









-
Double-stranded RNA-binding proteins (dsRBPs) represent a class of proteins that specifically recognize and interact with double-stranded RNA (dsRNA) structural domains. They play diverse and critical roles in cellular processes, including sensing viral infection and initiating innate immunity, RNA editing, RNA interference (RNAi) pathway, RNA splicing and translation, and RNA stability[1,2]. Among dsRBPs, HYPONASTIC LEAVES1 (HYL1) is one of the most extensively characterized members in plants. Previous studies have established that HYL1 functions critically in the MicroRNA (miRNA) pathway, serving as a key cofactor for DICER-LIKE 1 (DCL1) during the processing of primary miRNA transcripts (pri-miRNAs) into mature miRNAs[1,3]. In addition, HYL1 contributes to the subsequent transfer of mature miRNAs to the ARGONAUTE1 (AGO1)-containing RNA-induced silencing complex (RISC), thereby facilitating either cleavage of target mRNAs or inhibition of their translation[4]. Consistent with its essential role, Arabidopsis hyl1 mutants exhibit special developmental morphotypes, including reduced stature, hyponastic leaves, diminished root growth rate, and impaired gravitropism[5,6]. HYL1 also contributes to abiotic-stress adaptation by modulating miRNAs responsive to drought, cold, and pathogen challenge, including Mdm-miR156, Mdm-miR160, and Mdm-miR172[7,8]. Although current studies have confirmed the conserved function of HYL1 in modulating miRNA expression across plant species[9], its potential involvement in the regulation of additional classes of non-coding RNAs has not been fully clarified.
Long non-coding RNAs (lncRNAs) are RNA transcripts longer than 200 nucleotides that generally lack functional open reading frames (ORFs), and thus produce little or no protein[10]. At an earlier time, lncRNAs were dismissed as mere 'transcriptional noise'[11] until Nature and other journals reported that the lncRNA Xist plays an essential role in X chromosome inactivation[12]. Since then, lncRNAs have been viewed in a completely new light. Accumulating evidence shows that they participate in regulation at multiple levels, including epigenetic control (e.g., dosage compensation, chromatin modification, and genomic imprinting)[13−15], transcriptional regulation[16,17], and post-transcriptional regulation, such as mRNA degradation, competing endogenous RNA (ceRNA) activity, translational regulation, and alternative splicing regulation[18−21].
In recent years, genome-wide identification of lncRNAs across numerous plant species has revealed their critical regulatory roles in various biological processes. Notably, lncRNAs have been directly implicated in both abiotic and biotic stress responses. For abiotic stress responses, in rice(Oryza sativa), a comparative analysis between the cold-tolerant cultivar Kongyu131 and the cold-sensitive cultivar Dongnong422 identified 566 differentially expressed lncRNAs, providing valuable molecular insights for enhancing cold resistance[22]. For biotic stress, the lncRNA MSTRG.12742.1 was reported to positively regulate resistance to grape downy mildew[23]. Beyond stress responses, lncRNAs serve crucial functions in plant developmental processes, including the regulation of leaf morphogenesis and fruit maturation. In peach (Prunus persica), transcriptome profiling across three developmental stages revealed 575 differentially expressed lncRNAs, which were associated with key physiological and metabolic changes during ripening[24]. Likewise, Wang et al. identified 3,571 differentially expressed lncRNAs across four fruit developmental stages in apple, further elucidating their regulatory influence on ripening-related genes, including IAA32, SAUR36, and PA2[25]. Furthermore, in Arabidopsis thaliana, construction of CircularRNA (circRNA)/lncRNA-associated ceRNA (competing endogenous RNA) networks in leaves have further illuminated regulatory circuits underlying leaf development[26]. There are also studies identified 5,279 lncRNAs in apple peel during light-induced rapid anthocyanin accumulation, providing important insights into the regulatory role of lncRNAs in anthocyanin biosynthesis[27].
Although lncRNAs are known to participate in diverse levels of gene regulation, their functions related to leaf development and water transport in apple are still unclear. In the present work, it is demonstrated that MdHYL1-knockdown (RNAi) transgenic apple plants exhibited significantly impaired water transport capacity in both shoots and roots, including leaf size reduction and a dwarfed growth habit. The findings indicated that MdHYL1 plays a broader regulatory role beyond miRNA processing. However, it is unclear whether lncRNA acts as an eTM in this process. Thus, we performed genome-wide transcriptomic profiling to systematically identify and characterize lncRNAs in MdHYL1-knockdown plants. The integrated analysis uncovered potential endogenous target mimic (eTM) interactions between lncRNAs and miRNAs, providing new perspectives on the functional significance of lncRNAs in MdHYL1-mediated regulation of leaf development and water transport in apple.
-
Two-month-old GL-3 (serving as the wild-type transgenic background), and MdHYL1 RNAi transgenic apple lines[7] were cultivated under controlled greenhouse conditions. Plant materials were prepared through the following sequential process: first, tissue-cultured seedlings were rooted for 8 weeks in 1/2MS medium (for more details refer to Shen et al.[8]). Following root establishment, plants were acclimatized by transferring to soil and maintained in a growth chamber for a month. The acclimatized seedlings were then moved to greenhouse conditions, where they were cultivated for an additional month. Mature leaves from two-month-old GL-3 and MdHYL1 RNAi apple transgenic line (#5) were subsequently collected for genome-wide transcriptome analysis. GL-3 and two individual MdHYL1 RNAi apple transgenic lines (#2 and #5) were used for Reverse Transcription Quantitative PCR (RT-qPCR).
RNA library preparation
-
Total RNA was isolated using CTAB, with its quality and quantity verified by NanoDrop ND-1000 and Agilent 2100. Following the removal of ribosomal RNA with a Ribo-Zero™ kit from Illumina, the remaining RNA was fragmented and converted into double-stranded cDNA. Adapters were then ligated, and fragments of an appropriate size were chosen using AMPureXP beads. After treatment with heat-labile UDG, the samples were amplified via PCR under these conditions: an initial denaturation at 95 °C for 3 min; eight cycles of 98 °C for 15 s, 60 °C for 15 s, and 72 °C for 30 s; followed by a final extension at 72 °C for 5 min. The final library, with an average insert size of around 300 ± 50 bp, underwent paired-end sequencing on an Illumina HiSeq 4000 system.
Processing of RNA-seq data
-
The primary pipeline for lncRNA prediction involved alignment to the reference genome (
https://github.com/moold/Genome-data-of-Hanfu-apple , HFTH1), transcriptome assembly, lncRNA identification, coding potential prediction, and subsequent filtering.Following quality assessment and filtering, high-quality reads were mapped to the apple reference genome (HFTH1) using HISAT2 (v2.2.1)[28]. SAMtools v1.9[29] was used to convert, sort, and index BAM files. Expression levels for both lncRNAs and mRNAs were quantified with featureCounts v2.0.1[30]. Gene lengths were calculated using the GenomicFeatures package v1.42.3[31]. Fragments Per Kilobase of transcript per Million mapped reads (FPKM) was calculated using the formula: FPKM = Read counts / (Total mapped reads in millions × Exon length in kilobases).
Differential expression analysis was performed using the DESeq2 v1.40.2 package[32], transcripts with an adjusted p-value < 0.05 and |log2(Fold change)| > 1 were considered to be significantly differentially expressed. Additionally, the pheatmap v1.0.12 package was used to create heatmaps of expression patterns[33]. while Gene Ontology (GO) enrichment analysis was carried out using the clusterProfiler package v4.14.6[34].
Prediction of lncRNAs and targeted mRNAs
-
LncRNA transcripts were first assembled with StringTie v2.1.7[35] before being filtered using gffcompare v0.11.2[35], based on class codes 'u', 'x', 'i', 'j', or 'o', which means intergenic regions, exonic overlap on the opposite strand, intronic transcripts, multi-exon transcripts with splice junctions, and same-strand exon overlaps, respectively. Only transcripts exceeding 200 base pairs (bp) in length, with at least two exons were retained for subsequent analysis. The coding potential of these transcripts was then assessed using independent software tools: CPC2[36], PLEK v2.1[37], and CNCI v2[38]. Candidate transcripts were only kept if they were consistently identified as non-coding by all three analytical methods. These retained transcripts were then subjected to Pfam domain alignment using PfamScan. Following the removal of low-expression transcripts, the remaining transcripts were aligned to the NONCODE v6_Arabidopsis database to differentiate between known and novel lncRNAs.
For target prediction, cis-acting genes were defined for target prediction as protein-coding genes located within a 100 kb window upstream or downstream of an lncRNA's locus, reflecting potential positional regulation. Trans-acting targets were identified based on significant expression correlations between lncRNAs and coding genes across samples.
