








-
Traditional pest management strategies, specifically reliance on chemical pesticides have enormously led to the emergence of resistance in insect pests, environmental degradation, and nontarget effects on food safety[1]. Agricultural insect pests are highly adept at adapting to both natural and human-induced stressors, consistently overcoming population bottlenecks and developing resistance to intensive management strategies. Agricultural insects refer to both pests and beneficial insect species that have a direct or indirect impact on agricultural systems/space. The overuse and exploitation of insecticides have exacerbated adaptive resistance mechanisms in pest populations, rendering many conventional control strategies less effective[2]. This has resulted in a significant need for environmentally friendly, sustainable, and tailored approaches that can minimize the reliance on chemical-based interventions. In a wake of recent scientific breakthroughs, genomics has grown as a revolutionary field, providing unprecedented insights into the biology, evolution, and adaptation of insect pests. Leveraging whole-genome sequencing and transcriptomic analyses, researchers can decode the genetic blueprint of insect pests, identifying the genes and pathways responsible for key adaptive traits, including insecticide resistance, immune response, reproduction, and host plant specialization[3]. Briefly, genomics is a field of science that dwells on the study of an organism's entire set of genetic material, known as the genome. Genomics, as a key component of the omics field, has driven a paradigm shift in insect pest studies across multiple levels, including molecular, comparative, population, structural, epigenetic, and translational landscapes. The ability to perform genome-scale analyses enables researchers to address critical questions that were previously intractable using small numbers of neutral genetic markers[4]. Through the 5,000-insect genome project, several complete genomes have been accomplished. Through searches on PubMed we found over 78,910 related publications (Fig. 1a). Over the years, several insect genomes have been generated, ranging from contig-level to complete genomes, with contig- and chromosome-level assemblies dominanting (Fig. 1b). These advancements in insect pest genomics have enabled the tracking of resistance-linked alleles, the prediction of outbreaks, and a deeper understanding of evolutionary dynamics in response to environmental fluctuations.
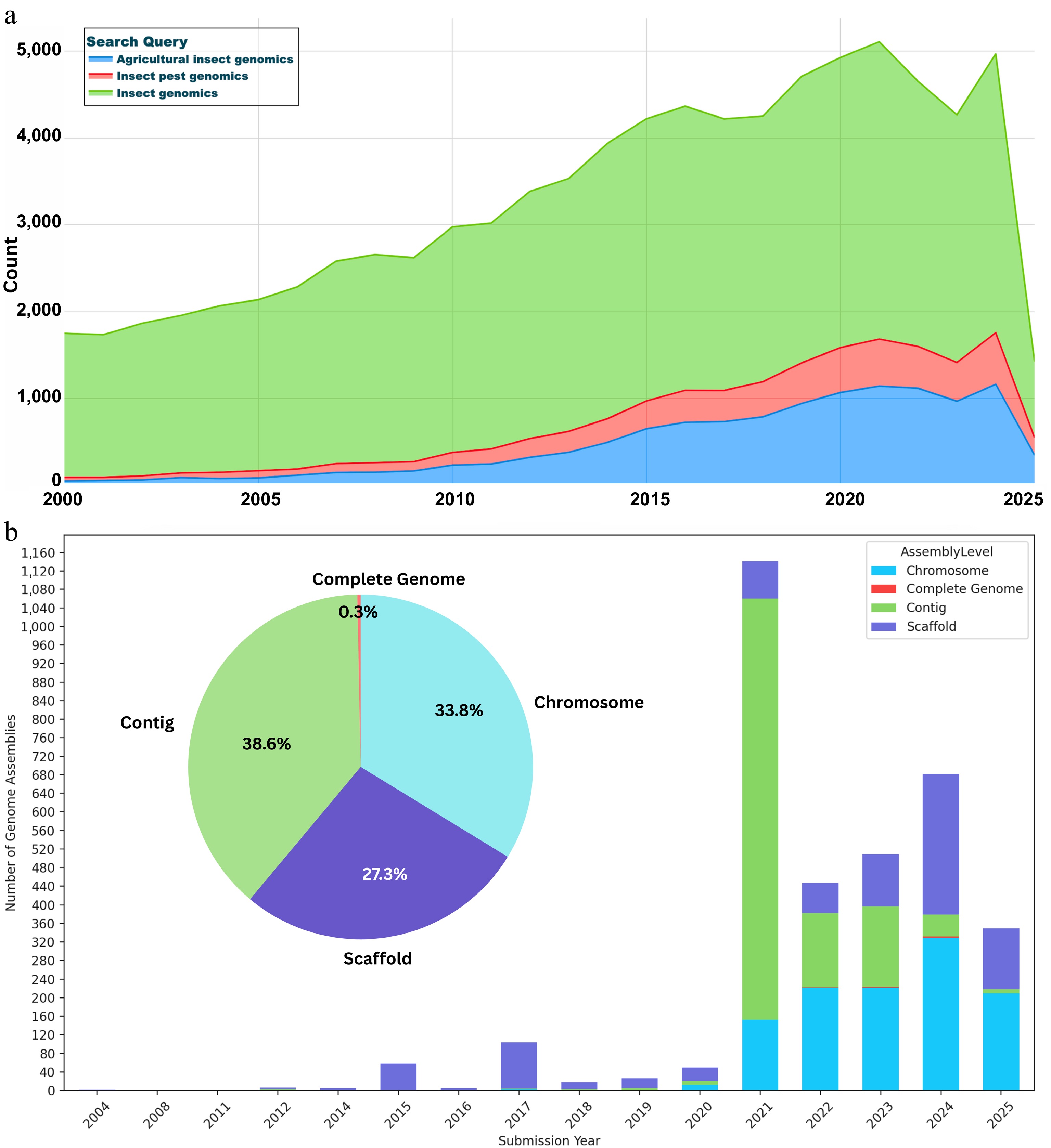
Figure 1.
