pathogenesis, and I.A. blocking of miRNA-34a-5p and miRNA-181-5p effectively ameliorated OA progression. In addition to cartilage, miRNAs in synoviocytes and synovial fluids also play important roles in OA. For example, miRNA-27b-3p in synoviocytes (upregulation), and miR-140-3p/5p in synovial fluids (downregulation), contribute to synovial fibrosis in knee OA[60,61].
Though a large number of miRNAs have been identified to participate in OA pathogenesis, the poor penetration of cartilage makes it difficult to apply miRNA in clinical use. Recently, a modified PEGylated polyamidoamine (PAMAM) dendrimer was developed and, in this system, cartilage ECM can be penetrated for sustained delivery of miRNA, thereby accelerating the delivery of miRNA to chondrocytes embedded in the cartilage ECM. miR-141/200c was encapsulated in the dendrimers for OA therapy and showed a promising protection against OA in a rat model[62].
In addition to therapeutic value, miRNA also has diagnostic effects in preventing OA. For instance, serum levels of miR-126-3p have been recently identified as twice as high in OA knee than in non-OA knee patients, and show promise as novel biomarkers in OA[63].
lncRNA
-
While miRNAs predominantly function through sequence complementarity to repress target genes, lncRNAs employ more intricate and complicated regulatory strategies, and participate in multiple layers of gene expression controlling, spanning chromatin remodeling, transcriptional modulation, and RNA processing regulation, thus impacting different cell processes like apoptosis and inflammation[64]. To investigate the expression difference of lncRNAs in OA and normal cartilage, van Hoolwerff et al. established lncRNA-mRNA co-expression networks by using paired RNA-seq data from 98 human cartilage samples (lesion vs intact cartilage samples from 65 knees and 33 hips)[65]. They effectively identified 5,053 robustly expressed lncRNAs in paired lesion and intact cartilage, and 191 of them were differentially expressed[65]. Further investigations are needed to elucidate the potential therapeutic targets within these 191 differentially expressed lncRNAs. Intriguingly, to profile the lncRNA changes in obesity-associated OA, Nanus et al. stratified synovial tissue donors into three experimental cohorts: non-OA controls with normal BMI (n = 6, BMI: 22.9 ± 0.6), OA patients with normal weight range (n = 8, BMI: 23.3 ± 0.3), and obese OA cases (n = 8, BMI: 39.8 ± 3.6). RNA-seq analysis of the synovium demonstrated distinct inflammatory transcriptional profiles in fibroblasts from the three cohorts. Comparative analysis identified 19 differentially expressed lncRNAs between normal-weight OA and non-OA patient fibroblasts, and an additional 19 lncRNAs showed differential expression when comparing obese OA patients to their normal-weight OA counterparts[66]. Among them, lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was identified to be up-regulated in normal-weight OA patients, and demonstrated further upregulated levels in obese OA patients. Therefore, MALAT1 may be a therapeutic target for obese OA patients[66]. In addition to cartilage, Wang et al. identified that LncRNA H19 in subchondral osteocytes contributed to subchondral bone sclerosis and inhibition of LncRNA H19 in subchondral bone alleviated murine OA[67].
-
m6A methylation represents a reversible RNA epi-transcriptomic modification that post-transcriptionally regulates gene expression without changing DNA sequence[68]. Like DNA methylation, m6A methylation is also driven by m6A methyltransferases (writers: Mettl family, WTAP, and VIRMA, Fig. 2), recognized by m6A binding proteins (readers: IGF2BPs, YTHDCs, and YTHDFs, Fig. 2), and removed by m6A demethylases (erasers: FTO and ALKBH5, Fig. 2). RNA methylation constitutes a versatile epi-transcriptomic mechanism that directs the molecular dynamics of gene expression through selective modulation of RNA-protein interactions[68]. Recently, mounting evidence indicates that RNA methylation is actively implicated in OA.