Establishment of the miRNA–mRNA–lncRNA interaction network
-
To investigate the potential role of lncRNAs as endogenous target mimics (eTMs), psRNATarget (
www.zhaolab.org/psRNATarget ) was employed. The sequences of miRNA were obtained from our previous study[7]. Putative interactions between miRNAs and lncRNAs, as well as between miRNAs and mRNAs, were identified separately. LncRNAs and mRNAs sharing common miRNA binding sites were considered as candidate ceRNA pairs. The network was visualized in Cytoscape.Protocols for RNA extraction and RT-qPCR
-
The total RNA was isolated using the CTAB method, consistent with our previous studies. For mRNA analysis via RT-qPCR, first-strand cDNA was synthesized by reverse transcription using the HiScript II 1st Strand cDNA Synthesis Kit. This was followed by quantitative PCR (qPCR) amplification with the HiScript II One Step RT-qPCR SYBR Green Kit. For miRNAs, expression levels were measured using a previously described stem-loop RT-qPCR protocol[39]. Each experiment included three biological replicates, and all primer sequences are listed in Supplementary Table S1.
-
Our prior research has already shown that the RNA-binding protein MdHYL1 is crucial for increasing apple's tolerance to various abiotic and biotic stresses, such as cold, drought, and pathogen infections[8]. MdHYL1 is the apple ortholog of Arabidopsis thaliana HYL1, a gene well established as essential for microRNA (miRNA) biogenesis and developmental regulation. Functional studies revealed that knockdown of MdHYL1 expression in apple leads to inhibited leaf development, phenocopying the Arabidopsis mutant hyl1. Additionally, two independent MdHYL1 RNAi lines (#2 and #5) exhibited significantly reduced height and hydraulic conductivity in both shoots and roots (Fig. 1). To uncover the regulatory mechanisms underlying leaf developmental defects and impaired water transport in MdHYL1 RNAi apple plants, integrated mRNA and noncoding RNA transcriptome analyses were performed.
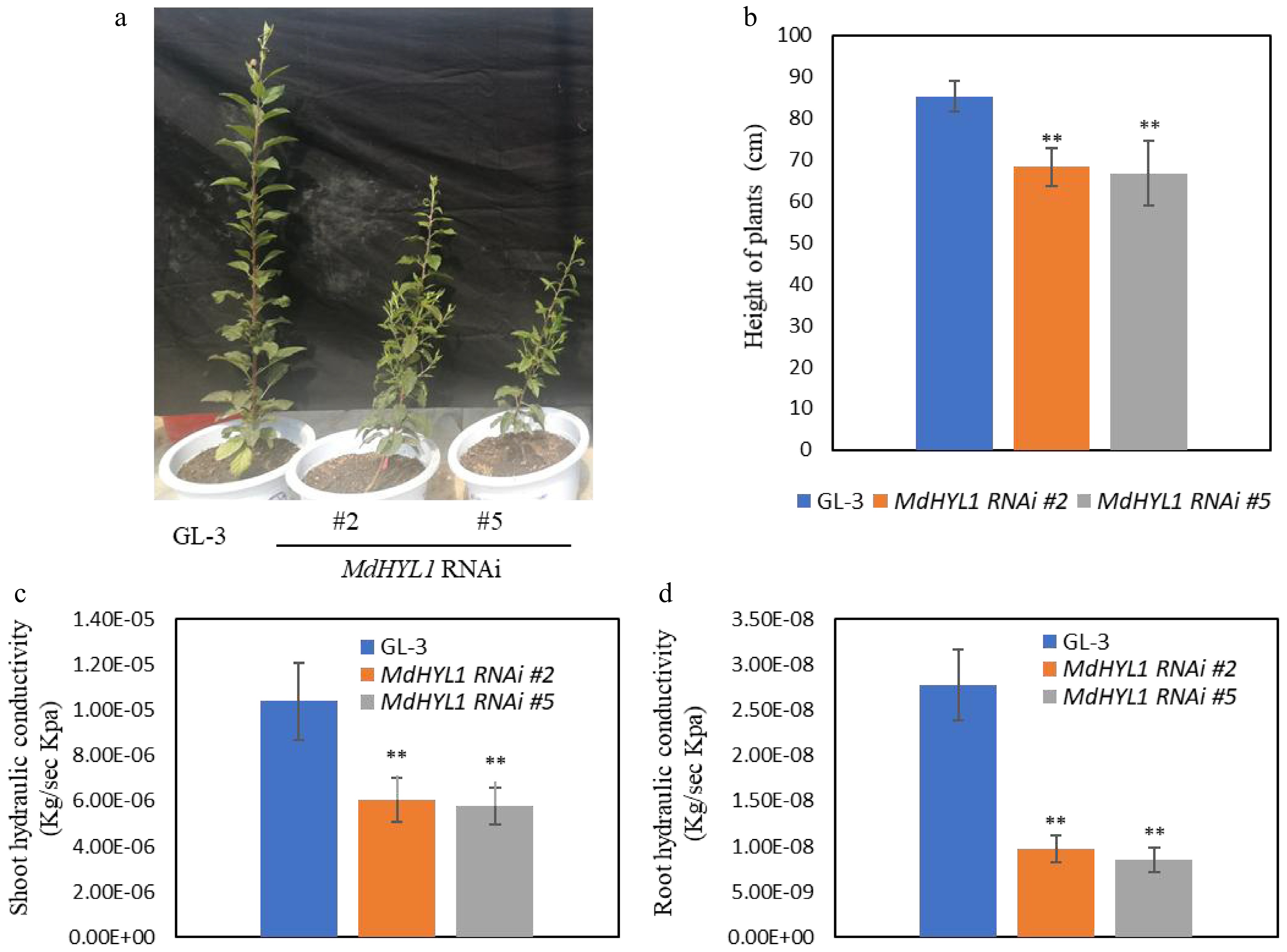
Figure 1.
MdHYL1 RNAi transgenic apple plants showed dwarfing phenotypes and reduced water transport capacity. (a) The morphology of wild-type GL-3 and two independent MdHYL1 RNAi plants (#2 and #5). (b) The plant height between GL-3 and MdHYL1 RNAi plants (#2 and #5). (c), (d) Hydraulic conductivity of shoot and root in GL-3 and MdHYL1 RNAi transgenic apple seedlings. The two-tailed t-test analyzed the differences between groups, all data are expressed as the mean ± SD (n = 6). *, p < 0.05; **, p < 0.01.
RNA sequencing in MdHYL1 RNAi plants and GL-3
-
To investigate the role of lncRNAs in leaf developmental defects and impaired water transport of MdHYL1 RNAi plants, ssRNA-seq (strand-specific RNA sequencing) was performed on leaves from MdHYL1 RNAi (#5) and GL-3 apple lines with three biological replicates per genotype. Paired-end sequencing on the Illumina platform generated 108,750,904, 109,553,184, and 107,591,190 raw reads from the three replicates of MdHYL1 RNAi, and 108,151,660, 101,209,678, and 104,253,686 raw reads from the three replicates of GL-3 (Table 1). After quality trimming, 107,590,616, 108,320,772, 106,396,686, 107,530,470, 100,269,134, and 103,303,754 clean reads were retained. The GC content ranged from 42.22% to 42.86%. Data quality was consistently high, with Q30 > 91% and Q20 > 96% across all samples. Alignment of clean reads to the Malus domestica HFTH1 v1.0 reference genome yielded mapping rates of 93.1%–94.3%, confirming the reliability of the sequencing data for subsequent analyses.
Table 1. Statistical summary of RNA sequencing reads.
MdHYL1 RNAi_1 MdHYL1 RNAi_2 MdHYL1 RNAi_3 GL-3_1 GL-3_2 GL-3_3 Raw reads 108,750,904 109,553,184 107,591,190 108,151,660 101,209,678 104,253,686 Clean reads 107,590,616 108,320,772 106,396,686 107,530,470 100,269,134 103,303,754 Clean base (G) 16.137259 16.247269 15.958526 16.128176 15.039191 15.494131 Error rate (%) 0.0014 0.0013 0.0013 0.012 0.008 0.0013 Q30 (%) 94.19 94.26 94.175 91.13 93.705 94.605 Q20 (%) 98.035 98.055 98.035 96.8 97.87 98.2 GC_content (%) 42.525 42.22 42.315 42.64 42.6 42.86 Mapped reads 101,105,892 102,019,938 100,375,286 100,113,566 94,297,562 97,071,070 Mapping rate (%) 0.939727792 0.941831711 0.943406132 0.931025095 0.940444564 0.939666433 Genome-wide identification of lncRNAs in MdHYL1 RNAi plants and GL-3
-
A comprehensive pipeline to identify lncRNAs was established (Fig. 2a). Following a series of stringent filtering steps, 14,948 transcripts were obtained. Coding potential was assessed using four methods: CPC2, PLEK2, and CNCI, which respectively identified 4,431, 4,156, and 4,373 transcripts with non-protein-coding potential (Fig. 2b). Only transcripts that were consistently identified as non-coding by all three predictive methods were retained for further analysis. These retained transcripts were then subjected to Pfam domain alignment using PfamScan. Low expressed transcripts were subsequently removed, the remaining transcripts were aligned to the NONCODE v6_Arabidopsis database to distinguish between known and novel lncRNAs. In total, 768 expressed lncRNAs were identified, including 762 novel lncRNAs and six known lncRNAs: MSTRG.194.2, MSTRG.24867.1, MSTRG.30213.4, MSTRG.449.2, MSTRG.504.1, and MSTRG.6070.1 ('MSTRG' denotes StringTie-assembled transcript IDs). These known lncRNAs mated three entries in the NONCODE v6_Arabidopsis database: NONATHT002169.1 (enriched in mature pollen of Arabidopsis thaliana), NONATHT000930.1 (enriched in flowers, cotyledons, and roots), and NONATHT001131.1 (enriched in flowers).