Trends in insect genomics research and genomic resources. (a) Publication trends in insect genomics from PubMed. The number of publications per year (as of 13 April 2025) was retrieved using the search query (insect genomics/insect genome/insect genome sequencing) in PubMed. The trend highlights the growing academic focus on insect genomics over the past two decades, with notable surges coinciding with the adoption of next-generation and third-generation sequencing technologies. (b) Cumulative count of publicly available nuclear genomes of insects, dominated by agro-economical insects, classified as complete, chromosome, scaffold, and contig-level genomes. Data were retrieved from the NCBI Assembly database (as of 12 April 2025) by searching for nuclear genomes associated with insect species classified as agricultural pests.
-
This review explores the transformative potential of genomic technologies in insect pest research, emphasizing their impact on pest management strategies. We highlight key advancements, including genome sequencing (GS), which has enabled the generation of high-quality pest genome assemblies; comparative genomics, which reveals the evolutionary relationships and species-specific adaptations; and population genomics, which tracks genetic diversity and the spread of resistance alleles. Additionally, we examine emerging fields such as pangenomics, which investigates genetic variation within species; epigenomics, which elucidates the role of epigenetic mechanisms in pest adaptation; and structural genomics, which enhances our understanding of the protein structures that are critical for insecticide development. Furthermore, we discuss museum genomics, which leverages historical specimens to study long-term evolutionary and ecological changes; mitochondrial genomics, which has a role in evolutionary biology; and translational genomics, which bridges fundamental genomic discoveries with practical pest control applications. 'Genomics is here today, and it will be there tomorrow': this statement encapsulates the transformative and enduring impact of genomic research on evolutionary biology. These genomic studies will help in safeguarding increased productivity, hence contributing to Sustainable Development Goal 2 (SDG 2) of the United Nations (
www.un.org/sustainabledevelopment/hunger ) in achieving a world free from hunger and a better world. By integrating various genomic approaches, this review provides a perspective in a simplified format on how genomics is revolutionizing pest management in addressing critical agricultural challenges worldwide. While it is not feasible to comprehensively cover all insect taxa, this review highlights notable advances and emerging trends in insect genomics. -
Genome sequencing has revolutionized the study of agricultural insect pests by providing a comprehensive view of their genetic makeup. It serves as the foundation for understanding key biological traits, including adaptation to host plants, insecticide resistance, and reproductive strategies. Advances in sequencing technologies, particularly next-generation sequencing (NGS) platforms like Illumina, and third-generation sequencing (TGS) methods such as PacBio and Oxford Nanopore, have facilitated the generation of high-quality reference genomes for numerous insect pest species[5]. Currently, genome assembly is a critical step following sequencing, involving the reconstruction of raw sequence reads into contiguous sequences that represent the insect genome. Genome assembly primarily employs two approaches: de novo assembly, which reconstructs genomes without relying on a reference, and reference-guided assembly, which leverages the genome of a closely related species as a template[6]. At present, several tools used for nuclear genome assembly have been developed with similar algorithms that involve aligning sequence fragments against one another, then identifying and merging overlapping sections[7]. Over 39 TGS-related tools for genome assembly are publicly available[8]. Other methods are haplotype-based genome assembly methods, which are known to be computationally intensive and difficult[9]. Short-read sequencing technologies like Illumina produce high-accuracy reads but often result in fragmented assemblies due to difficulties in resolving repetitive regions. Conversely, long-read sequencing technologies such as PacBio and Nanopore generate longer sequence reads that improve genomes' contiguity and structural resolution, particularly in highly repetitive insect genomes. Leveraging on these long reads, the near-complete assemblies of some chromosomes are now possible, also known as telomere-to-telomere (T2T) assembly in insects such as Oenanthe javanica (Apiales: Apiaceae) and Bombyx mori (Lepidoptera: Bombycidae)[10−12] among the few T2T insect assemblies that are publicly available. Hybrid approaches, which combine short- and long-read sequencing, have been widely used to generate high-quality assemblies[13], as seen in the genome projects of Helicoverpa armigera (Lepidoptera: Noctuidae)[14] among others. As of 2025, over 7,400 insect genome assemblies have been deposited in public databases, including more than 2,647 chromosome-level, 2,500 scaffold-level, and 2,300 contig-level assemblies, according to the National Center for Biotechnology Information (NCBI). This rapid expansion has been fueled by the increasing use of long-read sequencing technologies such as PacBio; Oxford Nanopore, which produce contiguous read sequences that are typically ≥ 10 kb in length; and high-throughput chromatin conformation capture (Hi-C) scaffolding, particularly since 2015, which have enabled the generation of highly contiguous and few complete genome assemblies (Fig. 2). However, despite these technological advances, taxonomic representation remains uneven, with genomic resources disproportionately concentrated in a few major insect orders, most notably Diptera, Hymenoptera, Hemiptera, and Lepidoptera, while many ecologically and economically significant groups remain under-represented (Fig. 2).
