









-
Terpenoids are a structurally diverse group of natural products with broad applications in pharmaceuticals, personal care, and agriculture[1]. Lycopene, a naturally occurring tetraterpenoid belonging to the carotenoid family, initially received limited attention due to its lack of provitamin A activity, unlike β-carotene. However, its strong antioxidant capacity and health benefits—such as antitumor effects and protection against prostate and cardiovascular diseases[2−5]—have expanded its use in cosmetics, nutraceuticals, and food products[6,7]. As demand for lycopene grows, developing cost-effective production methods has become a key research focus. Current approaches include plant extraction, chemical synthesis, and microbial fermentation. Plant extraction is limited by low yield, instability, and complex purification[8,9], while chemical synthesis faces issues with residual chemicals and lycopene degradation under heat and oxidative conditions[10]. In contrast, microbial fermentation has emerged as a promising alternative, enabled by advances in metabolic engineering and synthetic biology. This approach offers a more sustainable, environmentally friendly, and economically viable means of lycopene production[11]. To achieve this strategy, it is necessary to select efficient microbial hosts. Escherichia coli (E. coli) is one of the most widely used microbial hosts for heterologous terpenoid biosynthesis due to its well-characterized genetics, rapid growth, and ease of manipulation. Comparative lineage analyses reveal that K-12 strains outperform B strains for terpenoid production: their central carbon metabolism preferentially channels pyruvate and glyceraldehyde-3-phosphate into isoprenoid precursors, whereas B strains divert flux toward phosphoenolpyruvate synthesis and gluconeogenesis[12−14].
To increase the production of lycopene, increasing the intracellular supply of its precursors—isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP)—is a key strategy. These universal building blocks for terpenoid biosynthesis can be synthesized either via the endogenous methylerythritol phosphate (MEP) pathway or a heterologously introduced mevalonate (MVA) pathway (Fig. 1). Enhancing the supply of these precursors is crucial for improving terpenoid yields[15].
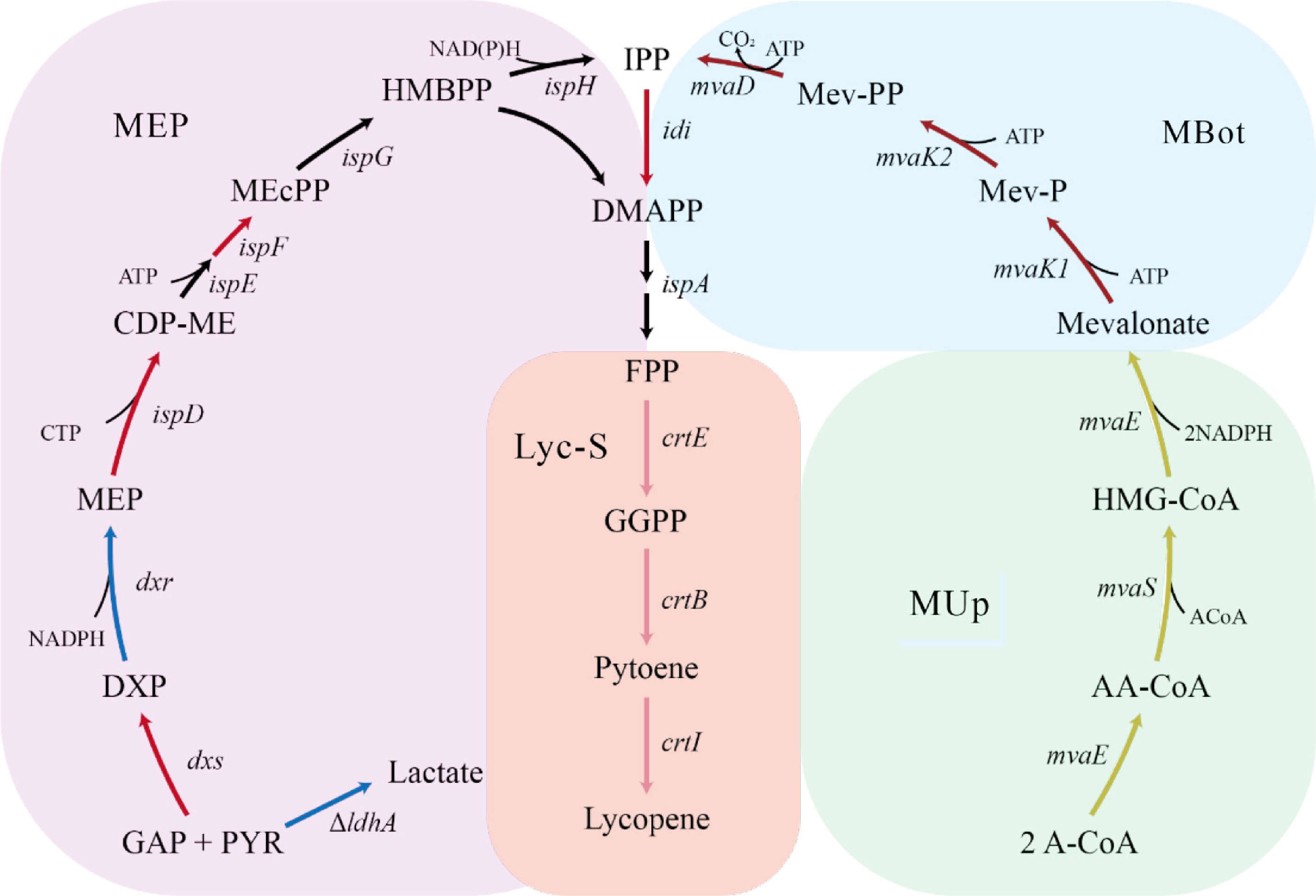
Figure 1.
Metabolic pathway for lycopene biosynthesis. GAP, glyceraldehyde-3-phosphate; PYR, pyruvate; DXP, 1-deoxy-D-xylulose-5-phosphate; MEP, methylerythritol-4-phosphate; CDP-ME, 4-diphosphocytidyl-2-C-methyl-D-erythritol; MEcPP, 2-C-methyl-D-erythritol-2,4-cyclodiphosphate; HMBPP, 2-Chydroxymethylbutenyl-4-diphosphate; IPP, isopentenyl pyrophosphate; DMAPP, dimethylallyl pyrophosphate; A-CoA, acetyl-CoA; AA-CoA, acetoacetyl-CoA; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; Mevalonate, mevalonic acid; Mev-P, mevalonate-5-phosphate; Mev-PP, mevalonate-5-pyrophosphate; FPP, farnesyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate; Phytoene, phytoene. Enzyme abbreviations: dxs, DXP synthase; dxr, DXP reductoisomerase; ispD, ispE, ispF, MEcPP synthase complex; ispG, MEcPP lyase; ispH, HMBPP reductase; idi, IPP isomerase; ispA, FPP synthase; crtE, GGPP synthase; crtB, phytoene synthase; crtI, phytoene desaturase; mvaE, cetoacetyl-CoA thiolase; mvaS, HMG-CoA synthase; mvaK1, mevalonate kinase; mvaK2, mevalonate-5-phosphate kinase; mvaD, mevalonate-5-pyrophosphate decarboxylase.
Within the MEP pathway, dxs encodes the rate-limiting enzyme and is frequently targeted to boost pathway flux[16,17]. Overexpression of idi helps balance the IPP/DMAPP ratio, further enhancing terpenoid biosynthesis[18]. Under normal growth conditions, both dxs and idi are expressed at low levels, and their co-overexpression further promotes terpenoid production[19,20]. Ajikumar et al. employed a modular metabolic engineering approach to overexpress dxs, idi, ispD, and ispF, which increased taxadiene production approaching 1 g/L in E. coli[21]. In addition to its role in isoprenoid biosynthesis, the MEP pathway also contributes to the oxidative stress response in E. coli[22].