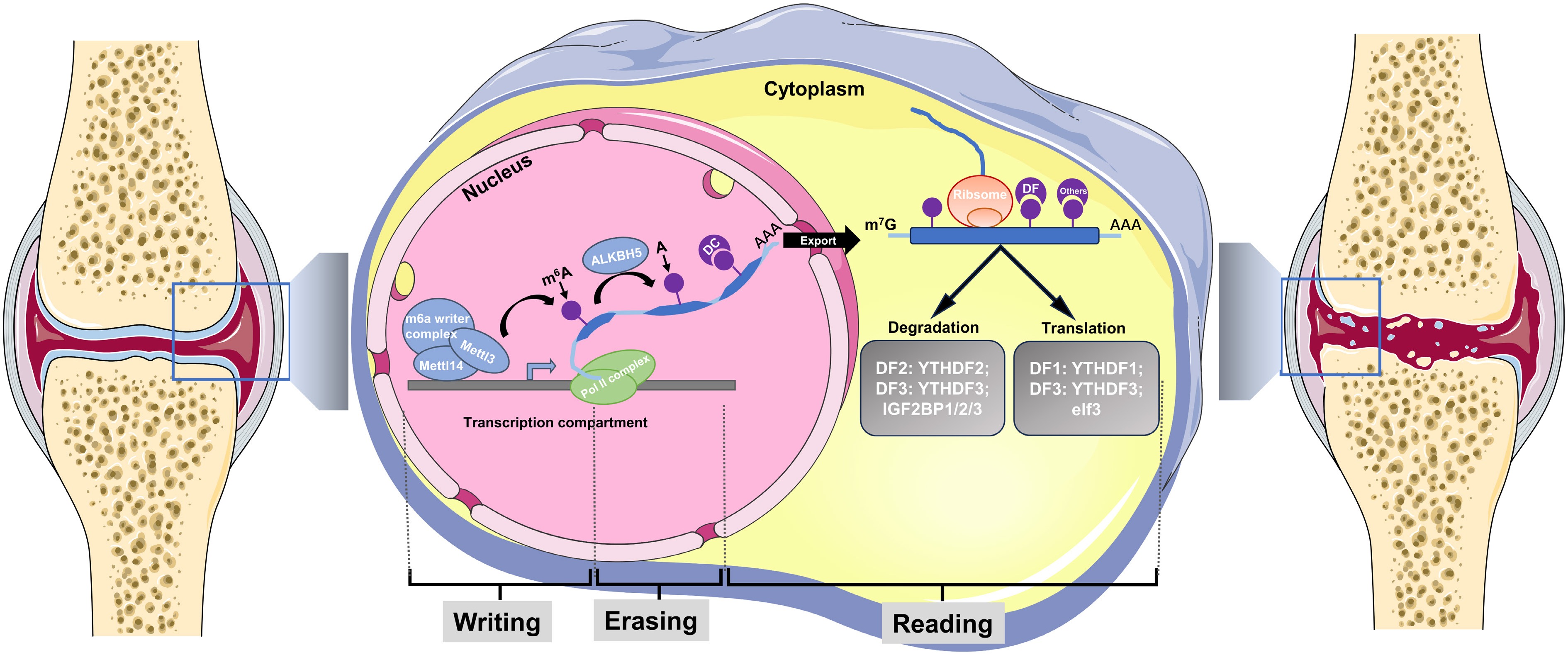
Figure 2.
Schematic diagram of RNA methylation in OA. OA is featured by cartilage degeneration, inflammatory synovium, periarticular bony changes (subchondral bone cyst, subchondral bone loss and sclerosis, osteophytes formation), accompanied by senescence of chondrocytes and cartilage debris. During OA progression, the function and fate of cells within the joint is regulated by N6-methyladenosine (m6A) methylation. The m6A mRNA 'life cycle': The m6A mRNA starts in the nucleus from in the nucleus during transcription. The m6A writer complex (box 1), which comprises the core methyltransferase-like protein 3 (METTL3) and its adaptors, is located in the nucleus, where it adds m6A co-transcriptionally. The m6A erasers are also largely localized in the nucleus. The main m6A eraser acting on m6A in mRNAs is ALKBH5. Fat mass and obesity-associated protein (FTO) has recently been found to preferentially target m6Am, not m6A, with its major target being m6Am in small nuclear RNAs (snRNAs). While in the nucleus, m6A can bind specific nuclear reader proteins, mainly YTHDC1 (DC1), which may affect splicing or other nuclear processes such as mRNA export. Upon mRNA export to the cytoplasm, m6A binds to specific reader proteins that affect the stability, translation and/or localization of the mRNA. In the cytoplasm, the m6A readers YTHDF1 (DF1), YTHDF3 (DF3), the eukaryotic translation initiation factor eIF3, and METTL3 all favour the translation of m6A mRNAs. YTHDF2 (DF2) and DF3 mediate the degradation of m6A mRNAs, while the insulin- like growth factor 2 mRNA-binding proteins (IGF2BP1/2/3), and the synaptic regulator FMRP (a polyribosome-associated RNA-binding protein known to have a central role in neuronal development and synaptic plasticity) enhance m6A mRNA stability. Am, methylated A; m6Am, N6, 2′-O- dimethyladenosine.