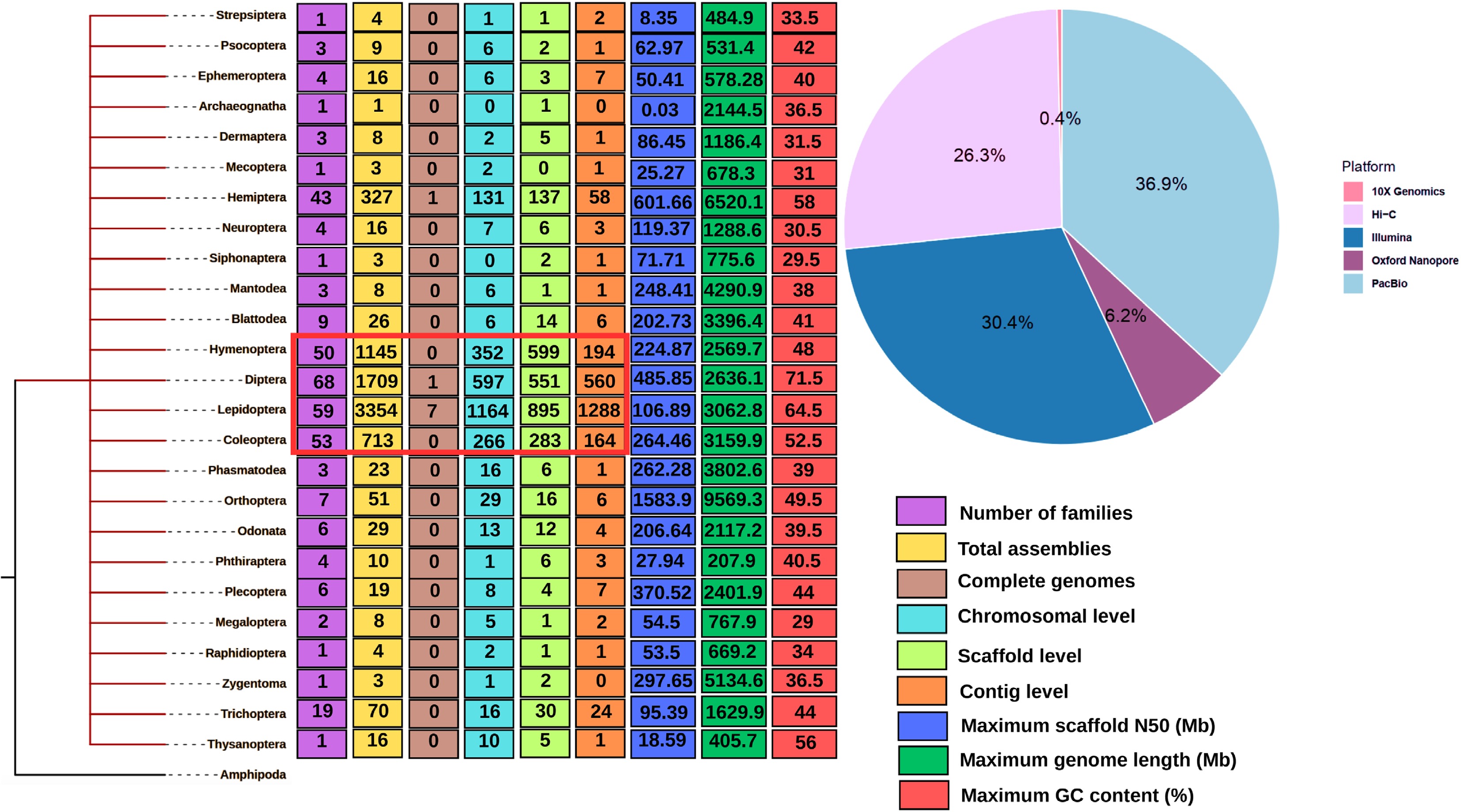
Figure 2.