The upstream MVA module (MUp), comprising mvaE and mvaS, synthesizes mevalonate, a key intermediate[23]. The downstream MVA module (MBot), including mvaK1, mvaK2, and mvaD, is also essential for efficient pathway operation. mvaK1 is recognized as a regulatory bottleneck[24], and replacement with high-activity isozymes, such as mvaK1 from Staphylococcus aureus (S. aureus), can substantially boost pathway flux in protoilludene production[25,26]. However, the MVA pathway typically involves the expression of multiple heterologous genes, traditionally delivered via plasmid-based systems. These systems are often plagued by genetic instability, antibiotic dependence, and increased metabolic burden on the host[19]. With the emergence of genome editing technologies, genomic integration of the MVA pathway has become a feasible alternative[27]. This approach improves genetic stability, reduces antibiotic usage, and shows better performance in long-term cultivation compared to plasmid-based systems[28,29].
In this study, modules of MVA from different sources were integrated into the genome of E. coli to evaluate their effects on lycopene production. The individual contributions of the MEP and MVA pathways to lycopene biosynthesis were also quantified. The genome-integration strategy developed in this study provides a practical and environmentally friendly approach for industrial-scale terpenoid production, addressing key limitations of plasmid-based expression systems. Moreover, these findings provide valuable insights and a technical reference for future efforts in microbial terpenoid biosynthesis.
-
The bacterial strains used in this study are listed in Table 1. E. coli MG1655 was engineered for lycopene biosynthesis, while E. coli DH5α competent cells (Shanghai Wedi Biotechnology Co., Ltd., China) were used for recombinant plasmid construction. Strain activation and construction were carried out in LB medium (NaCl 10.00 g/L; yeast extract 5.00 g/L; tryptone 10.00 g/L) with solid medium supplemented with 1.8% (w/v) agar. Cultures were incubated at 37 °C for 12 h. Lycopene synthesis was conducted in fermentation medium (FJM) composed of tryptone (15.00 g/L), yeast extract (12.00 g/L), NaH2PO4 (2.23 g/L), Na2HPO4 (11.55 g/L), NaCl (2.50 g/L), Tween 80 (5.00 g/L), glycerol (20.00 g/L), MgSO4 (0.50 g/L), and L-arabinose (1.50 g/L). MgSO4, glucose, and L-arabinose were added as sterile stock solutions.
Table 1. Strains constructed in this study.
Name Description Origin DH5α F− φ80 lac ZΔM15 Δ(lacZYA-argF) U169 endA1 recA1 hsdR17(rk−, mk+) supE44 λ− thi− gyrA96 relA1 phoA Lab stock MG1655 Wild-type Lab stock MG1655-pTrcBIE MG1655 harbouring pTrc-crtBIE This study MG1655-pGexBIE MG1655 harbouring pGex-crtBIE This study AME MG1655 ΔAraA:: Ptrc-dxs-idi-ispDF containing pGex-crtBIE This study PME MG1655 containing pcrEG-TrcMEP, pGex-crtBIE This study PAME AME containing pcrEG-TrcMEP This study MGT7 MG1655 ΔAraB::T7 RNAP This study MGT7E MGT7 ΔlpxM::T7 lac MvaES This study MGT7EE MGT7 ΔaraA::T7 lac MvaES This study MGT7E-pRSF-SaMBot MGT7E harbouring pRSF-SaMBot This study MGT7E-pRSF-ScMBot MGT7E harbouring pRSF-ScMBot This study MGT7E-pRSF-SpMBot MGT7E harbouring pRSF-SpMBot This study MGT7E-pRSF-PTrcSpMBot MGT7E harbouring pRSF-PtrcSpMBot This study MGT7E-pRSF-PGexSpMBot MGT7E harbouring pRSF-PgexSpMBot This study MGT7E-pRSF-PVegSpMBot MGT7E harbouring pRSF-PvegSpMBot This study MGT7E-pRSF-pJ23119SpMBot MGT7E harbouring pRSF-PJ23119SpMBot This study MGT7EKSa MGT7E ΔldhA::T7 lac SaMBot This study MGT7EKSp MGT7E ΔldhA::T7 lac SpMBot This study MGT7EKSc MGT7E ΔldhA::T7 lac ScMBot This study MGT7EKSaSp MGT7E ΔldhA::T7 lac SaSpMBot (mvaK1 from S.aureus and mvaK2/mvaD from S. pneumoniae) This study MGT7EKScSp MGT7E ΔldhA::T7 lac ScSpMBot (erg12 from S.cerevisiae and mvaK2/mvaD from S. pneumoniae) This study MGT7EKSaSc MGT7E ΔldhA::T7 lac SaScMBot (mvaK1 from S.aureus and mvaK2/mvaD from S.cerevisiae) This study MGT7EEKSp MGT7EE ΔldhA::T7 lac SpMBot This study MGT7EEKSc MGT7EE ΔldhA::T7 lac ScMBot This study MGT7EEKScSp MGT7EE ΔldhA::T7 lac ScSpMBot (erg12 from S.cerevisiae and mvaK2/mvaD from S. pneumoniae) This study MGT7EKSc dM MGT7EKSc Δdxr This study MGT7EKSp dM MGT7EKSp Δdxr This study MGT7EKScSpdM MGT7EKScSp Δdxr This study AMET7EKScSp MGT7EKScSp ΔAraA::Ptrc-dxs-idi-ispDF This study Plasmid constructions
-
Primers and sequencing services were provided by Beijing Qingke Biotechnology, China. Plasmid homologous recombination was performed using the Uniclone One Step Seamless Cloning Kit. Primers were designed to contain a 15–25 bp homologous sequence to ensure the efficiency of homologous recombination. The lycopene biosynthesis genes crtB, crtI, and crtE of Pantoea ananatis (P. ananatis) were synthesized by Jiangsu Genecefe Biotechnology Co., Ltd., China. Host strains used for heterologous gene sourcing included Enterococcus faecalis (E. faecalis, Ef), S. aureus (Sa), and Bacillus subtilis (B. subtilis, Bs), all maintained as laboratory stocks. Streptococcus pneumoniae (S. pneumoniae, Sp; GenBank: GCA_000007045.1) was kindly provided by Prof. Jie Feng of the Institute of Microbiology, Chinese Academy of Sciences. Saccharomyces cerevisiae (S. cerevisiae, Sc; GenBank: GCA_000146045.2) was provided by Dr. Dan Liu of China Agricultural University. All gene sequences used in this study are listed in Supplementary Table S1.
The crtB-crtI-crtE (crtBIE) module was assembled sequentially into pTrc99a and pGex-4T-2, with ribosome binding sites (RBS) inserted between stop and start codons to yield pTrc99a-crtBIE and pGex-crtBIE. The MEP pathway genes (dxs, idi, ispDF) were cloned into pTrc99a to construct pTrc99a-MEP. The upstream MVA module (MUp) containing mvaE and mvaS was amplified from E. faecalis genomic DNA and cloned into pCDFDuet to produce pCDF-MUp. Three downstream MVA modules (MBot) were constructed in pRSFDuet, each incorporating genes from different donor species. The SaMBot module consisted of mvaK1, mvaK2, and mvaD from S. aureus, along with idi from E. coli. The ScMBot module included erg12, erg8, and erg19 from S. cerevisiae, together with idi from E. coli. The SpMBot module comprised mvk, mvd1, and mvaK2 from S. pneumoniae, in combination with idi from E. coli. The schematic diagram for constructing the pcrEG vector employed in the CRISPR system is presented in Supplementary Fig. S1. All plasmids constructed in this study are summarized in Supplementary Table S2. The PAM sequences for CRISPR were predicted using the online tool CRISPR RGEN Tools. The PAM sequences corresponding to the target gene loci used in this study are listed in Supplementary Table S3.