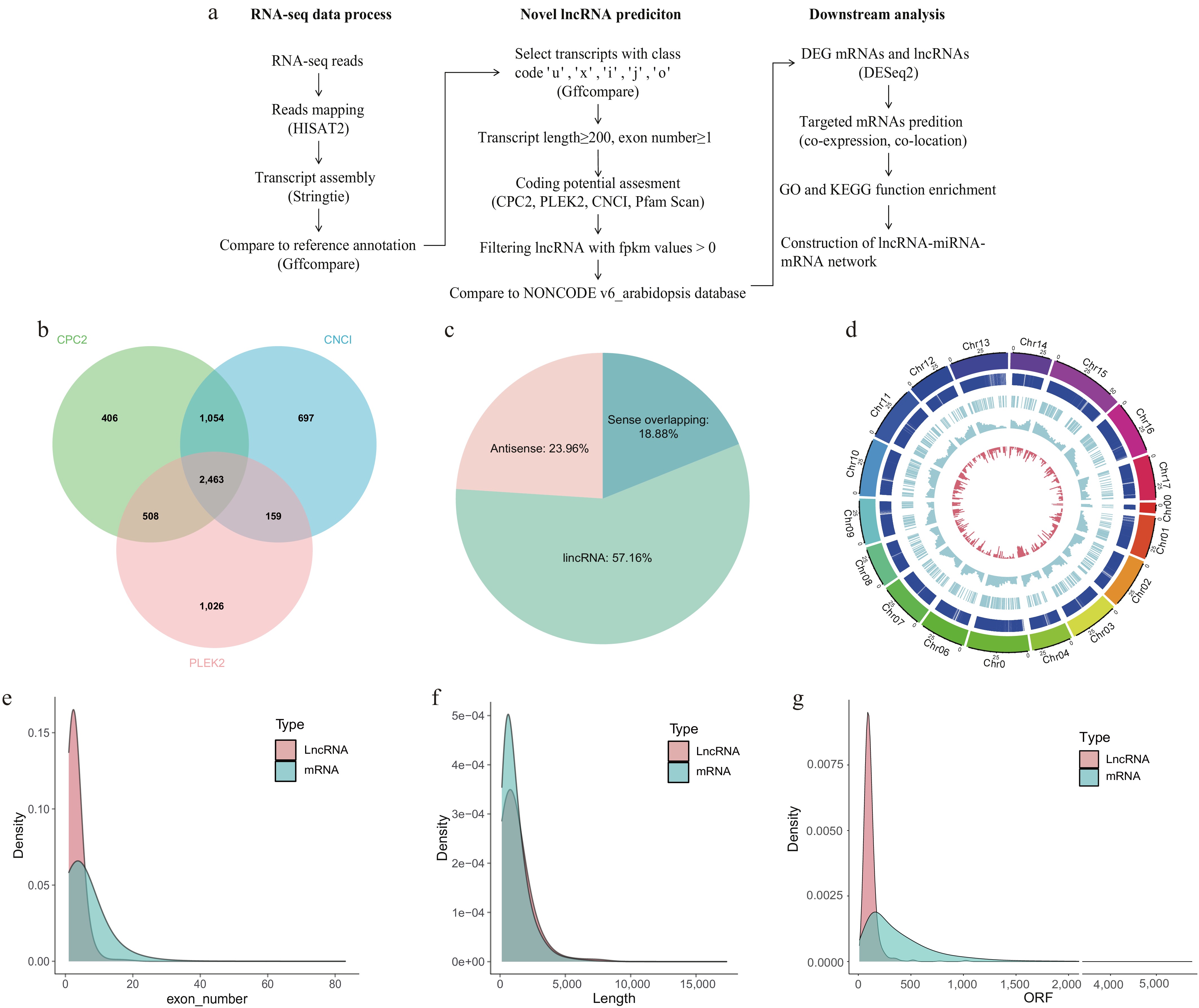
Figure 2.
Overview of lncRNA identification and characterization. (a) Pipline for RNA-seq data processing, novel lncRNA prediction, and downstream functional analyses. (b) The number of predicted lncRNAs supported by CPC2, CNCI, and PLEK2. (c) Classification of lncRNAs into different lncRNAs types. (Sense intronic lncRNAs were not found.) (d) Chromosomes 0−17 are displayed in different colors as the outermost thick track. The second track (outer to inner) shows mRNA locations, with each vertical dark blue line representing one gene. The third track indicates lncRNA locations. The fourth and fifth tracks display the abundance of mRNAs (blue columns), and lncRNAs (red columns), respectively, within 2 Mb physical bins across each chromosome. (e) The density of exon numbers, (f) sequence lengths, and (g) ORF lengths for mRNAs and lncRNAs.
A prior study classified lncRNAs into four categories[25], including: (i) long intergenic lncRNAs (lincRNAs), located between genes without overlapping any exonic sequences; (ii) antisense lncRNAs, transcribed from the opposite strand of protein-coding genes; (iii) sense intronic lncRNAs, originating from intronic regions on the same strand but not overlapping with exons; and (iv) sense overlapping lncRNAs, which partially or fully overlap with exonic regions on the same DNA strand. Among them, 184 were antisense lncRNAs (23.96%), 439 were lincRNAs (57.16%), and 145 were sense-overlapping lncRNAs (18.88%) (Fig. 2c). No sense intronic lncRNAs were detected, which is consistent with earlier reports[25]. To further examine genomic distribution, Circos plots were created to show the chromosomal locations of both lncRNAs and mRNAs throughout the apple genome (Fig. 2d). The results showed that lncRNAs were distributed across all 18 chromosomes. The inner histogram revealed a lower overall density of lncRNAs compared to mRNAs. Most lncRNAs and mRNAs exhibited similar distribution patterns along the chromosomes, suggesting a potential correlation between them.
The structural characteristics of the identified lncRNAs and mRNAs, including transcript length, open reading frame (ORF) length, and exon number, are summarized in Fig. 2e–g. Among lncRNAs, 66.7% contained two exons, with an average of 2.86 exons per transcript (Fig. 2e). The average transcript length of lncRNAs was 1,159.22 nt, with the majority (60.93%) ranging from 200 to 1,000 nt. By contrast, mRNAs exhibited a similar average transcript length of 1,182.67 nt, with most (64.77%) ranging from 200 to 1,300 nt, suggesting potential parallels in transcriptional processing (Fig. 2f). In terms of coding potential, the ORF length of most lncRNAs was ≤ 200 nt, whereas mRNAs exhibited a much broader ORF length distribution and were significantly longer (Fig. 2g). Collectively, these results indicate that, compared with mRNAs, lncRNAs generally have fewer exons and shorter ORFs, yet their transcript lengths are comparable to those of mRNAs, consistent with previous findings[22,40]. In the context of apple's perennial growth habit and complex developmental cycles, these features may facilitate rapid and flexible responses to environmental and physiological cues.
Differentially expressed genes in MdHYL1 RNAi plants and GL-3
-
The interaction between lncRNAs and mRNAs is a key mechanism through which lncRNAs exert their functions in biological processes. To explore the specific lncRNAs and mRNAs involved in MdHYL1 RNAi transgenic plants and GL-3, transcript expression levels were normalized as FPKM (fragments per kilobase of transcript per million mapped reads). A minimum FPKM threshold was applied to define expressed transcripts[41]. Since transcripts with FPKM = 0 were excluded during lncRNA filtering, the following classification is based on expressed lncRNAs. In both GL-3 and MdHYL1 RNAi lines, two lncRNAs were classified as non-expressed (FPKM < 1 in both conditions), 10 lncRNAs as mixed expression (FPKM > 5 in one condition and < 5 in the other), and 756 lncRNAs as expressed under all conditions (FPKM > 5 in both conditions). In comparison, 22,384 mRNAs were categorized as non-expressed, 8,400 as mixed expression, and 13,893 as expressed in both conditions.
Differential expression analysis was then performed using DESeq2, with transcripts considered significant at |log2(Fold change)| > 1 and padj (adjusted p-value) ≤ 0.05. Hierarchical clustering and heatmaps illustrated distinct expression patterns of differentially expressed lncRNAs and mRNAs (Fig. 3a, b). Density plots of FPKM values indicated no significant differences in global expression distributions between GL-3 and MdHYL1 RNAi plants (Fig. 3c, d). In total, 28 lncRNAs showed differentially expressed, with 23 upregulated and five downregulated transcripts. Additionally, 813 mRNAs showed significant differential expression, with 449 upregulated, and 364 downregulated transcripts (Fig. 3e, f). The read countsand differentially expressed lncRNA, mRNA and miRNA are shown in Supplementary Table S2.