Phylogenetic tree representing 25 insect orders for which nuclear genome assemblies are publicly available in the NCBI database. The tree is rooted with Amphipoda, which, while not an insect order, serves as an appropriate outgroup for comparative purposes. Each order is annotated with the number of insect families that have at least one genome sequenced, as well as proportions reflecting the genome assembly's level such as contig, scaffold, and chromosome-scale. Additional annotations include the maximum scaffold N50, contig N50, and GC content observed for each order. Insect orders with dominant representation at higher assembly levels are highlighted in red. The pie chart alongside the tree summarizes the relative distribution of major sequencing technologies such as Illumina, PacBio HiFi, Oxford Nanopore, 10X Genomics, and Hi-C used across these genome projects.
High-quality genome assemblies of some notable insect species with benchmarking universal single-copy ortholog (BUSCO) completeness ranging from 94% to 100% have been achieved (Fig. 3). These assemblies have enabled the identification of gene families involved in insecticide detoxification, host plant adaptation, and immune responses[15]. For instance, whole-genome seequncing (WGS) has revealed extensive expansions in cytochrome P450, carboxylesterase (COE), and glutathione S-transferase (GST) genes in several pest species, which are directly associated with their ability to metabolize chemical insecticides[16]. Additionally, genome assemblies have facilitated the characterization of chemosensory genes, including odorant receptors (ORs) and gustatory receptors (GRs), which are crucial for host plant selection and feeding behavior in various agricultural insects[17]. Moreover, well-assembled genomes have provided insights into structural variations that contribute to adaptation. By mapping sequences to reference genomes, WGS detects a range of genetic variations, including single nucleotide polymorphisms (SNPs), structural variations (SVs), insertions and deletions (InDels), and copy number variations (CNVs)[18]. Accurate genome assemblies also serve as references for population genomics studies, enabling researchers to track resistance alleles and genetic diversity in field populations. As more pest genomes become available, comparative analyses will continue to refine our understanding of the genetic mechanisms underlying pest resilience, reinforcing the role of WGS as an indispensable tool in agricultural entomology.
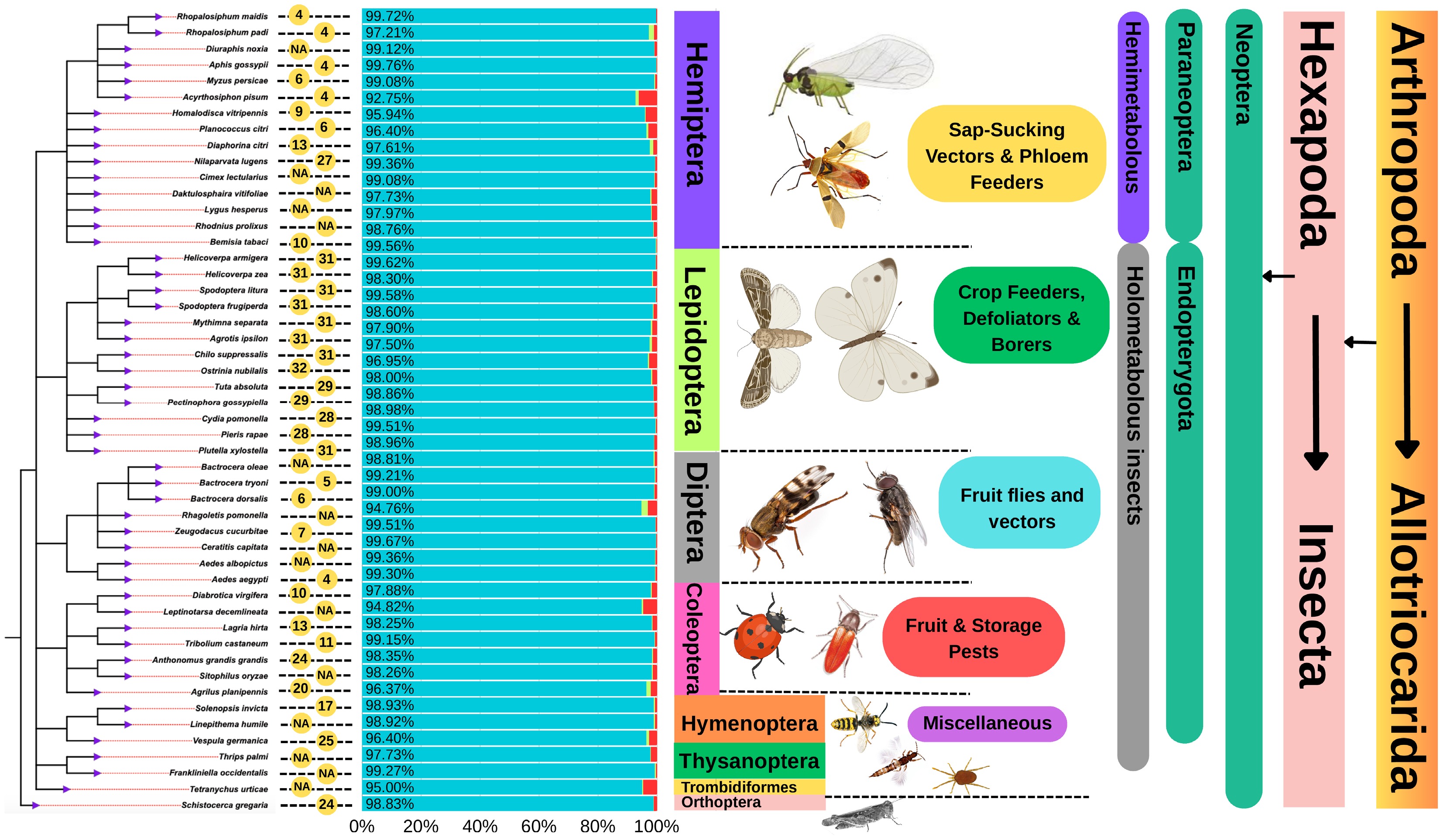
Figure 3.