Flask batch culture protocol and lycopene extraction
-
Strains were precultured overnight in LB at 37 °C, 220 r/min, then fermentation broth was prepared by transferring 1 mL of the pre-culture to a 100-mL FJM in 250 mL flasks. When OD600 reached approximately 0.25, 10 mM L-arabinose was added. When OD600 reached approximately 1.0, 0.2 mM IPTG was added, and cultivation continued at 30 °C, 220 r/min. As described by Yoon et al., lycopene was extracted and measured from E. coli using their established protocol[30]. Briefly, 1 mL of culture was centrifuged at 13,000 rpm for 3 min, and the cell pellet was washed with distilled water. The cells were then extracted with acetone at 55 °C in the dark for 15 min, followed by a second centrifugation. Lycopene content was quantified by measuring absorbance at 474 nm using a UV-2100 spectrophotometer (Unico, Shanghai, China). Each sample was subjected to three biological replicates simultaneously. Concentrations were calculated based on a standard curve (y = 0.2532x + 0.0073, R2 = 0.9999; Supplementary Fig. S2).
Preparation of competent cells and genome editing
-
Competent cells were prepared using the calcium chloride method. Overnight cultures were inoculated (1% v/v) into 50 mL LB medium and grown at 37 °C with shaking at 220 rpm until the OD600 reached 0.2–0.3. L-arabinose was then added to a final concentration of 10 mM to induce expression, and cultivation continued until the OD600 reached approximately 0.6. Cells were chilled on ice for 30 min, harvested by centrifugation at 4 °C and 4,200 rpm for 6 min, and washed twice with ice-cold 0.1 M CaCl2. The final pellet was resuspended in 0.1 M CaCl2 containing 20% glycerol and stored at –80 °C. Genome editing was performed using the CRISPR/Cpf1 two-plasmid system, as described by Liu et al.[27]. Competent cells harboring the pEcCpf1 plasmid were electroporated with 500−1,000 ng of pcrEG and plated on selective media, followed by incubation at 37 °C for 24 h. Colonies were screened by colony PCR and confirmed by Sanger sequencing. Positive clones were cured of pcrEG plasmids by inducing with 0.1 mM rhamnose and subsequently cured of pEcCpf1 using 5 g/L glucose and 10 g/L sucrose.
Growth curve analysis
-
Single colonies were inoculated into 5 mL of FJM medium supplemented with appropriate antibiotics and cultured overnight at 37 °C with shaking at 220 rpm. The overnight cultures were diluted to an initial OD600 of 0.5 and then inoculated (1:100) into 100 mL of fresh FJM in 250 mL Erlenmeyer flasks. Cell growth was monitored by measuring OD600 at 3-h intervals using a UV-2100 spectrophotometer (Unico, Shanghai, China).
Intracellular redox potential and ATP assay
-
The intracellular redox potential was measured using the Alamar Blue assay, with E. coli MG1655 as the negative control. Cultures grown to OD600 ≈ 0.6 were diluted 103−104-fold and mixed at a 1:1 ratio with fully reduced Alamar Blue reagent in 96-well plates (n = 6). Plates were incubated in the dark at 37 °C for 4−6 h. Absorbance at 570 and 600 nm was recorded to calculate redox changes.
Intracellular ATP levels were determined using a commercial ATP assay kit (Beyotime Biotechnology, Shanghai, China), following the manufacturer's instructions. Cultures at OD600 ≈ 0.6 were sampled in triplicate, and MG1655 was used as a reference strain.
Statistical analysis
-
Descriptive statistics and hypothesis testing were performed using GraphPad Prism 10. All experiments were independently repeated at least three times, and results are presented as mean ± standard deviation (SD). Differences among groups were analyzed using one-way repeated measures ANOVA. Post hoc multiple comparisons were conducted using Duncan's multiple range test with a significance level of α = 0.05.
-
To construct an initial strain capable of lycopene biosynthesis, the exogenous genes crtB, crtI, and crtE were cloned into the expression vectors pTrc99a and pGEX-4T-1, resulting in two lycopene biosynthesis expression constructs, pTrc99a-crtBIE and pGex-crtBIE. These constructs were transformed into E. coli MG1655 and subjected to shake flask fermentation. As shown in Fig. 2a, the strain harboring pGex-crtBIE produced 4 mg/L of lycopene, significantly higher than pTrc99a-crtBIE (p < 0.0001). Thus, pGex-crtBIE was selected as the lycopene biosynthetic module for subsequent experiments.
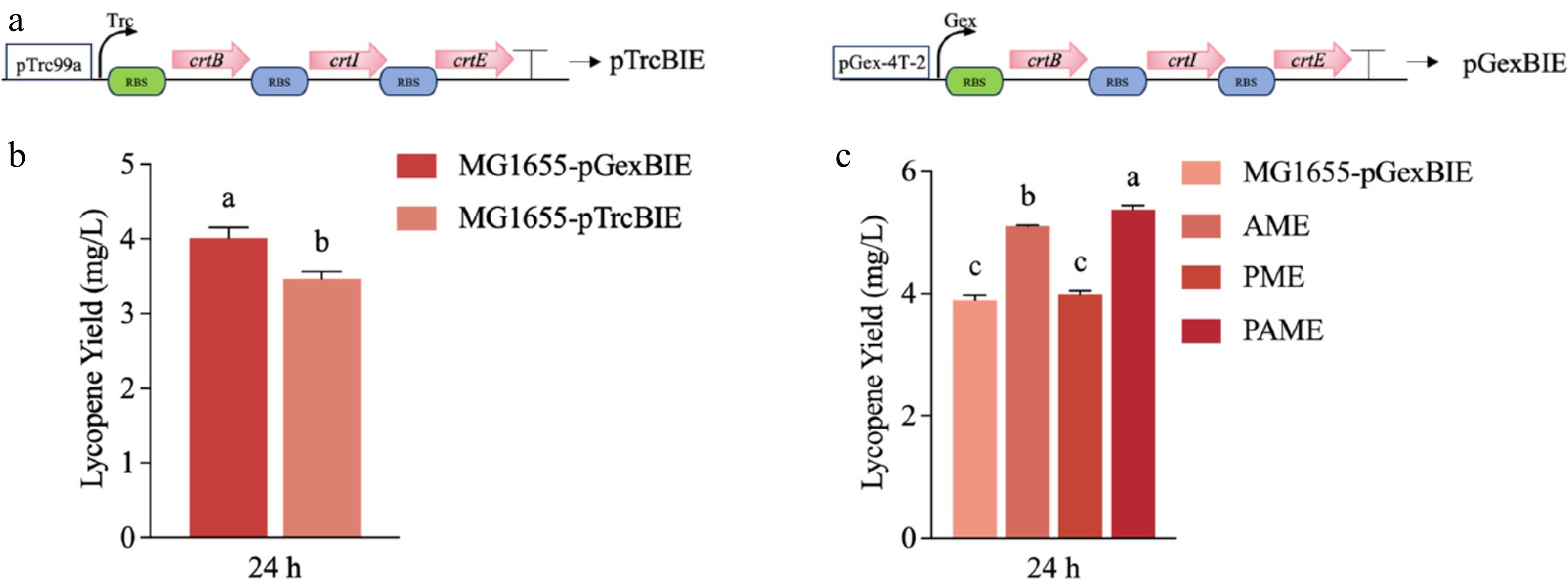
Figure 2.
Construction of the lycopene-synthesis module. (a) Genetic circuit diagrams of pTrcBIE and pGexBIE. Trc and Gex denote the Trc promoter and the Gex promoter, respectively, and a single arrow denotes one gene oriented 5′-3′. (b) Lycopene-producing capacities of pTrcBIE and pGexBIE in MG1655. (c) Effects of different MEP-pathway enhancement strategies on lycopene production. Values are means ± SD. Different lowercase letters (a, b, c) above the bars indicate significant differences among groups (p < 0.05).