m6A writers
-
m6A methylation is enzymatically installed by METTL3-METTL14-WTAP trimeric complexes at transcript-specific consensus sequences (Fig. 2). The m6A writer complex contains diverse proteins. Generally, the overall m6A modification is found to be elevated more than five times in IL-1β-treated chondrocytes (20 μM) than the control group, and the dysregulation of m6A modification in chondrocytes led to the aberrant expression of genes involved in ECM synthesis and degradation, which accelerates the development and progression of OA[69]. METTL3−METTL14 heterodimer enzymatically generates m6A modifications, with METTL3 providing catalytic activity and METTL14 conferring RNA-binding capacity and allosteric regulation. Xiao et al. discovered that Mettl3 expression and m6A methylation levels were significantly increased in human degenerative spinal endplate cartilage tissue. They further identified the underlying mechanism that tension stimulation inhibited the ability of human endplate chondrocytes to synthesize extracellular matrix by inhibiting Mettl3-meidated SOX9 production[70]. However, the expression of Mettl3 in human OA knee cartilage is controversial. Sang et al. discovered that Mettl3 was downregulated in knee OA cartilage and IL-1β induced in vitro OA model[71], while recently Xiong et al. found that Mettl3 is upregulated in human OA cartilage[72]. The inconsistency of Mettl3 expression in human knee OA cartilage indicates that studies with a larger sample size are urgent to reveal the exact changes of Mettl3 in OA cartilages. Intriguingly, Mettl3 is downregulated in TNF-α-stimulated chondrocytes and TMJ OA cartilage (fibrocartilage). The bioinformatic analysis identifies that the main m6A motifs (GGACU) of Mettl3 can bind to Bcl2 mRNA. The in vitro study confirms that Mettl3 can regulate Bcl2 to inhibit the chondrocyte apoptosis and autophagy. Inhibition of Mettl3 by small molecule (S-adenosylhomocysteine, SAH, 9μM) facilitates the TNFα-induced chondrocytes apoptosis and autophagy, and aggravates cartilage degeneration in mice MIA-induced TMJ OA model (SAH: 10 mg/kg, twice a week)[73]. The above in vitro and in vivo experiment is conducted in murine ATDC5 cell lines and murine TMJOA model, respectively. Further investigations on human hyaline chondrocytes are needed to elucidate the DMOA-potential of SAH. And the roles of the methionine cycle still need to be investigated as the methionine cycle may also be a future therapeutic target in OA.
In addition to the Mettl family, WTAP is another crucial m6A methyltransferase. An et al. identified that WTAP significantly increased in osteoarthritic cartilage and TNFα-treated chondrocytes. By MeRIP (methylated RNA immunoprecipitation)-sequencing analysis, the Wnt/β-catenin pathway inhibitor FRZB showed the most significantly downregulated expression with a high m6A modification, and was identified as the WTAP-regulated downstream target. In OA chondrocytes, WTAP activated Wnt/β-catenin pathway by inducing FRZB m6A hypermethylation, and inhibiting joint WTAP by I.A. injection of siWTAP attenuated DMM-induced OA in mice[74].
Besides chondrocytes, FLSs (fibroblast-like synoviocytes) also participate in OA pathogenesis. Chen et al. documented that the upregulation of Mettl3 contributed to elevated m6A levels and impaired autophagy in both human and murine OA FLS. Mechanistically, Mettl3 can combine with ATG mRNA and regulate autophagy and cellular senescence. Inhibition of Mettl3 effectively suppresses the senescence of FLS and decelerates OA development in DMM-induced OA models[75]. In addition to FLS, macrophagic mettl3 also plays an important role in OA pathogenesis[76]. miR-1208 derived from hucMSCs-EVs can target macrophagic Mettl3, and prevent macrophages from secreting inflammatory cytokines and degrading chondrogenic ECM. Recently, Kou et al. revealed that exosomal FTO derived from MSCs contributed to m6A demethylation of the downstream LncRNA GLCC1, enhancing the cancer aggressiveness of acute myeloid leukemia. Numerous MSC-derived exosomes are implicated in OA pathogenesis, and further investigations need to be conducted to illustrate the epigenetic (m6A) enzymes within the exosomes, and to develop the corresponding therapeutic strategies for OA[77].
m6A erasers
-
The m6A demethylases ('erasers') modulate m6A epi-transcriptome by catalyzing the removal of methyl groups from m6A, thereby converting it back to adenosine. ALKBH5 is the most common m6A eraser (demethylases). The m6A demethylase ALKBH5, functioning as a key eraser enzyme, regulates RNA metabolism by altering RNA stability, transport, and processing, while also modulating diverse cellular pathways. In chondrocytes, loss of ALKBH5 accelerates MSC senescence and leads to spontaneous OA-like phenotype in Prx1Cre; Alkbh5fl/fl mice[78]. Mechanistically, ALKBH5 targets CYP1B1 by the m6A reader IGF2BP1, and protects against senescence. Additionally, ALKBH5 can also directly erase the m6A modification of lncRNA HS3ST3B1-IT1, stabilize the HS3ST3B1-IT1, and elevate the maternal gene HS3ST3B1 expression[79]. In OA chondrocytes, the decrease of ALKBH5 gives rise to the downregulation of lncRNA HS3ST3B1-IT1 and HS3ST3B1, and subsequently decreases chondrocyte viability, induces chondrocyte apoptosis, and downregulates extracellular matrix deposition. Thus, targeting ALKBH5/HS3ST3B1 can be another therapeutic option in OA treatment.
m6A readers
-
A primary regulatory function of m6A modification involves the recruitment of specific RNA-binding proteins to methylated mRNAs. The YTH domain-containing proteins are the first m6A-binding proteins to be discovered and provide a mechanistic basis for understanding the effects of m6A in mRNA. ALKBH5, a Fe(II)/α-ketoglutarate-dependent dioxygenase, serves as the predominant demethylase responsible for reversing N6-methyladenosine modifications in mRNAs[80]. As introduced above, the m6A reader Mettl3 is upregulated in OA chondrocytes. As the m6A reader, IGFBP2 recognizes the RNA methylation and mediates and stabilizes m6A modification of STAT1, subsequently promotes STAT1 mRNA stability, and finally increases the STAT1 expression in OA chondrocytes. IL-1β induced nuclear translocation of STAT1, subsequently enhancing ADAMTS12 transcriptional activation and protein expression, thus contributing to OA progression[81]. The inhibition of Mettl3-IGF2BP2-STAT1-ADAMTs12 axis by inhibiting Mettl3 (I.A. injection of mettl3 inhibitor SAH) effectively attenuated ACLT-induced rat OA progression.