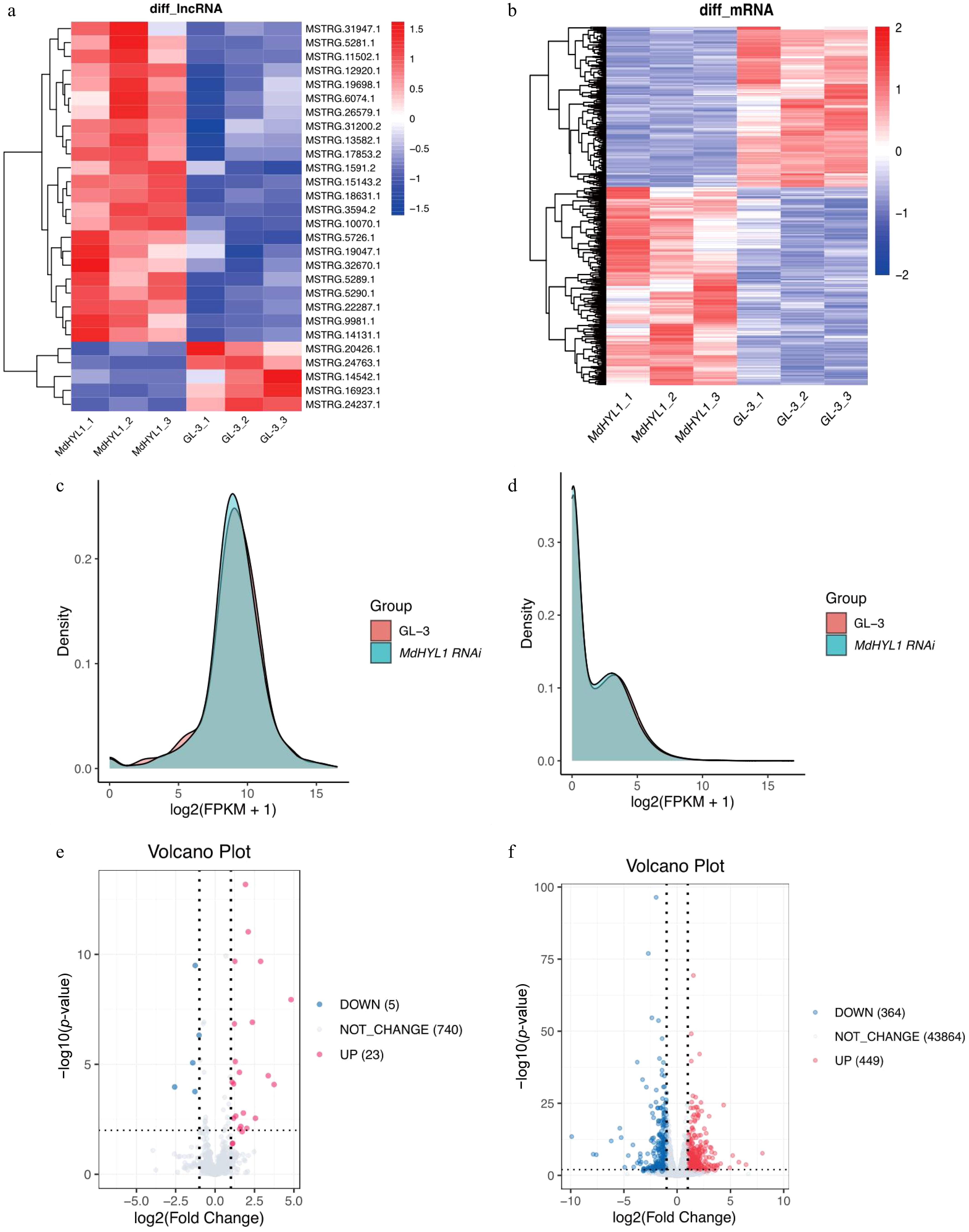
Figure 3.
Differential expression of lncRNAs and mRNAs in MdHYL1 RNAi and GL-3 plants. Heatmap of differentially expressed (a) lncRNAs and (b) mRNAs. Rows are clustered using hierarchical clustering. Density distribution of expression levels for (c) lncRNAs, and (d) mRNAs in MdHYL1 RNAi and GL-3. Volcano plot of differentially expressed (e) lncRNAs, and (f) mRNAs, showing upregulated (red), downregulated (blue), and non-significantly changed (gray) lncRNAs and mRNAs. Differential expression was defined as |log2(Fold change)| > 1 and adjusted p-value < 0.05.
Functional prediction of lncRNA targets of MdHYL1
-
LncRNAs are widely known for regulating gene expression via both cis- and trans-acting mechanisms[42]. In this study, potential lncRNA target genes and their functions were predicted using two strategies: genomic proximity for cis-acting and expression correlation for trans-acting. For cis-acting predictions, lncRNAs located within 100 kb of mRNAs were analyzed, resulting in 12 potential target genes. Among them, only two target genes did not overlap with trans-acting prediction, including HF12279 (enriched in apoplast) and HF25993 (enriched in potassium and calcium ion transport). For trans-acting targets prediction, 736 potential target genes co-expressed with lncRNAs. were identified, using Pearson correlation coefficient (PCC) > 0.8 and p-value < 0.01 as filtering thresholds. The correlation analysis and detailed description of target genes are shown in Supplementary Table S3 and S4.
To elucidate the biological functions of lncRNA target genes in MdHYL1 RNAi apple plants, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed. Differential expression analysis showed that lncRNA target genes were significantly enriched in pathways related to biological process (Fig. 4a, b). Four key biological pathways were the main focus: plant-type secondary cell wall biogenesis, flavonoid metabolism, lignin biosynthesis, and water transport. As depicted in Fig. 4c, several target genes were found to be involved in the flavonoid metabolic process, such as MdMYB12 (HF02692), MdbHLH (Basic helix-loop-helix DNA-binding family protein, HF05675), and MdCHS (Chalcone and stilbene synthase family protein, HF00720), which exhibited inhibited expression in MdHYL1 RNAi plants than GL-3. Within the lignin biosynthesis pathway (Fig. 4d), key structural genes such as MdCYP98A3 (cytochrome P450, family 98; HF17045), and the transcriptional regulator MdMYB85 (HF01424), both predicted targets of lncRNAs, exhibited significant upregulation in MdHYL1 RNAi plants than GL-3. Furthermore, comparative transcriptome analysis demonstrated that genes associated with secondary cell wall biosynthesis and modification were upregulated in MdHYL1 RNAi plants compared with GL-3 (Fig. 4e). These included: MdIRX1 (cellulose synthase family protein; HF18140), MdIRX6 (COBRA-like extracellular glycosyl-phosphatidylinositol-anchored protein family; HF09371), MdFLA12 (FASCICLIN-like arabinogalactan-protein 12; HF06217), MdEXPA1 (expansin A1; HF07525). Notably, pronounced differential expression of lncRNA target genes directly implicated in water transport mechanisms were also identified (Fig. 4f), including: MdSIP1;1 (small and basic intrinsic protein 1A; HF29379), MdPIP2;5 (plasma membrane intrinsic protein 2;5; HF04828), and MdTIP1;3 (tonoplast intrinsic protein 1;3; HF24165).