To increase lycopene yield, the endogenous methylerythritol-phosphate (MEP) pathway was augmented by either chromosomal integration or plasmid-based overexpression of dxs, idi, and ispDF, yielding the engineered strains AME (chromosomal integration), PME (plasmid expression), and PAME (combined chromosomal and plasmid overexpression). Fermentation results showed that lycopene production was elevated in all engineered strains (Fig. 2b), with AME showing the highest increase of approximately 2 mg/L. Relative to the chromosomally engineered CIBTS1557 strain (Yang et al.; 3.9 mg/L with co-overexpressed ispGH), our MEP-augmented chassis achieved 6.0 mg/L—a ~50% improvement that likely reflects optimized ribosome binding site strengths enhancing translational efficiency[31]. This improvement is likely attributable to the use of a stronger RBS set that elevates translational efficiency of the MEP-pathway enzymes. In contrast, plasmid-based overexpression of MEP genes showed minimal effect, indicating that genome-level enhancement is more effective than plasmid-based expression. However, the limited yield improvement suggests that further enhancement requires the introduction of an exogenous pathway.
Comparison of MVA downstream modules from different sources
-
Given that intracellular carbon flux restricts the MEP pathway's capacity for lycopene synthesis[32], and incorporating the MVA pathway into E. coli has been shown to significantly improve terpenoid production[14,30,33]. This study aimed to enhance precursor availability by introducing a heterologous MVA pathway. To assess the compatibility of MVA downstream modules (MBot) from different organisms, MBot from S. cerevisiae (ScMBot), S. aureus (SaMBot), and S. pneumoniae (SpMBot) were introduced into the host via the plasmid pRSFduet. It is worth noting that the araB gene in E. coli MG1655 was replaced with T7 RNA polymerase, enabling L-arabinose-inducible expression of MVA genes under the T7 promoter to prevent metabolic burden from toxic intermediates. The obtained strain was designated as MGT7 (Fig. 3a). The regulatory characteristics of this system (Supplementary Fig. S3) were validated using a GFP reporter plasmid (pet28a-sGFP). The results demonstrated minimal leaky expression in the absence of L-arabinose and repression of expression in the presence of ≥ 3 g/L glucose, confirming excellent regulation.
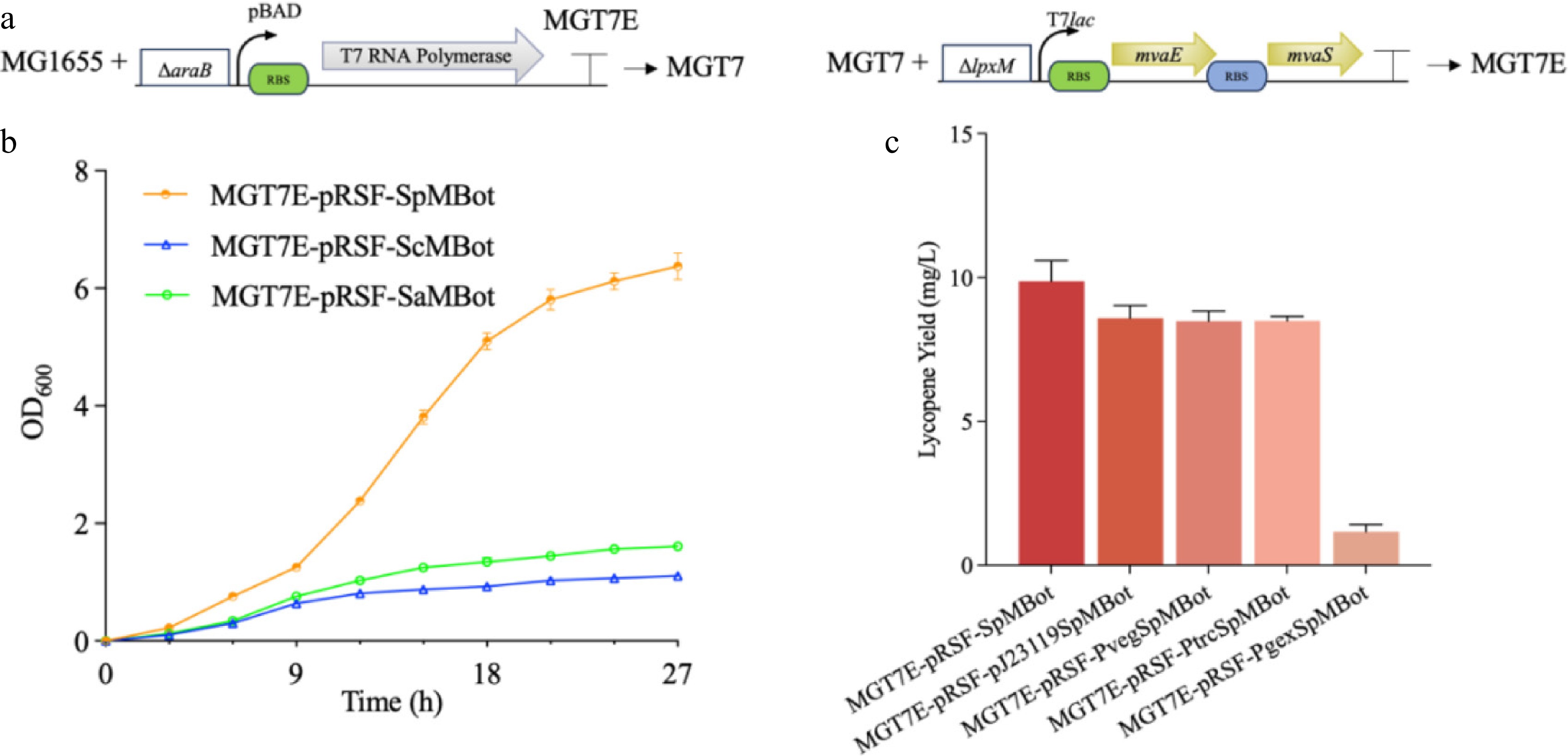
Figure 3.
Genomic integration of MUp and establishment of a plasmid-based MBot overexpression system. (a) Schematic depiction of the genomic integration sites and genetic circuits in the MGT7 and MGT7E strains. Green and blue rectangles indicate distinct RBS regions; ΔaraB denotes replacement of the chromosomal araB gene with T7 RNA polymerase; ΔlpxM indicates substitution of the lpxM gene with the MUp module. T7lac represents the T7-lac promoter, and each arrow represents a single gene oriented 5′-3′. (b) Effects of plasmid-based overexpression of MBot modules from different sources within pRSFduet on lycopene production. (c) Influence of promoter optimization for SpMBot in pRSFduet on lycopene synthesis.
Subsequently, by replacing the chromosomal lpxM gene—a built-in on/off switch for E. coli endotoxic activity and a commonly targeted locus in previous studies—with the MUp module in MGT7, the new strain MGT7E was constructed (Fig. 3a). Building on these observations, plasmid-based expression of these modules was then evaluated. Only strains expressing SpMBot retained normal growth and lycopene production (Fig. 3b), while those expressing ScMBot and SaMBot exhibited growth inhibition and failed to produce lycopene. Therefore, further plasmid-level optimization focused on SpMBot. Among the tested promoters, the T7 promoter showed relatively higher activity (Fig. 3c), likely due to tight regulation and low background expression, and was incorporated into the following expression constructs.