-
Different epigenetic modifications have close crosstalk during gene transcription and post-transcription regulation[82]. Increasing evidence indicates that, in mammals, different epigenetic modifications would coordinate or compete with each other and orchestrate an epigenetic regulatory framework to maintain the transcription and post-transcription homeostasis[82]. For instance, when LncRNA and miRNA share the same recognition elements, both of them could competitively bind to miRNA, forming a competitive endogenous mRNA regulatory network[83]. Also, in human and murine osteoarthritic cartilage, the lncRNA IGFBP7-OT and its maternal gene, IGFBP7, are upregulated and positively correlated with cartilage degeneration. In chondrocytes, the overexpression of lncRNA IGFBP7-OT enhanced the expression of IGFBP7, and inhibited cell viability, promoted cell apoptosis, and reduced ECM components (Col2a1), thus accelerating MIA-induced OA progression. Mechanistically, lncRNA IGFBP7-OT suppresses the hypermethylation of IGFBP7 by interacting with DNMT1/3a at the IGFBP7 promoter, and upregulates the IGFBP7 expression in chondrocytes. Intriguingly, the upregulation of lncRNA IGFBP7-OT is found to be mediated by Mettl3-mediated m6A modification. The identification of LncRNA IGFBP7 OT-DNMT1/3a-IGFBP7 axis not only provides a novel therapeutic target for OA treatment, but also reveals the precise epigenetic regulatory network among RNA methylation (m6A modification), non-coding RNA, and DNA methylation (m5C)[84].
-
Recently, with the development of sequencing technology, more attention has been focussed on epigenomes in OA chondrocytes. By conducting genome-wide association studies (GWAS) in Europeans, multiple osteoarthritis risk loci (> 90 independent signals) have been identified to date. The OA-related SNP rs6516886 (T > A; MAF = 0.29) on 21q21.2 showed that the major T allele confers risk for both knee and hip osteoarthritis in Europeans[85]. Previously, genotype at rs6516886 was identified to be correlated with methylation levels (six CpG sites) by a genome-wide CpG study, and cartilage-derived DNA samples from 87 patients. The T risk allele at rs65168886 coordinates methylation remodeling across six CpGs in its 90 kb locus (cg00065302, cg05468028, cg18001427, cg20220242, and cg24751378), with concordant hypermethylation (cg00065302−cg24751378) and telomeric hypomethylation (cg16140273), forming an allele-specific epigenomic domain. Further analysis of the associated region uncovered several genes, including tLTN1 (coding E3 ubiquitin-protein ligase/listerin), RWDD2B (coding RWD domain-containing protein 2B), USP16 (coding ubiquitin carboxyl-terminal hydrolase 16), CCT8 (coding chaperonin containing TCP1 subunit 8), and MAP3K7CL (coding MAP3K7 C-terminal–like protein). Therefore, these five genes may be associated with OA pathogenesis, and further investigation of the correlation of these genes with OA is important and promising for OA treatment.
In addition, OA is gradually becoming a global disease, and how the whole body epigenome changes during OA is also being actively investigated. Recently, Prof. Ali's team generated a circulating OA miRNome by collecting plasma from early- and late-stage OA patients[86]. Among all 215 differentially expressed miRNAs (DE-miRNAs) in OA progression, 97 (45.1%) exhibited consistent dysregulation across more than 85% early-stage OA patients, compared to late-stage OA patients. The panel of these seven circulating miRNAs (has-miR-335-3p/199a-5p/671-3p/1260b/191-3p/335-5p/543) may be promising therapeutic targets or diagnostic parameters in early radiographic knee OA patients[86]. Further research is needed to elucidate the functional mechanisms of the discovered miRNAs in OA pathogenesis, and to evaluate their potential as diagnostic markers for OA.
-
Currently, 11 epigenetic modulatory drugs have been identified and six of them are clinically used. 5-Aza, also known as Decitabine, is one of the most promising epigenetic DMOADs. Targeting DNMT, the most promising epigenetic DMOAD is Diacerein. Approved by the FDA in 2008, Diacerein has been widely used to treat OA in clinics. Long-term clinical randomized controlled trials are required however to evaluate the therapeutic effects of Diacerein against OA. 5-Aza, also known as Decitabine, was approved by the FDA in 2010 for treating myelodysplastic syndromes and is currently in clinical trials for acute myeloid leukemia. However, multiple side effects, including neutropenia, anemia, gastrointestinal effects, and spermatogenesis impairment, occur after using 5Aza, limiting the clinical application of 5Aza to OA. In the future, controlled release of 5aza by biomaterials might be an option for local delivery of 5aza in OA joints. Melatonin is a hormone that helps us sleep and has not been approved to treat OA in clinics. Animal studies shed light on the therapeutic potential of melatonin in OA. In addition to DNA methylation, targeting histone modifications are another clinically available epigenetic intervention options. Trichostatin A and Panobinostat are two HDAC inhibitors that are clinically used in hematologic disorders and show therapeutic effects in mice OA models. Some traditional Chinese herb extract like Honokiol also participates in histone acetylation and has long been used clinically in China. In the future, a combination of molecular docking and artificial intelligence helps researchers discover more potential epi-DMOAD from traditional Chinese herb extracts.