Phylogenetic tree of 50 representative insect species with publicly available genome assemblies, based on data retrieved from the NCBI and Insectbase databases. The tree is rooted with Schistocerca gregaria (Orthoptera) and annotated with key genomic and taxonomic information. Yellow circles indicate the number of chromosomes per species (NA denotes unavailable chromosome data). BUSCO bar graphs accompany each species, with blue bars representing complete BUSCOs, light green indicating duplicated BUSCOs, and red showing missing BUSCOs, reflecting genomes' completeness and quality. The tree is further annotated with hierarchical classifications, including phylum, subphylum, clade, class, and family; developmental type (e.g., hemimetabolous or holometabolous); and body morphology. Taxonomic classifications have been adapted from publicly available sources, including Wikipedia and NCBI Taxonomy.
-
Comparative genomics, which is a holistic approach, provides a platform for researchers to compare two or more genomes or sequences, enabling the discovery of similarities and differences among the genomes or sequences. It involves the comparison of genetic information within and across organisms to analyze the identification, evolution, structure, and function of genes, proteins, noncoding regions and regulatory elements[19]. Various techniques are employed in comparative genomics to unravel the evolutionary relationships and functional adaptations in insect pests. For instance, ortholog detection, using tools such as OrthoFinder, identifies gene counterparts across species to infer evolutionary conservation and divergence. Not only that, gene family expansion and contraction analysis, conducted with tools like CAFÉ, reveals lineage-specific adaptations linked to ecological specialization, such as detoxification or host plant adaptation. Furthermore, phylogenomic analysis, utilizing tools such as IQ-TREE and PhyloBayes, reconstructs evolutionary histories by analyzing genome-wide sequence data. For instance, three solanaceous feeding species, Phthorimaea absoluta, Keiferia lycopersicella, and Phthorimaea operculella (Lepidoptera: Gelechiidae) were found to cluster together[20]. Additionally, syntenic analysis with tools like MCScanX detects genome rearrangements, while positive selection analysis using such as PAML and HyPhy may identify genes under adaptive evolutionary pressure.
Through comparative genomics, some researchers have revealed evolutionary patterns in aphid chromosomes, where the X chromosomes have undergone extensive intra-chromosomal recombination while the autosomes have undergone rapid inter-chromosomal recombination[21]. Comparative genomics is used as a tool for identification of key evolutionary events that contributed to pest adaptation, and studies have also shown significant gene family expansions in phytophagous insects, particularly in Oncopeltus fasciatus (Hemiptera: Lygaeidae) and Riptortus pedestris (Hemiptera: Alydidae), with enzyme-encoding genes likely facilitating hemipteran–plant interactions and host adaptation[22]. Additionally, comparative analysis revealed differences in the regulation of most genes between Laodelphax striatellus and Nilaparvata lugens (Hemiptera: Delphacidae) under similar conditions, though sugar transporters and heat-shock proteins showed similar variations[23]. Several comparative studies have identified the expansion of gene families related to detoxification, chemoreception, nutrient metabolism, and transport systems in Colaphellus bowringi (Coleoptera: Chrysomelidae), Spodoptera frugiperda (Lepidoptera: Noctuidae), Drosophila melanogaster (Diptera: Drosophilidae), Helicoverpa assulta (Lepidoptera: Noctuidae), Helicoverpa armigera, etc. On the other hand, genetic comparisons of haplotype frequencies have revealed significant differences between geographically distant fall armyworm populations[24]. In the same vein, integrating RNA-sequencing of epithelial tissues with comparative genomics in Helicoverpa armigera revealed distinct tissue-specific expression patterns among the seven P-glycoprotein paralogs, suggesting a spatial division of labor[25]. Comparative genomics of C-type lectins (CTLs) in seven holometabolous insects revealed both conserved and species-specific expansions. Spodoptera litura (Lepidoptera: Noctuidae) had unique CTL clusters, while Drosophila melanogaster showed CTL-S gene expansions. Gene duplications contributed to variation, with rapid evolution in Drosophila melanogaster (Diptera: Drosophilidae) and Lepidopteran immune-related molecules (IMLs). Comparative expression analysis in Spodoptera litura (Lepidoptera: Noctuidae) showed that CTLs respond differently to viral and fungal infections, highlighting their immune roles[26]. Comparative genomics has further facilitated the identification of sex chromosomes, as demonstrated in Mythimna separata (Lepidoptera: Noctuidae), where researchers used a comparative analysis of male and female genomic samples[27].