Optimization of combinatorial MVA pathways
-
Plasmid-based expression of MVA downstream modules (MBot) from diverse origins conferred only a marginal increment in lycopene titer, a limitation that is further compounded by well-documented genetic instability inherent to plasmid-borne expression systems[19]. Therefore, these modules were integrated into the ldhA locus of the genome (Fig. 4a). On the one hand, ldhA encodes a key enzyme in the competing metabolic pathway, and thus its deletion could enhance the metabolic flux of the target pathway. On the other hand, previous studies have validated ldhA as an efficient integration site with favorable outcomes, and the results obtained in this study further confirmed that this locus exerted a significant positive effect on lycopene production (Fig. 4b)[29,31]. Among the resulting strains, MGT7EKSp exhibited the highest lycopene production capacity, reaching 36 mg/L—significantly higher than that of strains harboring MBot from other sources (Fig. 4b). In contrast, MGT7EKSa did not show a notable improvement in lycopene yield. From a fermentation time perspective, MGT7EKSc produced more lycopene than MGT7EKSp during the early fermentation phase (first 48 h) and reached its maximum yield within 48 h. This observation suggests that ScMBot may have higher catalytic activity than SpMBot, whereas SpMBot exhibits more sustained catalytic performance. Based on these findings, we further conducted combinatorial optimization of different pathways to explore their interactions and overall effects on lycopene production.
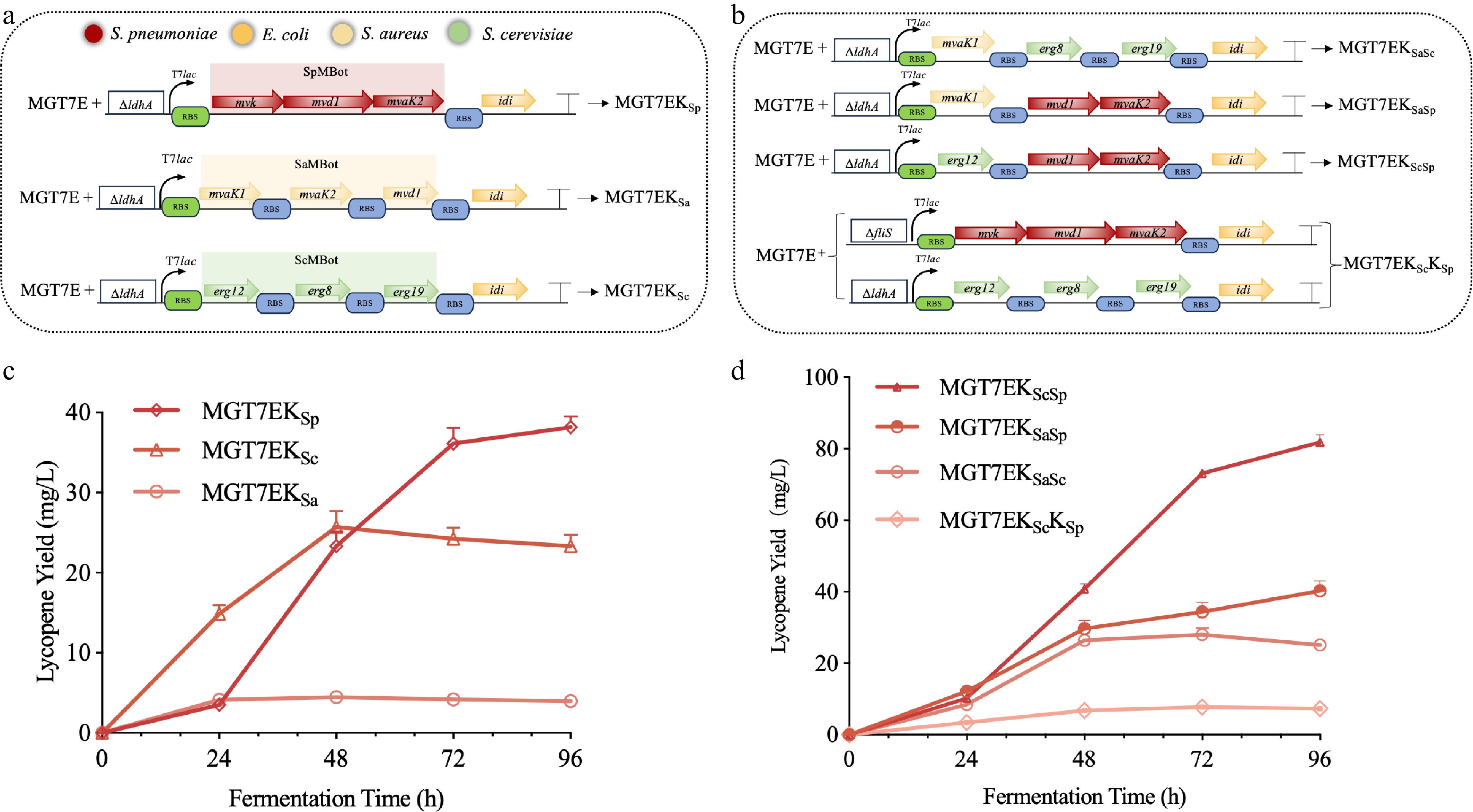
Figure 4.
Genomic integration and optimization of MBot modules from diverse origins. (a) Schematic representation of the genomic integration loci and genetic circuits for MBot modules from different sources. ΔldhA indicates replacement of the ldhA gene with the corresponding MBot cassette. Green and blue rectangles denote distinct RBS regions; T7lac represents the T7-lac promoter; each arrow represents a single gene oriented 5′-3′. Colors are keyed as follows: red—S. pneumoniae, orange—Escherichia coli, beige—S. aureus, green—S. cerevisiae. (b) Lycopene production profiles following genomic integration of MBot modules from the indicated sources. (c) Schematic overview of the distinct MBot optimization strategies. Square brackets denote simultaneous integration of two pathways into the fliS and ldhA loci of the MGT7E genome. (d) Lycopene-producing capacities of strains constructed via the indicated MBot optimization strategies.
As shown in Fig. 4c, optimization of MBot from different sources was performed using two strategies. Through a cross-optimization strategy, we obtained strains MGT7EKSaSp, MGT7EKScSp, and MGT7EKSaSc, while a parallel optimization strategy yielded strain MGT7EKScKSp. The hybrid MGT7EKScSp strain, combining S. cerevisiae erg12 with S. pneumoniae downstream enzymes, achieved 86 mg/L (35.8 mg/g DCW)—a 21-fold improvement over the parental strain and titre parity with multi-copy genomic integrants, yet without their propensity for recombination-mediated instability (Fig. 4d). This value is comparable to titers obtained with multi-copy genomic integration[29], while eliminating the elevated risk of intramolecular homologous recombination that progressively undermines genetic stability. Although the 86 mg/L titer was obtained in shake flasks, the resulting strain and operational parameters now provide a well-defined starting point for subsequent bioreactor characterization, which is scheduled as the next phase of this project.
This result strongly indicated that the erg12 gene from S. cerevisiae plays a key role in the MVA pathway and that its synergistic optimization can significantly boost lycopene production. This is consistent with previous findings that mvaK1, a key enzyme in the MVA pathway, directly influences pathway flux[24], and the fermentation titer peaked at approximately 72 h[34]. Conversely, the low yield of MGT7EKScKSp suggested that simply combining two pathways did not necessarily promote lycopene synthesis. This may be due to excessive accumulation of intermediates or overexpression of multiple enzymes, imposing a substantial metabolic burden on the strain and thereby hindering efficient lycopene biosynthesis.
Comparative survey of idi sources for balancing isoprenoid precursors in E. coli
-
As key intermediates in the terpenoid biosynthetic pathway, the equilibrium between IPP and DMAPP plays a critical role in modulating the conversion efficiency to downstream products[18]. Disruption of this balance not only reduces product synthesis efficiency but may also inhibit strain growth. To investigate the influence of idi, which catalyzes the interconversion between IPP and DMAPP, the idi gene in MGT7EKScSp was replaced with orthologs from S. cerevisiae (ScI), S. aureus (SaI), S. pneumoniae (SpI), B. subtilis (BsI), and E. faecalis (EfI), yielding strains MGT7EKScSpScI, MGT7EKScSpSaI, MGT7EKScSpSpI, MGT7EKScSpBsI, and MGT7EKScSpEfI (Fig. 5a). Fermentation results (Fig. 5b) showed that the E. coli idi present in the parental strain was the most effective. Replacement with orthologs from other organisms led to reduced yields, underscoring the importance of balanced isoprenoid precursor supply for efficient lycopene biosynthesis.