Table 3. Potential epi-DMOADs in the pipeline.
Drug name FDA approval date Clinical indication Epigenetic target Clinical use Decitabine/5Aza 2010 ( www.drugs.com/history/dacogen.html )Myelodysplastic syndrome; Leukemia Dnmt inhibition Diacerein 2008[98] Anti-inflammation Dnmt inhibition Melatonin Non approval (recommended as a
supplement, not for medical administration)Sleep disorders Tet inhibition Trichostatin A Non approval (Phase I Trial: NCT03838926) Relapsed hematologic malignancies HDAC inhibition Panobinostat Approved, but finally withdrawn ( www.drugs.com/history/farydak.html )Relapsed multiple myeloma HDAC inhibition Honokiol Non approval (traditional Chinese
medicine, widely used in China)Anti-bacterial Sirt3 activation Non-clinical
useDaminozide N/A N/A Kdm2/7 inhibition IOX2 N/A N/A DOT1L activation (hypoxia mimetic) S-adenosylhomocysteine N/A N/A Mettl3 inhibition (metobolites) β-aminopropionitrile N/A N/A Dnmt inhibition 2-hydroxyglutarate N/A N/A Tet inhibition Non-clinically used DMOADs
-
Some epigenetic interventions show promising anti-OA effects in animal studies but still have not gained the approval for clinical use. DNA methylation (DNMTs and TETs), and histone medication (methylation and acetylation) are the two most promising epigenetic targets, and several small molecules have been developed for OA treatment in animal studies. Among them, d-2-Hydroxyglutarate (2-HG) is a small molecule inhibiting TET1. Supplementation of 2-HG enhances the chondrogenic ability of primary human chondroprogenitors isolated from OA cartilage. I.A. injection of 2-HG twice a week increases cartilage area in murine OA knee joints. Since 2-HG is also an endogenous metabolite in human body fluids[87], local supplementation of 2-HG might be a safe and efficient way to treat OA, and further clinical trials are needed to verify the safety and DMOA effects of 2-HG.
-
The aging global population presents an unprecedented challenge worldwide. By 2040, the Chinese population aged over 60 years is estimated to reach 28%; meanwhile, by 2050, the US population aged over 60 years will grow from approximately 20% (current data) to 27%. The worldwide population of individuals aged 100 or older is projected to increase by over 100% before 2030, with estimates suggesting a rise to approximately 3.4 million by 2050 (
http://esa.un.org/unpd/wpp/index.htm ). Notably, OA is an aging-related disease, and about 73% of people living with OA are older than 55 years, the prevalence of OA has doubled since the mid-20th century due to a longer life span[88]. Therefore, elucidating the exact OA pathogenesis and identifying the effect of DMOADs is urgent due to the increasing number of the OA population. Epigenetic modification, as one of the most pivotal mechanisms of gene regulation during aging, participates in OA pathogenesis from various aspects. Epigenetic abnormality is regarded as a direct trigger to cell senescence. A recent study demonstrates that correcting pathological epigenetic abnormalities contributes to reversing aging-related phenotypes[89]. Therefore, updates on epigenetic alterations in OA have been summarized (Table 1), and a summary has also been given for the potential epigenetic targets and potential DMOADs (Table 2). Currently, 11 epigenetic modulatory drugs have been identified and six of them are in clinical use (Table 3). In addition, chondrogenesis shares a similar biological process with endochondral ossification. Some epigenetic medications for fracture healing should also be trialled for the development of DMOADs. In the future, clinical trials are urgently required to verify the DMOA-effects of these clinically approved epigenetic drugs. These results indicate that targeting epigenetic modifications in OA shows a bright potential for preventing OA and the development of DMOADs.Table 1. Epigenetic drugs and related targets in different animal studies.