Furthermore, genomic divergence has been increasingly examined through the lens of comparative genomics, incorporating SVs, gene architecture, regulatory sequence evolution, and three-dimensional (3D) chromatin organization. These dimensions have proven critical in shaping genomes' structure and functional plasticity, particularly in insects undergoing rapid ecological adaptation, as they directly affect changes in gene expression (Fig. 4). Structural variants such as insertions, deletions, and chromosomal inversions can disrupt or duplicate gene regions, leading to adaptive traits[28,29] such as insecticide resistance, as was the case in Leptinotarsa decemlineata (Coleoptera: Chrysomelidae)[30]. Similarly, variation in exon–intron structures and alternative splicing patterns modulate isoform diversity, influencing phenotypes across developmental and environmental space[31,32]. Beyond gene structure, changes in noncoding regulatory elements such as enhancer turnover and conserved noncoding elements (CNEs) located outside the protein-coding regions are crucial in fine-tuning gene expression paradigms (Fig. 4)[33,34]. Additionally, comparative genomics also focuses on studying the 3D genome architecture, with the reorganization of topologically associating domains (TADs) affecting the long-range regulatory interactions of the enhancers and promoters in the genome (Fig. 4)[35,36]. These genomic divergence dimensions are critically analyzed through an incorporation of several overlapping methods and tools that involve an integration of SVs that may affect genes' architecture and splicing dynamics that could inform regulatory sequence evolution (Fig. 5a). Together, these mechanisms reveal a nuanced and multilayered comprehension of genomic evolution in insect lineages, complementing classical gene family and phylogenomic analyses.
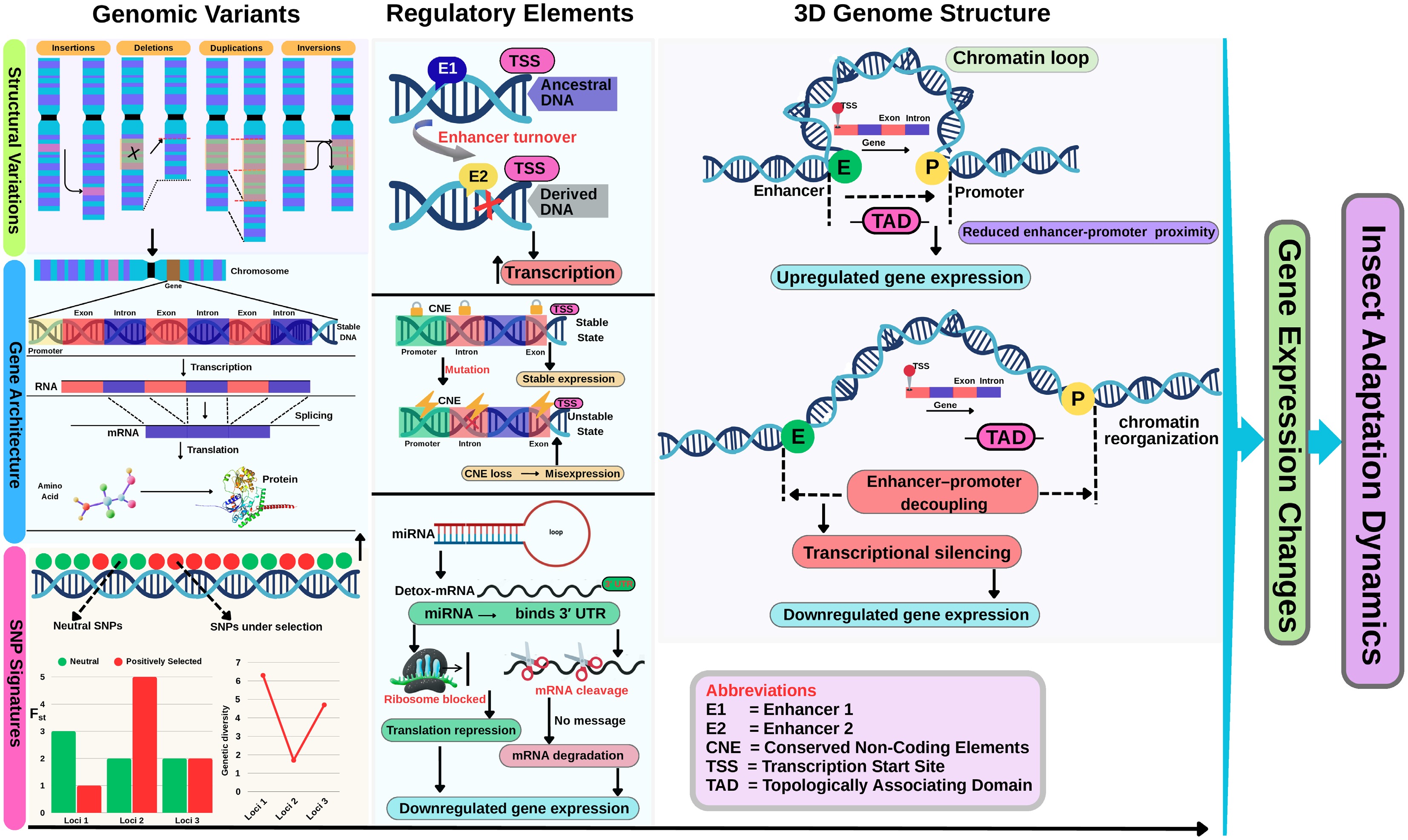
Figure 4.
Important dimensions driving genomic divergence and gene regulation in insects. This figure integrates three major layers. First, genomic variants such as structural variants (insertions, deletions, duplications, inversions) and SNPs can reshape genes' architecture by altering exon–intron structures, splicing patterns, and transcriptional activity. Second, regulatory