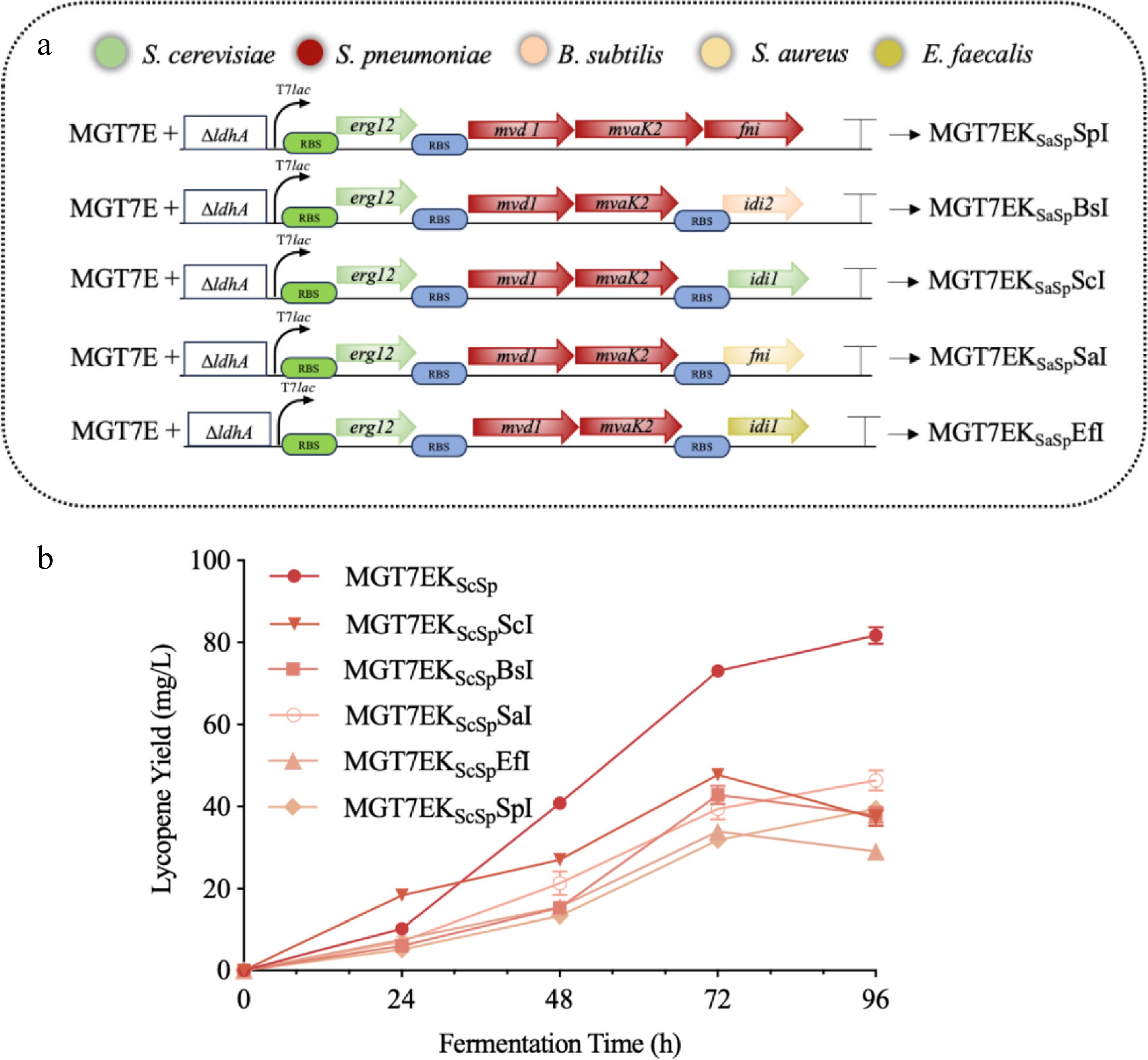
Figure 5.
Screening of idi orthologs from diverse sources. (a) Schematic diagram of the optimization strategies employing idi genes from multiple origins. ΔldhA denotes replacement of the ldhA gene with the corresponding MBot cassette. Green and blue rectangles indicate distinct RBS regions; T7lac represents the T7-lac promoter; each arrow represents a single gene oriented 5′-3′. Colors correspond to the following sources: green—S. cerevisiae, red—S. pneumoniae, peach—B. subtilis, beige—S. aureus, chartreuse—Enterococcus faecalis. (b) Lycopene production profiles for strains harboring idi variants from the indicated sources.
MUp enhancement and balancing of MBot modules
-
It was shown that the enhancement of the Mup module can effectively promote the metabolic efficiency of the MVA pathway[29,31]. In this study, an additional Mup module was further introduced into three high-yield strains to enhance IPP/DMAPP precursor supply and improve substrate availability. As shown in Fig. 6a, b, compared with the MGT7EKSc strain containing a single-copy Mup module, MGT7EEKSc exhibited a significant increase in product concentration, reaching 43 mg/L, indicating a positive effect of module amplification. In contrast, the yield of MGT7EEKSp was significantly reduced compared with its parental strain. This reduction was speculated to be associated with the relatively low catalytic activity of SpMBot during the first 48 h of cultivation, which may have prevented timely conversion of accumulated upstream intermediates, thereby causing metabolic stress accumulation, impairing cell growth, and ultimately suppressing product synthesis.
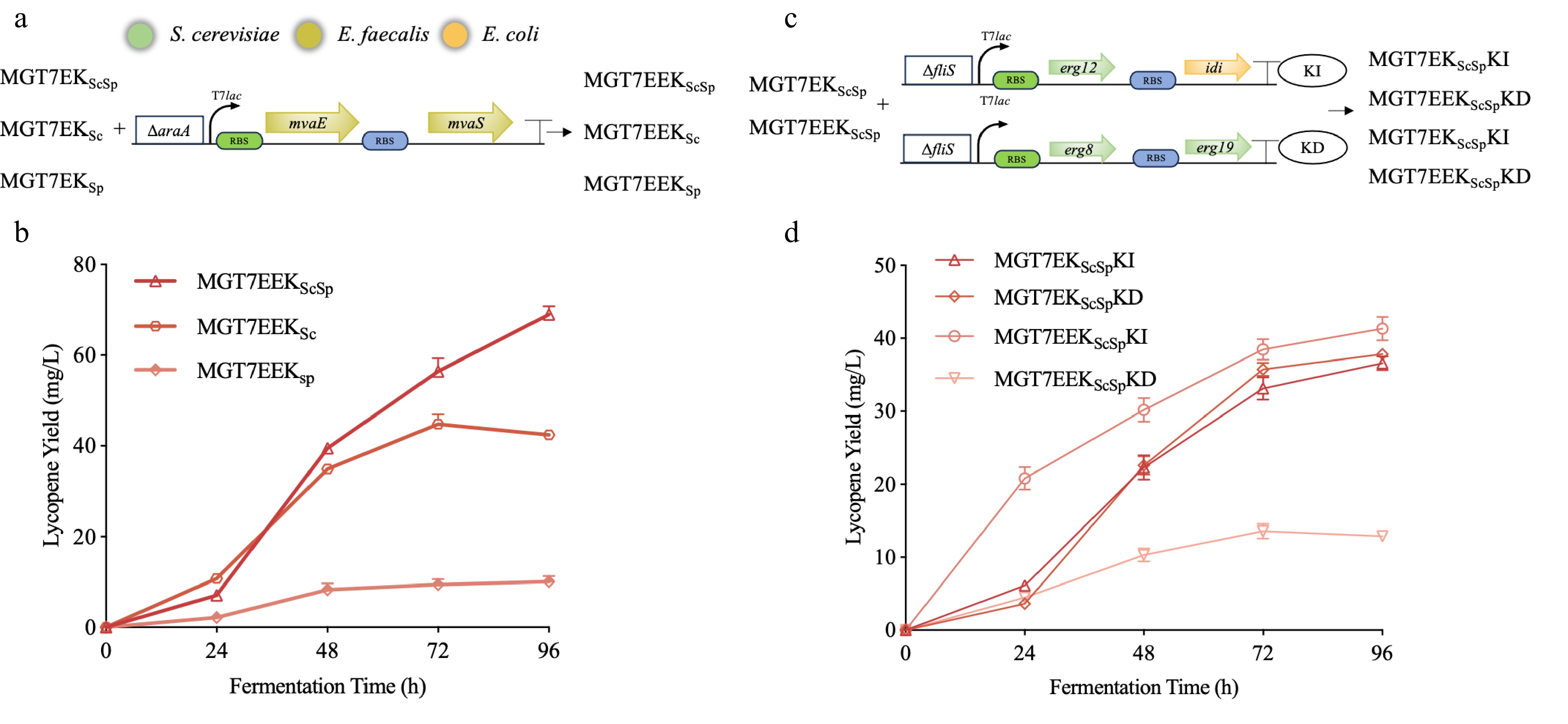
Figure 6.