Epigenetic regulation Animal model Intervention method Drug Ref. DNA methylation Dnmts (Dnmt1/3a) DMM-induced OA (mouse) I.P. injection Decitabine (5Aza) [14] Dnmts (Dnmt1/3a) DMM-induced OA (mouse) Oral gavage Diacerein [17] Dnmts (DNMT1/3a) PDE-induced TMJOA (rat) I.P. injection Decitabine (5Aza) [15] Dnmts (Dnmt1) Age-related OA (mouse) Subcutaneous injection β-aminopropionitrile (BAPN) [26] Dnmts (Dnmt3b) DMM-induced OA (mouse) Deletion of STAT3
in chondrocytesN/A [25] TET (TET1) DMM-induced OA (mouse) I.A. injection 2-hydroxyglutarate (2-HG) [30] Histone modification HDACs (Histone deacetylases) MIA-induced OA and DMM-induced OA model (mouse) Subcutaneous injection TSA (HDAC inhibitor) [34] HDACs (Histone deacetylases) ACLT-induced OA model (rat) I.A. injection Panobinostat (LBH589, HDAC inhibitor) [36] HDACs (Histone deacetylases) DMM-induced OA (mouse) I.P. Injection Panobinostat (LBH589) [35] Sirt3 (Histone deacetylases) DMM-induced OA(mouse) I.A. injection Honokiol (derived from Houpu) [45] Sirt6 (Histone deacetylases) DMM-induced OA (mouse) I.A injection tgg2-PP-MDL-800 NP [41] JMJD3/Kdm6b (Histone demethylase) ACLT-induced OA (mouse) I.A. injection GSK-J4 (Kdm6b inhibitor) [51] UTX/Kdm6a (H3K27 demethylase) Aging-related OA model (mouse) I.A. injection Lenti-virus UTX [52] Kdm6a (Histone demethylases) DMM-induced OA (mouse) I.A. injection GSK-J4 (Kdm6a inhibitor) [53] Kdm7a/b (Histone demethylases) DMM-induced OA (mouse) I.A. injection Daminozide [55] DOT1L (Histone demethylases) DMM-induced OA (mouse) I.A. injection IOX2 (2 Ref) [57] NSD1 (H3K36 methyltransferase) Age-related OA (mouse) I.A. injection adenovirus Osr2 [47] RNA methylation ALKBH5 (m6A demethylation) MIA-induced OA (mouse) I.A. injection AAV-IT1 [79] Mettl3 (m6A methylation) MIA-induced TMJ OA (mouse) I.A. injection S-adenosylhomocysteine (Mettl3 inhibitor) [73] Mettl3 (m6A methylation) DMM-induced OA (mouse) I.A. injection Sh-Mettl3 [72] Mettl3 (m6A methylation) ACLT-induced OA (rat) I.A. injection S-adenosylhomocysteine (Mettl3 inhibitor) [81] Mettl3 (m6A methylation) DMM-induced OA (mouse) I.A. injection rAAV9.HAP-1-si-METTL3 [75] Mettl3 (m6A methylation) DMM-induced OA (mouse) I.A. injection human umbilical cord MSCs-derived exsomes [76] WTAP (m6A methylation) DMM-induced OA (mouse) I.A. injection siRNA (si-WTAP) [74] Table 2. Epigenetic modifications and related signaling pathway in different OA subtypes.
Epigenetic regulation OA subtype (affected organ) Targets Trigger process Ref DNA methylation Knee OA Dnmt1/3a-PPARγ Inflammation and oxidative stress [14,17] Knee OA Dnmt1-α-Klotho-PI3K/Akt Senescence and mechanical force [26] Hip FAI (Pre-Hip OA) Dnmt3b-ABAT Inflammation and mechanical force [24] TMJ OA Dnmt1/3a-PPARγ Inflammation [15] Knee OA Phlpp1 Inflammation [21] Histone modification Knee OA Kdm7a/b-H3K79 Inflammation [55] Knee OA Sirt3-COX412 Mitochondria metabolism [45] Knee OA Sirt6-IGF1-Col2 ECM remodeling [40] Knee OA Sirt6-IL15/JAK3/ STAT5 Senescence [41] Knee OA DOT1L-Sirt1-Wnt Senescence [56,57] Knee OA Ezh2-TNFSF13b Inflammation [54] Knee OA FoxO1-PRG4 Inflammation and ECM remodeling [35] Knee OA H3K27me-Kdm6a-Wnt10a Osteogenesis (ossification) [53] Knee OA H3K36me-NSD1-OSR2 Senescence [47] RNA methylation Knee OA Mettl3-ATG7-SASP Senescence [75] Knee OA ALKBH5- HS3ST3B1-IT1- YTHDF2 Inflammation [79] Spine OA Mettl3-Sox9 Senescence and inflammation [70] TMJ OA Mettl3-Ythdf1-Bcl2 Apoptosis [73] Knee OA Mettl3-Dnmt1/3a-IGFBP7 ECM degradation and apoptosis [84] Knee OA Mettl3-IGF2BP2-STAT1 Inflammation [81] Knee OA Mettl3-ALKBH5-CYP1B1 Senescence and inflammation [78] Knee OA WTAP-YTHDF2-TIMP Inflammation [97] Knee OA WTAP-FRZB-Wnt/β-catenin Inflammation [74] Future directions
-
Recent advances in next generation sequencing (NGS) technologies make it possible for us to conduct investigations and achieve high throughput of cellular and molecular alterations at multiple levels (DNA/genome, RNA/transcriptome, epigenome, protein/proteome etc., as shown in Fig. 3). Therefore, it is necessary for researchers to take advantage of these novel techniques to elucidate the chromatin protein changes in OA (listed in Fig. 3). For example, CUT Tag is an enzyme-tethering technique reported in 2019 that adds antibodies to target the chromatin proteins (histones or transcription factors) between nucleosomes in the genome. With this technique, high-resolution sequencing libraries for profiling diverse chromatin components can be precisely achieved, which is an ideal tool for studying alterations in chromatin peaks. Compared with conventional ChIP seq, CUT Tag can supply similar high-quality chromatin profiles with a significantly lower number of cells (5,000−500,000 cells for CUT Tag vs 1−10 million cells for ChIP seq), without cell fixation[90]. CUT tag have been applied to osteoporosis studies[91], but has not been widely used in OA research. In humans, cartilage only contains 1% chondrocytes; meanwhile, the chondrocytes are embedded in the condensed extracellular matrix, making it difficult to achieve large numbers of primary chondrocytes in human samples. Therefore, CUT Tag is an ideal strategy for researchers to study the dynamic changes of chromatin proteins with a limited number of chondrocytes in human OA chondrocytes. Furthermore, multi omics strategies integrating CUT Tag with other omics technologies, such as transcriptomics, proteomics, and metabolomics, allows us to precisely understand the epigenetic landscape dynamics in OA. Also, with the increasingly frequent application of NGS to achieve high throughput in epigenomic studies in OA, researchers will obtain high volumes of sequencing readout. To precisely elucidate the underlying key epigenetic regulators and figure out the epigenic crosstalk frameworks, it is necessary for OA researchers to embrace the latest machine learning and AI computational biology approaches.