elements, including enhancers, conserved noncoding elements (CNEs), and microRNAs (miRNAs), modulate gene expression through multiple mechanisms; for instance, enhancer gain/loss affects transcription initiation, CNE mutations may lead to misexpression, and miRNAs regulate gene expression post-transcriptionally by repressing translation or mRNA degradation. Third, the 3D chromatin architecture shows how the genome folds into topologically associating domains (TADs), bringing enhancers and promoters into physical proximity to promote transcription. Disruption of these 3D structures can impair regulatory interactions and lead to altered expression. Collectively, these layers illustrate how genomic, regulatory, and structural mechanisms converge to drive adaptive and evolutionary responses in insect genomes, which directly influence their adaptation dynamics.
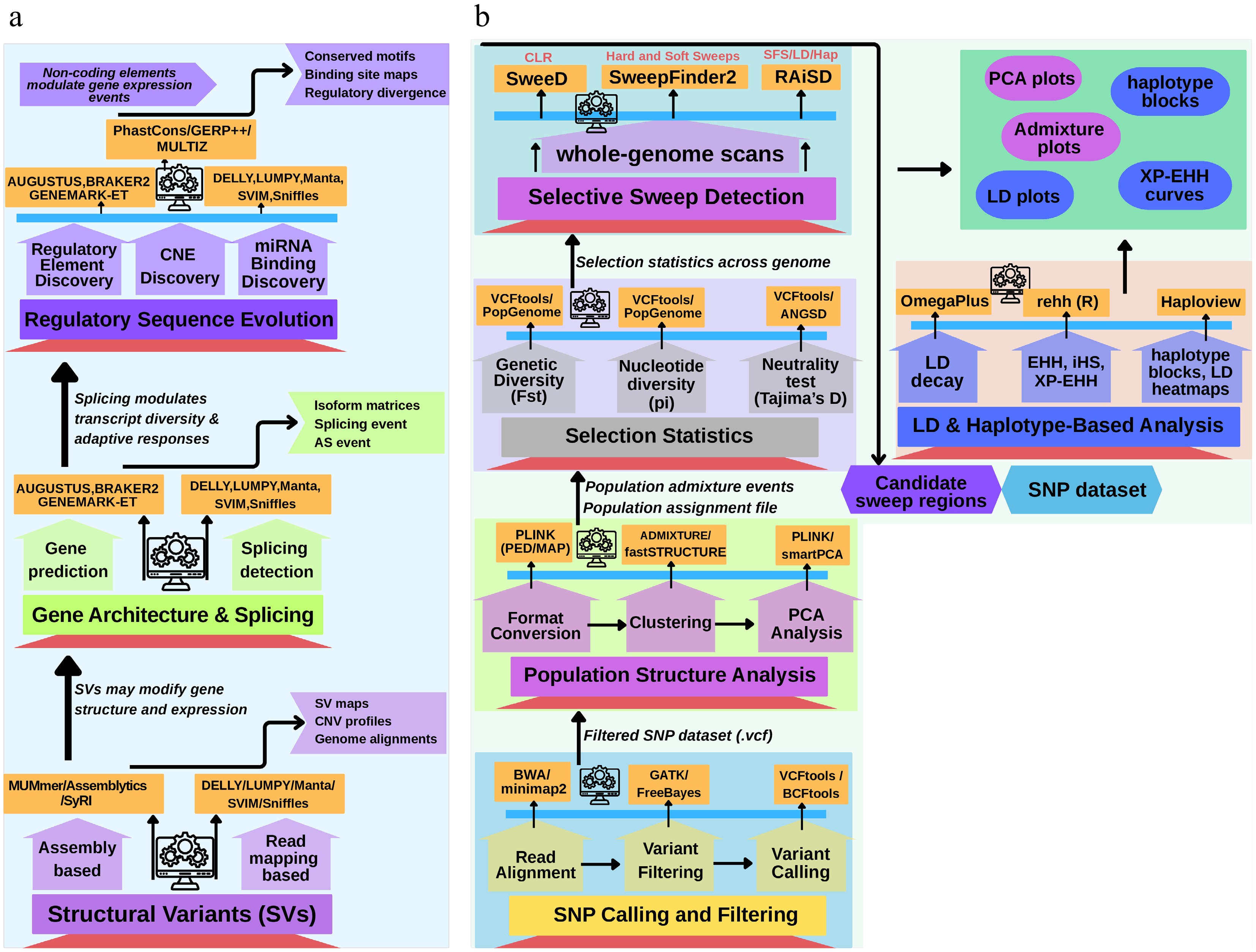
Figure 5.
Multi-scale genomic tools for detecting adaptive evolution in insect pests. This schematic illustrates a tiered framework highlighting the major genomic layers contributing to insect pests' adaptation, along with the corresponding analytical tools and biological outputs at each level. Starting with Panel (a), Tier 1 showcases how structural variants (SVs), detected using tools such as DELLY, Manta, Assemblytics, and MUMmer, can reshape genomes' architecture and influence gene expression, yielding structural variant maps and genome alignments. Tier 2 focuses on alternative splicing and exon–intron variation, where tools like rMATS, SUPPA2, MAJIQ, and IsoSeq uncover isoforms' diversity and splicing event profiles. Tier 3 concerns regulatory sequence evolution, including enhancer turnover, conserved noncoding elements (CNEs), and miRNA binding changes, analyzed with PhastCons, GERP, MEME, FIMO, miRanda, and TargetScan to reveal transcriptional rewiring and binding site alterations. Panel (b) focuses on the detection of adaptive evolution through single nucleotide polymorphisms (SNPs), using pipelines such as GATK, PopGenome, ANGSD, RAiSD, rehh, and ADMIXTURE, alongside various tools to infer selective sweeps, population divergence, and haplotype dynamics. Collectively, this integrative approach demonstrates how layered genomic methodologies uncover the complex mechanisms underlying pest evolution and resistance.