Enhancement of MUp and synergistic optimization of MBot. (a) Schematic of MUp enhancement in the MGT7EKScSp, MGT7EKSc, and MGT7EKSp. ΔaraA indicates replacement of the araA gene with the MUp module. Green and blue rectangles denote distinct RBS regions; T7lac represents the T7-lac promoter; each arrow represents a single gene oriented 5′-3′. Colors correspond to the following sources: green—S. cerevisiae, chartreuse—Enterococcus faecalis, orange—Escherichia coli. (b) Impact of MUp enhancement on lycopene production by different MBot modules. (c) Optimization strategies for MBot. KD and KI designate the corresponding modules in strain nomenclature; ΔfliS denotes replacement of the fliS gene with the indicated module. (d) Lycopene production profiles following MUp and MBot optimization.
In comparison, ScMBot displayed stronger catalytic activity during the same time frame, enabling MGT7EEKSc to efficiently convert upstream precursors, thereby alleviating metabolic burden and enhancing product yield. Moreover, MGT7EEKScSp showed no significant change in yield under enhanced upstream flux, further confirming that ScMBot possesses superior metabolic capacity to SpMBot during the initial production phase. This observation aligns with previous findings that ScMBot has higher early-stage catalytic efficiency than SpMBot. It was further speculated that this advantage may be attributed not only to the erg12 gene within ScMBot, but also to the enzymatic activities of erg8 and erg19.
To further investigate why MGT7EEKScSp did not exhibit a marked yield improvement, erg12 and idi were assessed for remaining optimization potential, and erg8 and erg19 were examined as possible rate-limiting factors. For this purpose, the erg12–idi module (KI) or the erg8–erg19 module (KD) was individually enhanced in the genomes of both MGT7EKScSp and MGT7EEKScSp, yielding four recombinant strains: MGT7EKScSpKI, MGT7EKScSpKD, MGT7EEKScSpKI, and MGT7EEKScSpKD (Fig. 6c). Fermentation results revealed that neither KI nor KD modification produced a significant yield improvement in MGT7EEKScSp (Fig. 6d), indicating that these modules were not key factors limiting its productivity.
Deletion and functional validation of dxr
-
The MEP pathway is essential in E. coli; its complete deletion has rarely been achieved in prior work. Although attempts have emerged in the past two years[35], whether the MEP and MVA pathways conflict with or complement each other remains unresolved. Therefore, in this study, the interaction between MEP and MVA pathways in E. coli was systematically investigated by overexpressing MEP genes and deleting the rate-limiting dxr gene in various MBot-integrated strains: MGT7EKSc, MGT7EKSp, MGT7EKSa, and MGT7EKScSp.
The dxr deletion was unsuccessful in MGT7EKSa, likely because SaMBot did not provide sufficient metabolic flux to compensate for the loss of dxr, resulting in lethal growth defects. Similarly, no positive clones were obtained for MGT7EKSc overexpressing MEP, likely due to toxic intermediate accumulation caused by ScMBot's strong catalytic activity combined with elevated MEP flux.
Functional validation revealed that the dxr deletion significantly impaired growth in all three viable strains. Without L-arabinose induction, cultures exhibited severe growth arrest, especially MGT7EKSc, which remained transparent even after 30 h, suggesting that dxr is essential for E. coli viability. Although growth partially recovered with arabinose induction, it remained below that of non-deletion controls (Fig. 7a). Fermentation analysis (Fig. 7b) showed marked decreases in lycopene yield post-dxr deletion, with MGT7EKScSpdm dropping from 86 to 10 mg/L. As dxr is an indispensable component of the MEP pathway that dominates terpenoid biosynthesis in E. coli, this study then performed a complementary experiment by overexpressing the entire MEP pathway to explore whether enhancing the flux of this pathway could rescue or promote lycopene accumulation. Unexpectedly, in contrast to the inhibitory effect of dxr deletion, MEP overexpression had no significant effect on lycopene synthesis (Fig. 7c).
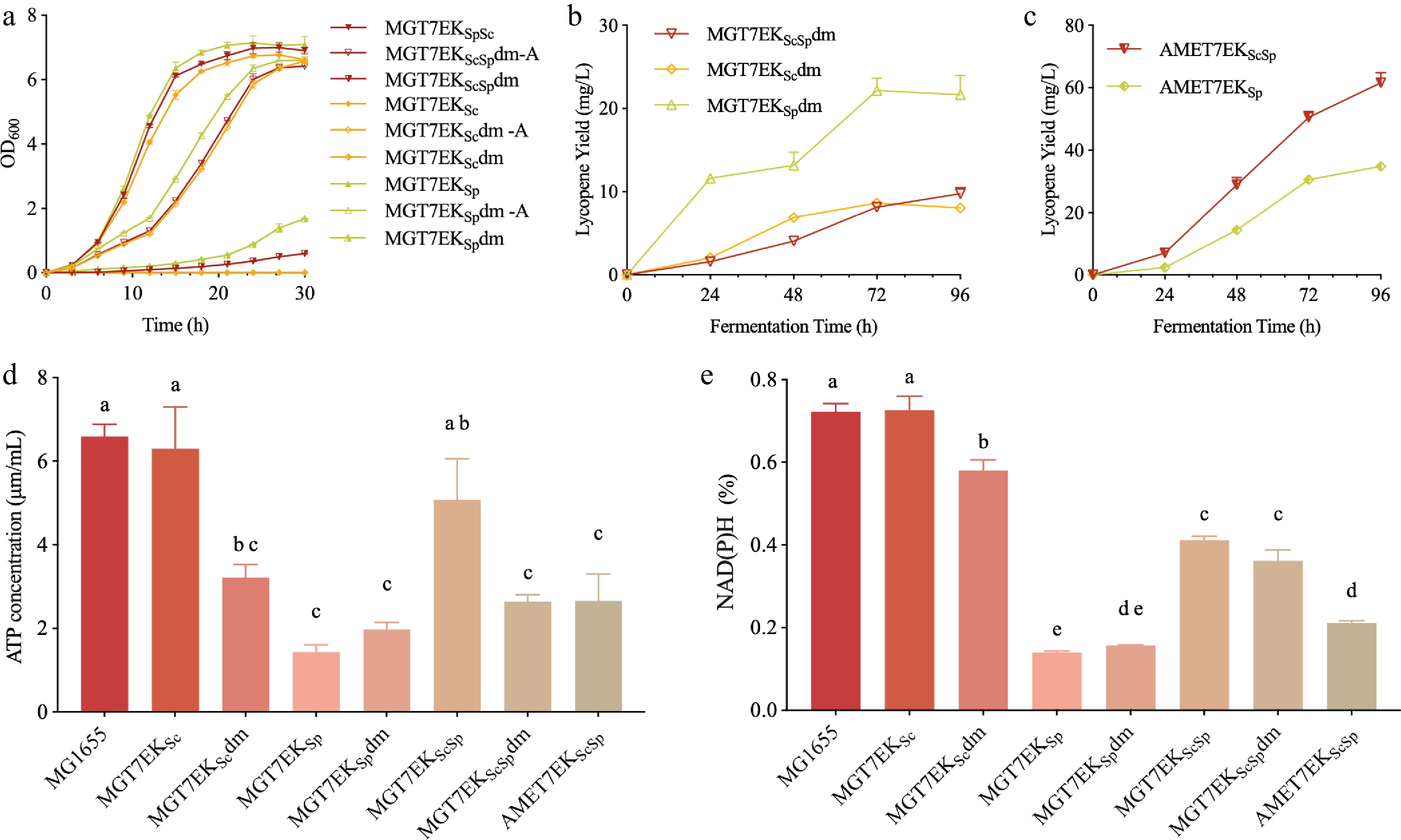
Figure 7.