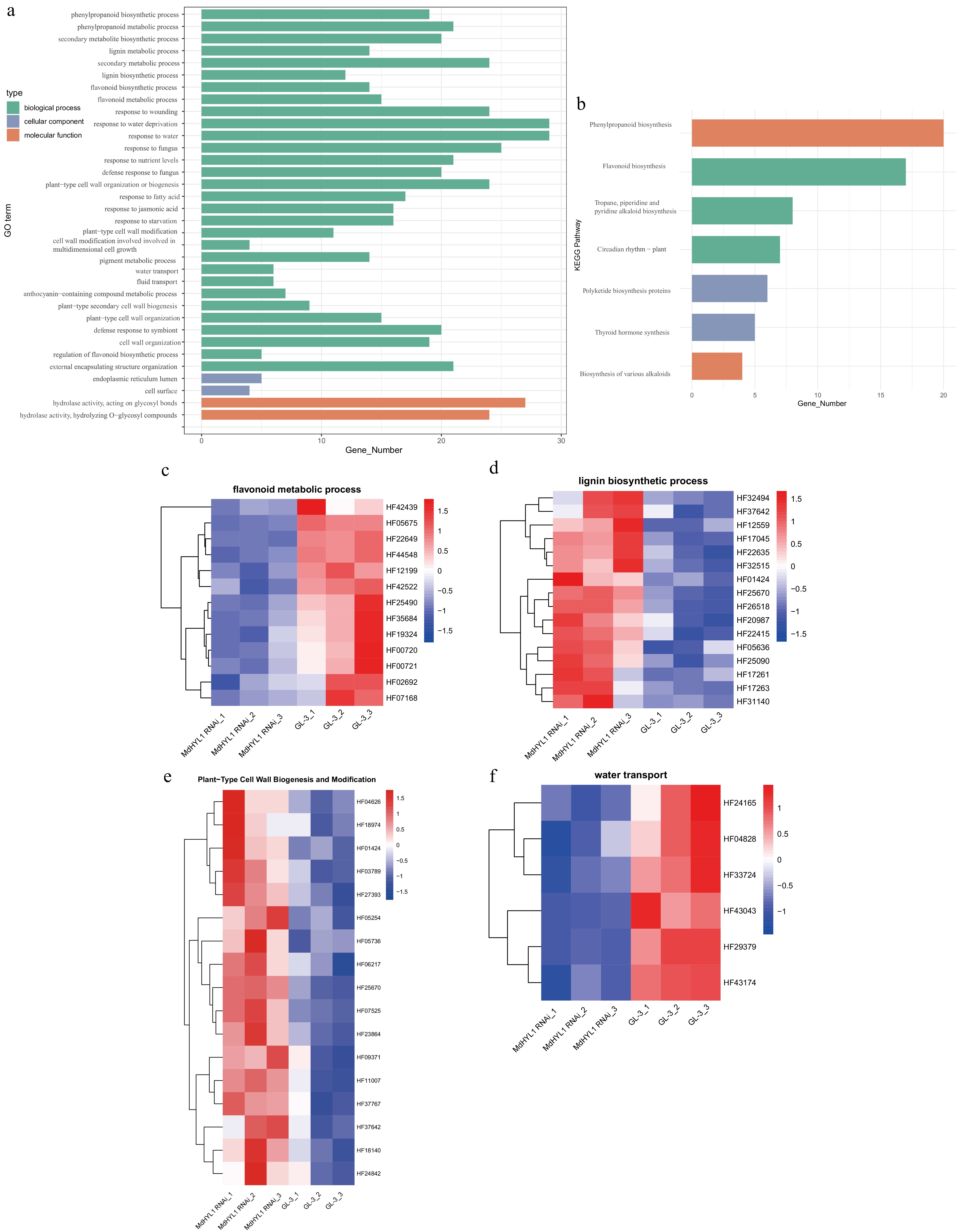
Figure 4.
Functional enrichment and expression of lncRNA target genes in MdHLY1 RNAi plants and GL-3. (a) GO enrichment of differentially expressed lncRNA targets. (b) KEGG enrichment of differentially expressed lncRNA targets. Heatmap showing expression patterns of target genes across MdHYL1 RNAi plants and GL-3 in (c) flavonoid metabolic process, (d) lignin biosynthetic process, (e) plant-type cell wall biogenesis and modification, and (f) water transport. Rows are clustered using hierarchical clustering.
LncRNA-mediated regulatory networks of key genes modulated by MdHYL1
-
To explore the potential relationships between lncRNAs and mRNAs, 13 target genes were selected from several biological pathways of interest involved in four key pathways: flavonoid metabolism, lignin biosynthesis, plant-type cell wall biogenesis and modification, and water transport. An mRNA–lncRNA co-expression network was constructed (Fig. 5) by analyzing the Pearson correlation coefficients (PCC) between lncRNAs and mRNAs. Only pairs with a PCC > 0.8, and an adjusted p-value < 0.01 were included. To facilitate systematic analysis, lncRNAs were named with the prefix 'MSTRG' in the present research.
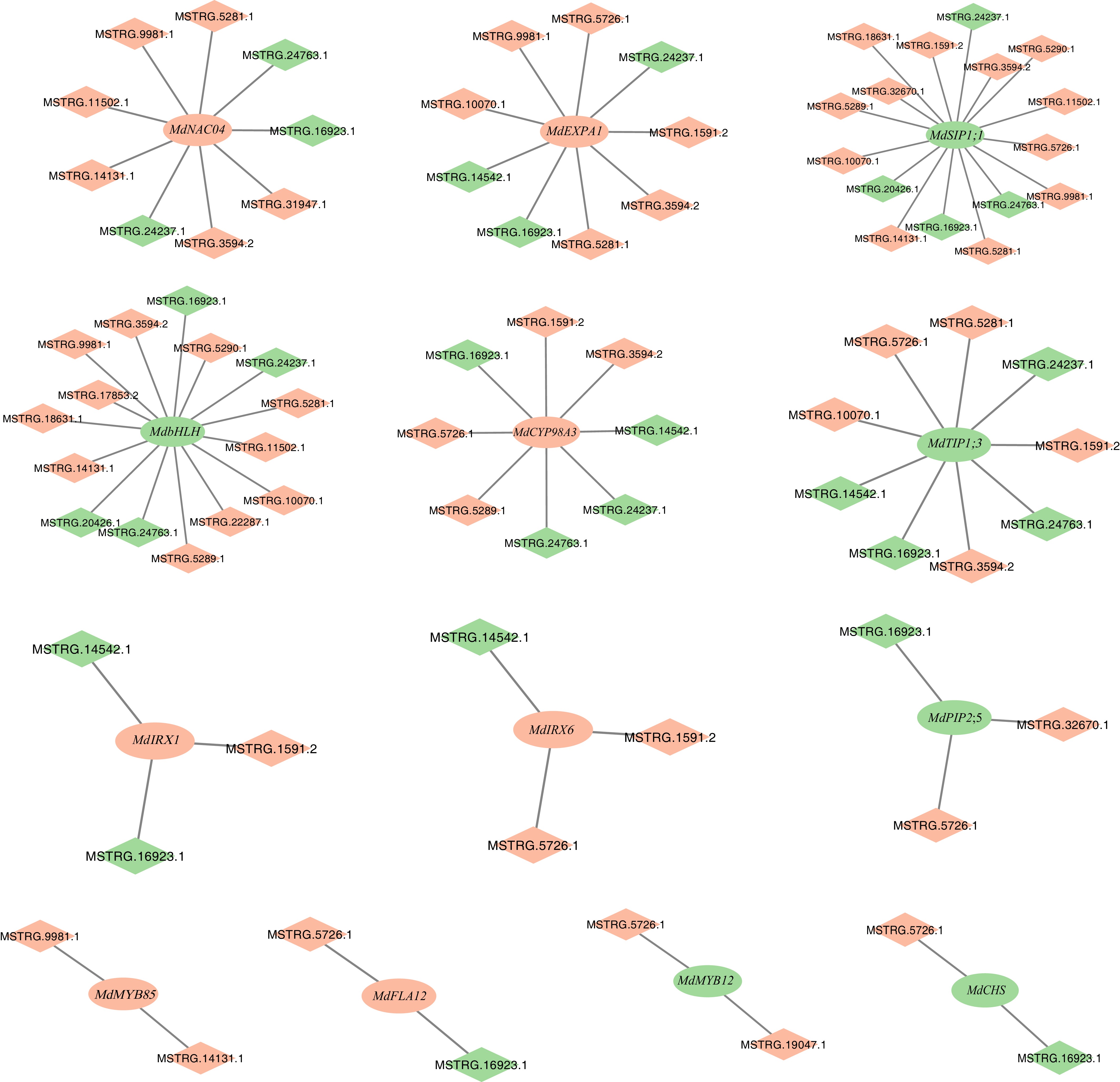
Figure 5.
mRNA–lncRNA regulatory networks centered on key genes involved in flavonoid metabolic process, lignin metabolic process, plant-type cell wall biogenesis and modification, and water transport. The rectangle represents lncRNAs, the diamond represents mRNA, green represents down-regulated genes in MdHYL1 RNAi apple plants than GL-3, and red represents up-regulated trend in MdHYL1 RNAi apple plants than GL-3. Ellipses represent mRNAs, diamonds represent lncRNAs.
In plant-type cell wall biogenesis networks, both the downregulated MSTRG.14542.1 and upregulated MSTRG.1591.2 were co-expressed with MdIRX1 (HF18140) and MdIRX6 (HF09371). The key genes MdMYB12 (as a flavonol-specific activator in the flavonoid biosynthesis pathway, HF02692) and MdCHS (encoding chalcone synthase, the enzyme catalyzing the first committed step of flavonoid biosynthesis, HF00720)[43,44], which were connected with lncRNAs, including MSTRG.5726.1, MSTRG.16923.1, and MSTRG.19047.1. It was also found that MdbHLH (Basic helix-loop-helix DNA-binding family protein, HF05675) connected with four downregulated lncRNAs, and 11 upregulated lncRNAs. Within the lignin biosynthesis network, MdMYB85 (a MYB transcription factor regulating secondary wall formation, HF01424) and MdCYP98A3 (which encodes a key enzyme in the lignin biosynthesis pathway, HF17045) showed no shared regulatory lncRNAs[45,46]. However, MdMYB85 was independently regulated by two lncRNAs, while MdCYP98A3 was associated with eight lncRNAs. The water transport-associated network, key aquaporin genes MdPIP2;5 (the first aquaporin identified as a major contributor to mesophyll conductance of CO2, HF04828), MdTIP1;3 (which encodes a TIP1-type aquaporin, HF24165), and MdSIP1;1 (an aquaporin that facilitates the selectively transports water and neutral substrates, HF29379)[47−49] were both linked to upregulated MSTRG.5726.1 and downregulated MSTRG.16923.1. This dual regulatory pattern suggests that lncRNAs may act as critical modulators of water transport processes in MdHYL1 RNAi plants.