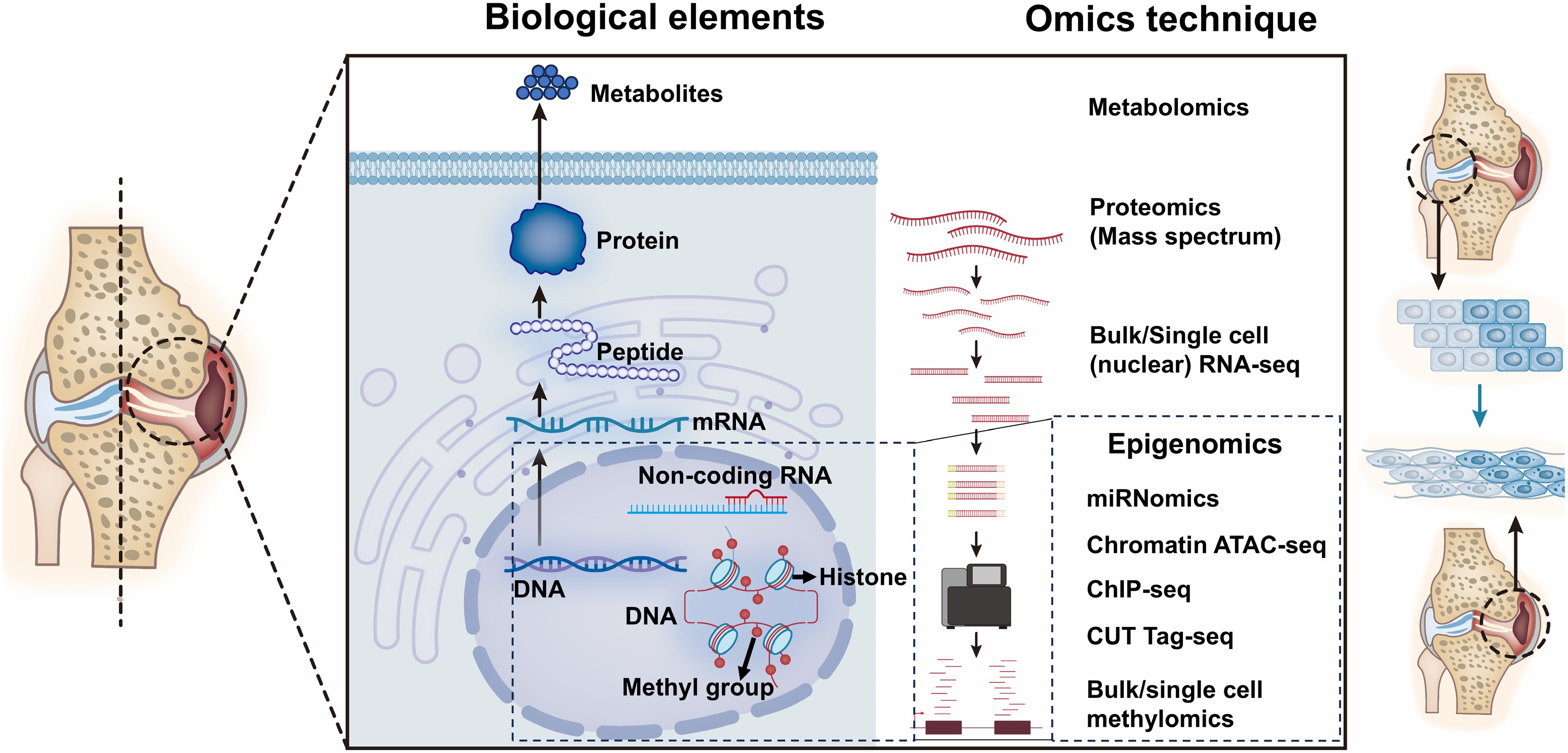
Figure 3.
Overview of current multi-omics techniques in OA epigenetic research. An overview of the advanced multi-omics techniques used in OA research. Cleavage under targets and tagmentation: CUT Tag; Assay for Transposase Accessible Chromatin: ATAC; Chromatin Immunoprecipitation: ChIP.