Role of the MEP pathway in MVA-mediated lycopene synthesis. (a) Growth curves of MGT7EKScSp, MGT7EKSc, and MGT7EKSp after deletion of the dxr gene; '–A' indicates medium supplemented with 0.2% L-arabinose, and 'dm' denotes dxr deletion. (b) Impact of dxr deletion on lycopene production. (c) Effect of MEP-pathway overexpression on lycopene synthesis. (d) Intracellular ATP levels in dxr-deleted and MEP-overexpressing strains. (e) Intracellular reducing-power levels in dxr-deleted and MEP-overexpressing strains.
To elucidate the underlying metabolic differences, intracellular redox status and ATP levels were assessed. Except for MGT7EKSp, dxr deletion significantly reduced NAD(P)H levels (Fig. 7e). ATP levels followed a similar trend (Fig. 7d), highlighting the importance of the MEP pathway in cellular redox and energy homeostasis. Thus, while MEP augmentation failed to boost lycopene titres, it proved essential for maintaining cellular bioenergetic capacity—indicating that dxr and other MEP nodes warrant preservation even when a heterologous MVA pathway is operational. These findings align with recent proposals that the MEP pathway serves as a sentinel for stress perception, signal transduction, and cellular protection across diverse organisms[22]. Collectively, these data indicate that, while the MEP pathway may not directly boost product formation in E. coli, it is vital for maintaining robust cellular growth. Supplementation of the medium with L-arabinose partially restored the growth of our engineered ∆dxr strain, laying the groundwork for future mechanistic dissection of the MEP pathway.
-
In this study, key MVA-pathway enzymes from diverse organisms were screened and optimized, assembled into a single operon, and integrated as a one-copy insertion into the chromosome, resulting in an antibiotic-free, repeat-free E. coli chassis that stably and efficiently produces lycopene. Shake-flask fermentations revealed that the lycopene titer of this single-copy integrant matches the values previously reported for strains harboring multiple genomic copies, while the engineered strain grew as robustly as the parental control, thus circumventing the conventional liabilities of plasmid dependence and genomic instability.
To clarify the interplay between the MVA and MEP pathways, this study engineered an MVA-complete background carrying either the dxr deletion or MEP overexpression. MEP overexpression did not enhance lycopene titers, whereas dxr loss severely impaired growth and dramatically decreased yield, underscoring that the MEP pathway is irreplaceable for basal metabolic balance and energy homeostasis. This aligns with emerging evidence that the MEP pathway functions as a biosensor for oxidative stress, coupling metabolic state to stress signalling and cellular defence across kingdoms—a role that may supersede its canonical function in isoprenoid precursor supply[35]. In summary, this study provides a more stable strategy for lycopene synthesis in E. coli and successfully constructs an E. coli strain that is deficient in the dxr gene yet capable of normal growth, offering a defined genetic context for the mechanistic dissection of the MEP pathway.
This work was supported by the Key R&D Program of Shandong Province, China (Grant No. 2024TZXD013), Provincial and Municipal Agricultural Subsidy Special Funds for the Construction of CAU-SCCD Advanced Agricultural & Industrial Institute, and Project of National Center of Technology Innovation for Comprehensive Utilization of Saline-Alkali Land (Grant No. GYJ2023006). We sincerely thank Prof. Jie Feng (Institute of Microbiology, CAS) and Dr. Dan Liu (China Agricultural University) for kindly providing microbial genomic DNAs and giving valuable advice on the experimental methods.
-
The authors confirm their contributions to the paper as follows: study conception and design: Xia W, Chen J; data collection: Xia W; analysis and interpretation of results: Xia W, Li W, Yao Q; draft manuscript preparation: Xia W, Li W; writing − review and editing: Li W, Yao Q, Chen J; supervision: Yao Q, Chen J; funding acquisition, methodology, supervision: Chen J. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/fia-0026-0017.
- Supplementary Table S1 Key gene sequences used in this study.
- Supplementary Table S2 Plasmids used in this study.
- Supplementary Table S3 PAM sequences corresponding to different genes.
- Supplementary Fig. S1 Schematic of pcrEG assembly. Dashed segments indicate regions that must be swapped between different pcrEG constructs; colored squares at the ends of each arc mark 15–20 bp homology arms used for fragment assembly.
- Supplementary Fig. S2 Standard curve for lycopene.
- Supplementary Fig. S3 MGT7-pet28a-GFP fluorescence intensity CT: without l-arabinose.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Agricultural University, Zhejiang University and Shenyang Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Xia W, Li W, Yao Q, Chen J. 2026. A metabolic optimization strategy based on modular genomic integration of the MVA pathway to enhance lycopene production in Escherichia coli. Food Innovation and Advances 5(2): 251−260 doi: 10.48130/fia-0026-0017 

A metabolic optimization strategy based on modular genomic integration of the MVA pathway to enhance lycopene production in Escherichia coli
- Received: 15 December 2025
- Revised: 28 January 2026
- Accepted: 31 January 2026
- Published online: 19 May 2026
Abstract: Lycopene, a bioactive tetraterpenoid antioxidant valued in food, pharmaceutical, and cosmetic sectors, remains constrained by low plant extractability and expensive chemical synthesis; microbial engineering now presents a cost-effective alternative. In this study, key genes from the methylerythritol phosphate (MEP) pathway—dxs, idi, and ispDF—were integrated into the genome of the chassis strain Escherichia coli MG1655 using a CRISPR-Cpf1-based system, resulting in MEP pathway-overexpressed strains. Additionally, the downstream module (MBot) of the mevalonate (MVA) pathway was optimized by introducing T7 RNA polymerase, mvaE, and mvaS from different species, including Saccharomyces cerevisiae, Streptococcus pneumoniae, and Staphylococcus aureus. Integration of the MEP genes improved lycopene production by 2 mg/L compared with the initial strain. Notably, fermentation performance varied significantly depending on the source of the downstream MBot module. The optimal combination—erg12 from Saccharomyces cerevisiae, mvaK and mvd1 from Streptococcus pneumoniae, and idi from Escherichia coli—achieved a lycopene titer of 86 mg/L in shake-flask cultures, representing a 21-fold increase compared to the parental strain. Paradoxically, dxr deletion to eliminate endogenous MEP flux precipitated a 7-fold drop in lycopene titre, whereas MEP overexpression failed to enhance production—revealing that MEP–MVA pathway synergy, rather than simple precursor supply, governs efficient carotenoid biosynthesis.