Through network analysis, 21 differentially expressed lncRNAs that participate in regulating the expression of 13 key target genes were identified. Among these lncRNAs, downregulated MSTRG.16923.1 and upregulated MSTRG.5726.1 appeared most frequently, suggesting their potentially strong regulatory roles. Combined with phenotypic observations, it was found that lncRNAs in MdHYL1 RNAi apple plants regulate several target genes, which may influence leaf development by affecting lignin biosynthesis and flavonoid metabolism. Notably, some lncRNAs were also directly linked to aquaporin genes, suggesting a role in modulating water transport efficiency. These findings highlight how lncRNAs may regulate multiple pathways that affect leaf development and water transport.
Regulatory role of lncRNAs mediated by MdHYL1 in the miRNA–mRNA-lncRNA network
-
MiRNAs are one kind of non-coding RNA which regulate gene expression after transcription. They mainly function by binding to the 3'UTRs (3'untranslated regions) of their target messenger RNAs (mRNAs), which leads to translation inhibition or the degradation of the mRNA. LncRNAs, which frequently contain multiple sites complementary to miRNAs, can function as ceRNAs (competitive endogenous RNAs). By binding to specific miRNAs, lncRNAs can reduce miRNA's ability to suppress its target mRNAs. Previous studies have highlighted the significant role of miRNAs in the regulatory landscape of MdHYL1 RNAi apple plants[7,8]. Based on the ceRNA hypothesis, the interactions among miRNAs, mRNAs, and lncRNAs mediated by MdHYL1 were explored. To investigate potential ceRNA roles of LncRNAs, psRNATarget (
www.zhaolab.org/psRNATarget ) was used to predict the miRNA binding site. When a miRNA was predicted to bind both lncRNA and mRNA, this overlapping interaction was considered a potential ceRNA interaction. In this context, the lncRNA may function as a molecular sponge, competitively binding the miRNA and thereby reducing its inhibitory effect on the mRNA, ultimately promoting mRNA expression.The results showed that out of 143 exhibiting differential expression between MdHYL1 RNAi plants and GL-3, a total of 37 miRNAs were predicted to interact concurrently with at least one lncRNA, and one mRNA. Based on these overlapping interactions, a miRNA–lncRNA–mRNA regulatory network was established (Fig. 6). The detail information of the network is shown in Supplementary Table S5. Examination of expression patterns allowed classification of the regulatory relationships into two major categories: (1) low expression levels of both lncRNA and mRNA corresponding to high expression of miRNA (Fig. 6b); and (2) high expression levels of both lncRNA and mRNA corresponding to low expression of miRNA (Fig. 6c). These two patterns are considered to represent the classical ceRNA regulatory model. Among them, nine miRNAs were identified as highly expressed, including PC-3p-128914_27, mdm-MIR10981a-p3_1ss20CT, PC-3p-856_3929, PC-3p-142997_23, PC-3p-74751_56, cca-MIR6112-p5_2ss15CG17AT, PC-3p-163000_19, PC-5p-110844_34, and PC-3p-79491_52. In contrast, ten miRNAs were identified as lowly expressed, including mdm-MIR2118a-p5_2ss9TG23TA, PC-5p-60653_71, PC-5p-54849_79, PC-3p-154744_20, mdm-MIR166d-p5, mtr-miR395g_1ss16GA, PC-3p-108186_35, mdm-MIR319d-p3, mdm-MIR2118c, and PC-3p-61565_70.
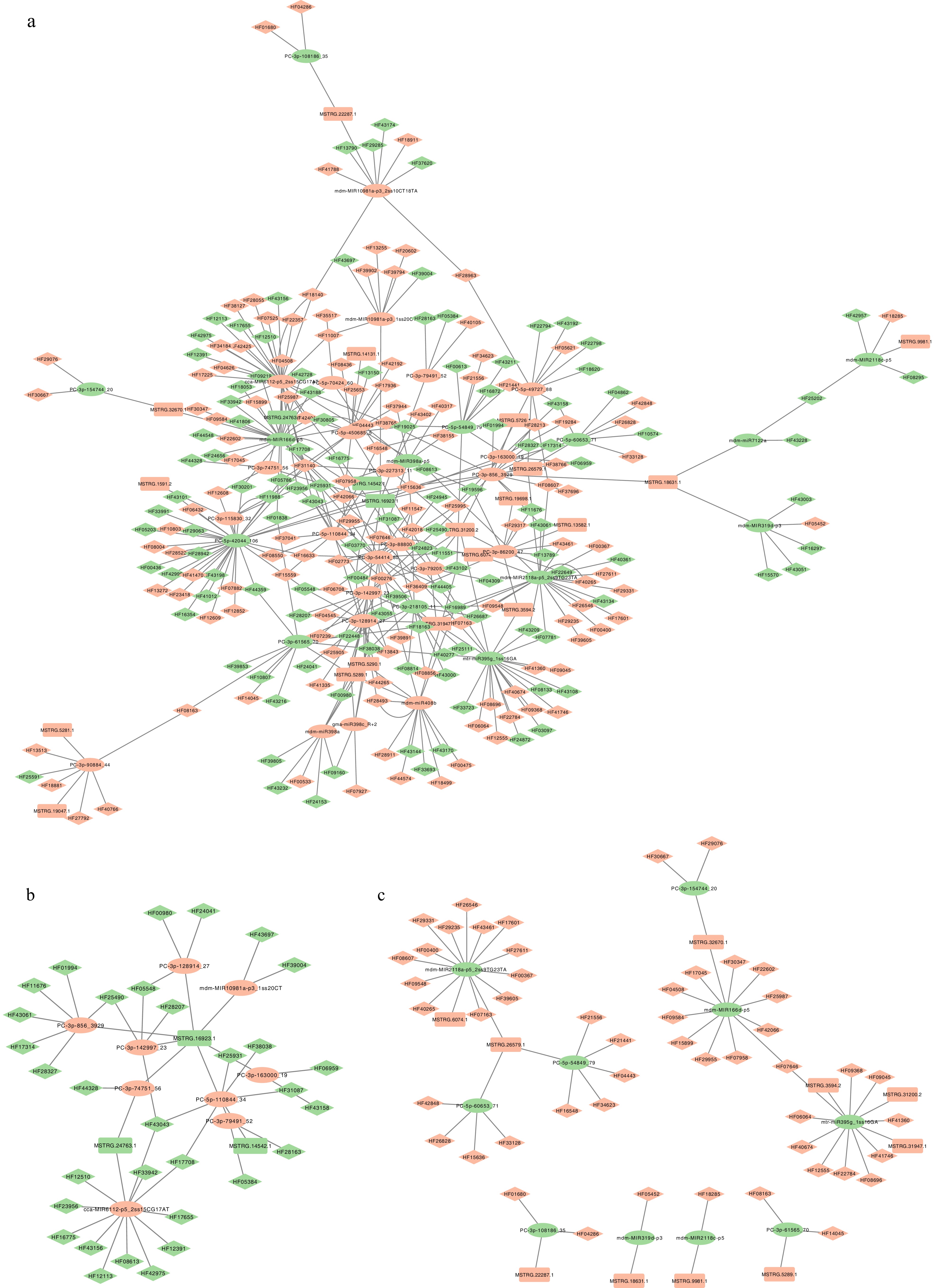
Figure 6.
Visualization of miRNA-mRNA-lncRNA regulatory network in MdHYL1 RNAi plants and GL-3. (a) Predicted comprehensive miRNA-lncRNA-mRNA interaction network. (b) Subnetwork with upregulated miRNAs and their downregulated targets. (c) Subnetwork with downregulated miRNAs and upregulated targets. The ellipse represents miRNA, the rectangle represents lncRNA, the diamond represents mRNA regulated in MdHYL1 RNAi apple plants than GL-3. Green represents down-regulated genes in MdHYL1 RNAi apple plants than GL-3, and red represents up-regulated genes in MdHYL1 RNAi apple plants than GL-3.
Among these miRNAs, the miR166 family (e.g., mdm-MIR166d-p5) is known to regulate developmental processes by targeting class III homeodomain leucine zipper (HD-ZIP III) transcription factors (TFs). These TFs are indispensable for determining leaf and root architecture, particularly in establishing adaxial–abaxial polarity, where miR166 restricts HD-ZIP III expression to the adaxial domain of the leaf primordium[50−52]. The miR319 family (e.g., mdm-MIR319d-p3) has been shown to regulate leaf margin growth, leaf curvature, and flowering time in various species, including Arabidopsis[53], tomato[54], rice[55], and poplar[56]. This regulation is primarily through its target, the TCP transcription factor family, which controls cell proliferation during leaf morphogenesis. In poplar, overexpression of miR395c was found to decrease ABA content in stems and leaves, increase stomatal aperture, and enhance water loss[57]. The miR395 family (e.g., mtr-miR395g_1ss16GA) may influence ABA biosynthesis and expression of MYB46 through the sulfur metabolism pathway during secondary xylem development[57]. Notably, MYB46 overexpression has been associated with leaf curling and lignin accumulation, indicating its role in modulating water transport and leaf development[58,59]. Collectively, these results indicate that lncRNAs may modulate miRNA activity by acting as ceRNAs, thereby regulate plant leaf development and water transport.