Currently, the most important epigenomic discoveries are based on clinical samples from Europe and US populations, which represents the epigenetic and epigenomic profiles of Caucasian populations[92,93]. But large-scale epigenomic profiles of joint tissues from Asian and African populations are still not reported. Therefore, another direction is teamwork and cooperation by epigenetic researchers from different countries to decipher the epigenomic profiles of different races and compare the differences of epigenomic profiles in different races.
As previously introduced, epigenetic changes are modulated by different epigenetic enzymes, including Dnmt, TET, Sirt, etc. The activity of most epigenetic enzymes requires a metabolite (amino acids, lactate, free fatty acids, and α-ketoglutarate) as a cofactor or substrate. For example, the histone deacetylase Sirt family requires NAD+ as a co-substrate, meanwhile, DNA methyltransferase needs S-adenosyl methionine (SAM) as a co-substrate because the SAM is the universal supplier of one-carbon group for the methylation process. Therefore, concentrations of metabolites can impact epigenic landscape by interacting with epigenetic enzymes, which is also a typical epigenetic profile that environmental alterations (metabolite concentration) impact gene expression. Further, many metabolites have been identified to be involved in metabolic-related diseases. For example, SAM was found to be highly accumulated in cachexia muscles, and resulted in the activation of DNMT3a, and the resultant hypermethylation of downstream REDD1[94]. Interestingly, SAM supplementation is recommended in the US, and attenuative effects were observed in OA patients with dietary SAM supplements[95]. However, the therapeutic mechanisms of SAM in OA are still in unecessary, and the SAM-DNMT axis might be a potential working signaling pathway. Besides SAM, many other metabolites are also dysregulated in OA conditions. Vitamin B1 was recently identified as a decreased metabolite in OA synovial fluid, and intra-articular supplementation of Vitamin B1 can alleviate OA in mice[96]. OA is a metabolic disorder, and there is still a lack of reasearch analysis of metabolite profiles in different cells (i.e., synovial macrophages, fibroblasts, chondrocytes, subchondral bone cells, and synovium fluid) in OA joints. Thus, future studies are required to decipher the key dysregulated metabolites in each cell, and targeting metabolites might be another epigenetic DMOA strategy for OA, particularly metabolic related OA.
In conclusion, studies of epigenetic modifications and epigenomic investigations in OA were reviewed in this study, as well as crosstalk among different epigenetic modifiers. The clinically-approved and pending epigenetic DMOADs (epi-DMOADs) from current research were also summarized. Future directions for researchers in this field are proposed to take more advantages of NGS and multi-omics techniques to focus on epigenetic changes between different races in OA patients, and investigate metabolite-related epigenetic profiles and related epigenetic therapeutic targets in OA.
This work was supported by the National Natural Science Foundation (NSFC) of China (82302728; 82100765), the Chinese Postdoctoral Science Foundation (2023M733087; 2024T170776), and the Nature Science Foundation of Jiangsu Province (BK201210149).
-
This is a retrospective review study, and ethical approval is not required.
-
The authors confirm their contributions to the paper as follows: conferring the structure of the manuscript: Zhu X, Chen T, Cao W; draft the manuscript and its outline: Zhu X, Ruan B, Chen T; manuscript revision: Zhu X, Wang B, Dong J, Cai D, Hu Y. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhu X, Chen T, Dong J, Ruan B, Wang B, et al. 2025. Targeting epigenetic modifiers in osteoarthritis: from biological insights to pre-clinical practice. Epigenetics Insights 18: e012 doi: 10.48130/epi-0025-0011 

Targeting epigenetic modifiers in osteoarthritis: from biological insights to pre-clinical practice
- Received: 04 January 2025
- Revised: 11 July 2025
- Accepted: 15 August 2025
- Published online: 25 September 2025
Abstract: Osteoarthritis (OA), characterized by the whole joint degeneration, is one of the most common joint diseases worldwide, and more than 37% people at 60 years of age or older will develop OA. So far, the exact mechanism of OA pathogenesis remains unclear, and the search for an effective disease-modifying approach for OA has been unsuccessful. Epigenetic alterations refer to heritable changes in chromatin organization and biochemical composition that do not involve changes to the DNA sequence itself. Recently, the roles of epigenetic modifications in ageing-related diseases have been widely studied, and numerous OA-involved epigenetic targets have been identified. The disruption of epigenetic information is regarded as a reversible cause of OA. In this review, the most recent studies on pathological processes and epigenetic changes relevant to OA, encompassing different subtypes (knee/hip/spinal, and temporomandibular joint-OA), various epigenetic modulations (DNA methylation, histone modification, non-coding RNAs, and RNA methylation), and therapeutic signaling pathways have been integrated. The current therapies targeting epigenetic machinery, future directions for epigenetic illustrations in OA, and potential epigenetic interventions and epigenetic disease modifying-OA drugs are also further described.
-
Key words:
- Osteoarthritis /
- Epigenetics /
- DNA methylation /
- Aging /
- Disease modifying osteoarthritis drugs