Confirming the expression results of RNA sequencing with RT-qPCR
-
To validate the reliability of the RNA-seq data, ten key protein-coding genes involved in lignin biosynthetic process, plant-type secondary cell wall biogenesis and modification, flavonoid biosynthetic process, and water transport were selected. The RT-qPCR analysis revealed distinct expression patterns between MdHYL1 RNAi plants and GL-3 controls (Fig. 7a). Specifically: MdCYP98A, MdNAC042, MdIRX1, MdIRX6, and MdMYB85 showed significantly higher expression levels in MdHYL1 RNAi plants compared to GL-3. In contrast, MdMYB12, MdCHS, MdSIP1;1, MdTIP1;3, and MdPIP2;5 was markedly suppressed in MdHYL1 RNAi plants relative to GL-3. Furthermore, six lncRNAs exhibiting the most robust predicted trans-regulatory relationships and ten miRNAs with high-confidence interaction predictions (via psRNATarget analysis) were selected for experimental validation by RT-qPCR. Among the lncRNAs, three (MSTRG.10070.1, MSTRG.14131.1, and MSTRG.1591.2) showed significant upregulated in MdHYL1 RNAi plants compared to controls, while the remaining three (MSTRG.14542.1, MSTRG.16923.1, and MSTRG.24237.1) demonstrated clear downregulated patterns (Fig. 7b). Further stem-loop qPCR confirmed miRNA dysregulation in MdHYL1 RNAi plants vs GL-3: downregulation of mdm-miR482a, mdm-miR398a, mdm-miR858, mdm-miR2118c, and PC-5p-60653_71; upregulation of mdm-miR11017-p5, cca-miR6112-p5, mdm-miR10996a, PC-5p-109892_34, and PC-3p-9232_479. Predicted targets of miRNAs based on mRNAs included MdMYB85 (mdm-miR482a), MdNAC042 (mdm-miR858), MdIRX6 (mdm-miR11017-p5), MdMYB12 (PC-5p-60653_71), and MdCHS (PC-5p-109892_34). Bioinformatic analysis of lncRNA-associated miRNAs further predicted additional target genes, including: MdIRX1 (targeted by mdm-miR398a) and MdBHLH (targeted by mdm-miR2118c). As shown in Fig. 7, the expression patterns from RT-qPCR aligned with the RNA-seq data, confirming the reliability and reproducibility of the high-throughput sequencing data.
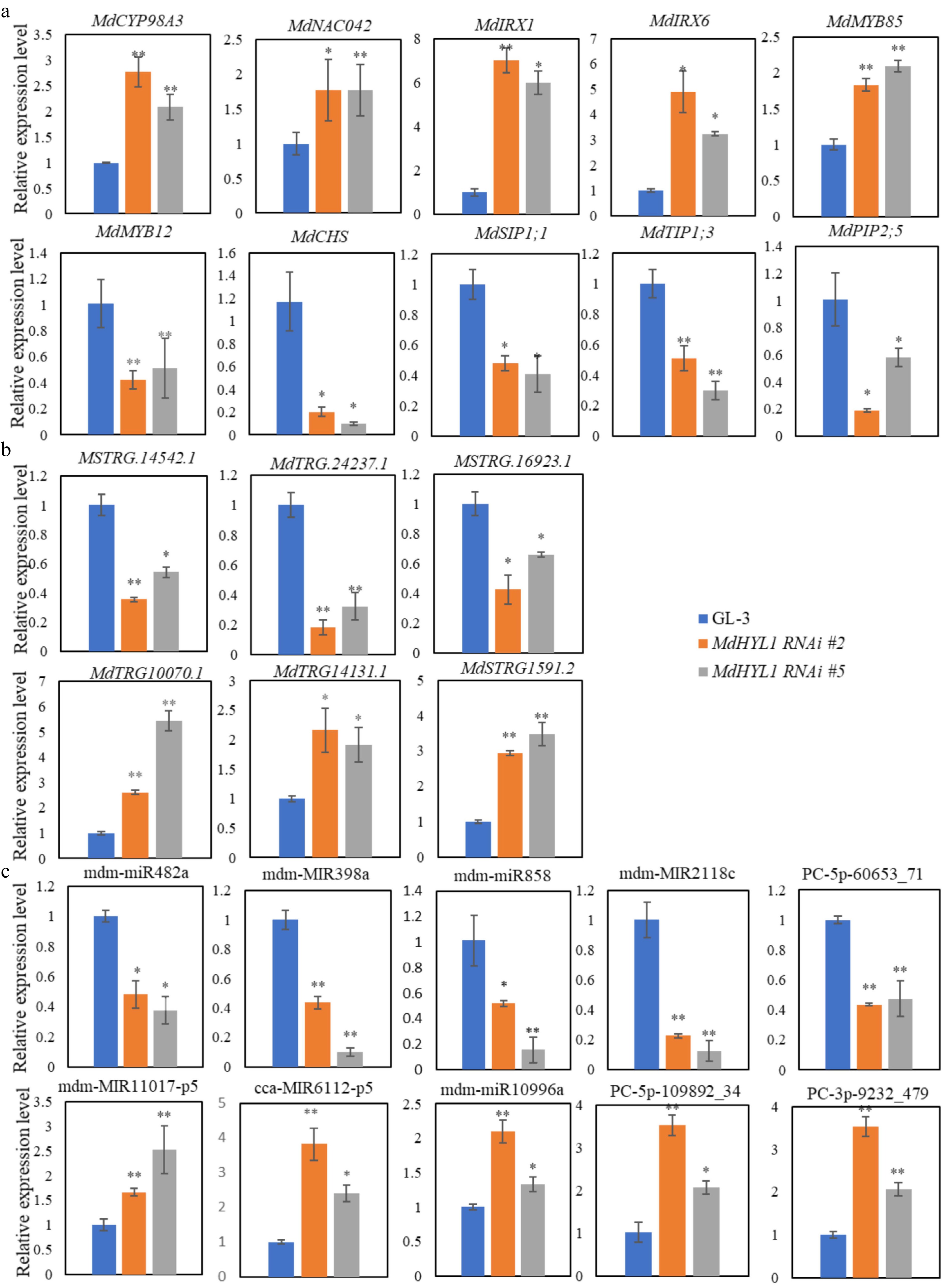
Figure 7.
Relative expression of (a) mRNAs, (b) lncRNAs, and (c) miRNAs with RT-qPCR confirmation in the GL-3 and MdHYL1 RNAi plants. The two-tailed t-test analyzed the differences between groups, * p < 0.05; ** p < 0.01.
-
Apple (Malus domestica) is one of the most cultivated and commercially important fruit crops, with a long history of domestication and a broad geographic distribution. LncRNAs, defined as transcripts longer than 200 nucleotides without protein-coding capacity, have emerged as important regulators of plant growth, development, and stress adaptation. Accumulating evidence demonstrates their functional significance in organ morphogenesis and environmental responses. For example, in leaf development, lncRNAs have been reported to: (1) modulate leaf shape determination in Arabidopsis thaliana[60]; (2) maintain leaf flatness in rice (Oryza sativa)[61]; and (3) associate with leaf developmental traits through single nucleotide polymorphism (SNP) variations in 951 identified long intergenic non-coding RNAs (lincRNAs) in maize (Zea mays)[62]. Furthermore, pathogen response studies revealed lncRNA upregulation in tomato (Solanum lycopersicum) following tomato yellow leaf curl virus infection[63]. Beyond developmental regulation, lncRNAs participate in abiotic stress adaptation, particularly in: (1) water transport regulation and drought resistance in Arabidopsis[64]; (2) stress response modulation in tomato[65]; (3) drought adaptation in cotton (Gossypium hirsutum)[66]; and (4) water use efficiency in upland rice (Oryza sativa)[67]. Despite these advances, the role of lncRNAs in regulating leaf development and water transport in apple remains poorly understood. In the present study, it was observed that MdHYL1 RNAi transgenic apple plants exhibited clear defects in leaf development and impaired water transport. Previous work has demonstrated that miRNAs play essential roles in MdHYL1 RNAi plants[2,7,8,68]. Given the ceRNA hypothesis, it was hypothesized that lncRNAs may also contribute to these processes by modulating miRNA activity. Therefore, strand-specific RNA sequencing (ssRNA-seq) was conducted on both MdHYL1 RNAi and GL-3 apple plants. This comprehensive transcriptomic analysis revealed a potential regulatory network involving lncRNAs in leaf development and water transport. The results indicate that lncRNAs, as crucial regulatory non-coding RNAs, may influence leaf development and water transport efficiency in apple by modulating miRNA activity or directly regulating target gene expression.
In total, 768 lncRNAs were identified, among which 762 were classified as novel lncRNAs based on alignment with the NONCODE v6_Arabidopsis database. These newly discovered lncRNAs provide an important molecular resource for future studies of plant biological processes. Compared with mRNAs, the identified lncRNAs displayed fewer exons and shorter open reading frames (ORFs), which is consistent with previous reports[22,25,40]. Differential expression analysis revealed 813 mRNAs, and 28 lncRNAs showing significant changes between MdHYL1 RNAi and GL-3 plants. Pearson correlation analysis predicted 736 potential target genes of these differentially expressed lncRNAs, and a regulatory network was subsequently constructed using Cytoscape. Functional enrichment analysis (GO and KEGG) demonstrated that these target genes were significantly associated with biological pathways, including lignin biosynthesis, flavonoid metabolism, water transport, and plant-type cell wall biogenesis and modification. For example, the target genes MdMYB85 and MdCYP98A3 are directly involved in lignin biosynthesis[45,46], suggesting that MdHYL1 may regulate lignin deposition—and consequently water transport efficiency through lncRNA-mediated mechanisms. A previous study found that lignin is predominantly deposited in the secondary cell wall, where it reinforces structural rigidity to sustain plant growth[69]. Meanwhile, studies have shown that excessive lignin deposition of bundle sheath (BS), and bundle sheath extension (BSE) cells in minor veins leads to cell wall thickening. Such lignified BS cells not only inhibit water transport through the apoplastic pathway but also restrict water flow across cell membranes, ultimately resulting in a reduction of leaf hydraulic conductance (K_leaf)[70].
In addition, the significant differential expression of flavonoid biosynthesis-related genes such as MdMYB12 and MdCHS, implies that disruptions in flavonoid metabolism may influence auxin transport and stomatal development, leading to reduced leaf size. These observations align well with earlier studies emphasizing the role of flavonoids in plant drought responses and developmental processes[43,44]. Furthermore, a miRNA–mRNA-lncRNA regulatory network based on the ceRNA hypothesis. was constructed By integrating expression level changes, several key miRNAs with functional significance were identified, including members of the miR166, miR319, and miR395, and miR2118 families[50,53,57,71], which have been widely reported to play roles in leaf development, water stress responses, and flavonoid metabolism. Notably, miR166 targets HD-ZIP III family genes and directly regulates water transport in plants[50−52]. MiR319 influences leaf morphology and anthocyanin accumulation through TCP transcription factors[53−56,72,73]. Interestingly, it was found that both the upregulated MSTRG.32670.1 and MdCYP98A3(HF17045) can bind to the downregulated miR166. MdCYP98A3 is enriched in the lignin biosynthetic process, and miR166 is related to the leaf phenotype. It is therefore proposed that MSTRG.32670.1 may function as an eTM, which links non-coding RNA interactions to lignin biosynthesis and leaf phenotype. Compared to previous studies that investigated individual components of post-transcriptional regulation, such as miRNA-target relationships or lncRNA functions in isolation, the present study presents an integrative framework that simultaneously incorporates lncRNAs, miRNAs, and mRNAs into a unified regulatory network. These well-established regulatory mechanisms further support the reliability and biological significance of the miRNA–mRNA–lncRNA network, demonstrating that lncRNAs may modulate plant leaf development and water transport efficiency, by binding miRNAs and affecting their regulatory control over target mRNAs. Finally, RT-qPCR validation of representative mRNAs, lncRNAs, and miRNAs confirmed the expression trends observed in RNA-seq, supporting the reliability of the transcriptomic data.
In this study, although a relatively small number of differentially expressed lncRNAs were identified through transcriptome analysis comparison between MdHYL1 RNAi transgenic apple plants and GL-3, it was observed that 90.5% (736 out of 813) of the differentially expressed mRNAs were correlated with these lncRNAs. This led to the construction of a complex miRNA–mRNA-lncRNA regulatory network. It is speculated that this may be attributed to the role of the upstream gene MdHYL1 in the processing of pre-miRNAs, thereby affecting miRNAs and mRNAs expression levels. Some key lncRNAs identified in this study likely function as ceRNA modules, interacting with multiple mRNAs by sharing common miRNA targets. This structural characteristic suggests that under MdHYL1 disruption, lncRNAs may act as central 'hub' regulators that amplify miRNA-mediated effects within the response pathway, ultimately influencing leaf development and water transport in MdHYL1 RNAi plants. Nevertheless, although the integrative multi-omics analysis has provided a preliminary regulatory model, the precise biological functions of these lncRNAs remain to be experimentally identified in future studies.
-
ssRNA-seq was performed on MdHYL1 RNAi apple plants to identify lncRNAs involved in their regulatory network. In total, 28 lncRNAs, and 813 mRNAs were differentially expressed. Through predictions of both cis-acting and trans-acting function, 736 target genes of these lncRNAs were identified, and a lncRNA–mRNA interaction network constructed. GO enrichment analysis revealed that these target genes were significantly involved in biological processes. Several of these targets were associated with flavonoid metabolism, lignin biosynthesis, plant-type secondary cell wall biogenesis, and water transport. Based on miRNA data, endogenous target mimics (eTMs) among the lncRNAs were predicted, suggesting that lncRNAs may directly regulate gene expression or competitively bind to miRNAs, thereby influencing leaf development and water transport. Notably, it was found that MSTRG.32670.1 may act as an eTM of miR166 to regulate MdCYP98A3 (HF17045), linking non-coding RNA regulation with lignin deposition and water transport. RT-qPCR was used to validate a selection of key transcripts, and the results confirmed the findings from the RNA-seq data.
Overall, this work offers novel insights into the functional involvement of lncRNAs in MdHYL1-mediated regulation of leaf development and water transport in apple, and establishes a theoretical framework that may guide future investigations of lncRNA-mediated regulatory mechanisms in apple, as well as other woody fruit species.
This work was supported by the National Natural Science Foundation of China (32402520, 32441069), Guangdong-Shenzhen Joint Fund for Basic and Applied Basic Research Science and Technology Research (2023A1515110171), Xinjiang Apple Research System of China (XJLGCYJSTX04), and Chinese Universities Scientific Fund (2452023067). We thank the High-Performance Computing (HPC) Center of Northwest A&F University (NWAFU) for providing the computational resources that supported this work.
-
The authors confirm their contributions to the paper as follows: conceived and designed the study: Guan Q, Li X; contributed to discussions about the work: Ma F; performed the experiments: Liu Z, Wang H, Shen X, Geng D; carried out the data analysis: Wen J, He J; drafted the manuscript: Wen J. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Jiale Wen, Jieqiang He
- Supplementary Table S1 Primers used in this study.
- Supplementary Table S2 Read counts and differentially expressed lncRNA, mRNA and miRNA.
- Supplementary Table S3 Predicted target of lncRNA by co-expression.
- Supplementary Table S4 miRNA–mRNA-lncRNAnetwork.
- Supplementary Table S5 Description of target genes.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wen J, He J, Wang H, Liu Z, Shen X, et al. 2025. Genome-wide identification and expression analysis of long noncoding RNAs in MdHYL1-mediated regulation of leaf development and water transport in apple. Fruit Research 5: e044 doi: 10.48130/frures-0025-0035 

Genome-wide identification and expression analysis of long noncoding RNAs in MdHYL1-mediated regulation of leaf development and water transport in apple
- Received: 30 June 2025
- Revised: 23 August 2025
- Accepted: 13 October 2025
- Published online: 15 December 2025
Abstract: HYL1 serves as a central component of the miRNA processing machinery, playing a vital role in coordinating plant development and adaptation to environmental stresses. In apple, MdHYL1 knockdown impairs leaf development and water transport, though whether this regulation involves miRNA/lncRNA networks remains unclear. To elucidate the regulatory mechanisms underlying leaf development and water transport mediated by MdHYL1 in apple, transcriptome-wide profiling of lncRNAs, miRNAs, and mRNAs in wild-type GL-3 vs MdHYL1 RNAi transgenic plants were conducted. In total, 28 lncRNAs and 813 mRNAs were detected as exhibiting significant differential expression, with lncRNAs predicted to regulate target mRNAs through cis- or trans-acting mechanisms. Functional annotation revealed these target genes to be enriched in flavonoid biosynthesis (MdMYB12, MdCHS), lignin metabolism (MdCYP98A3, MdMYB85), cell wall biogenesis/modification (MdNAC042, MdIRX1, MdIRX6), and water transport pathways (MdSIP1;1, MdTIP1;3, MdPIP2;5). RT-qPCR analysis further confirmed the co-expression networks between the identified mRNAs and lncRNAs. How MdHYL1 influences the regulatory functions of lncRNAs within the miRNA–mRNA–lncRNA network was subsequently examined. Members of the miR482, miR398, miR858, and miR2118 families were identifed as endogenous target mimics (eTMs) for specific lncRNAs. Notably, MSTRG.32670.1 may act as an eTM of miR166 to regulate MdCYP98A3, linking non-coding RNA regulation with lignin deposition and water transport. The present results uncover new mechanistic perspectives on how lncRNAs participate in MdHYL1-mediated regulation of leaf development and water transport in apple, while establishing a conceptual framework for future investigations of lncRNA functions in woody perennial crops.
-
Key words:
- Apple /
- lncRNA /
- MdHYL1 /
- Leaf development /
- Water transport