Population genomics: tracking insect evolution in real-time
-
Population genomics provides a powerful toolkit for uncovering intrinsic factors such as migration-driven population fluctuations, genetic diversity, adaptation strategies, delineation of evolutionarily significant units and the identification of cryptic species[4]. It provides a platform that integrates SNP, INDEL, SV, and CNV data into biologically meaningful, sensible, and applicable insights. However, the use of SNP datasets is a very common phenomenon in deciphering the dynamic population structure. Several statistical metrics, including the fixation index (Fst), nucleotide diversity (π), Tajima's D (D), linkage disequilibrium (LD), and selective sweeps are employed in exploring population genomics (Fig. 5b). Incorporating demographic inference methods such as coalescent theory and pairwise sequentially Markovian coalescent (PSMC)[37] enhances historical reconstructions of population bottlenecks and expansions, complementing metrics like Tajima's D. Beyond Fst and selective sweeps, population genomic approaches like genome-wide association studies (GWAS)[38] improve the detection of adaptive loci, while tools like STRUCTURE clarify ancestry and divergence timelines. In this vein, analyses of time-series data from over 6,000 European species revealed increasing diversity among freshwater insects but significant declines in terrestrial species, whereas studies across US research sites found no evidence of a continent-wide insect decline[39]. Investigating the trends in Ne of Ostrinia furnacalis (Lepidoptera: Crambridae) populations, scientists discovered that its population has undergone declines in the past 50 generations, with the largest negative NeS ~16 generations ago[40]. These findings indicate how powerful population genomics can reveal applicable insights. By leveraging the foundation of genomics and computational analyses, scientists can monitor shifts in allele frequencies, detect signatures of selection, and predict potential resistance mechanisms before they become widespread (Fig. 5b)[41]. The field of population genomics has provided insights into the role of chromosomal rearrangements in tailoring pest populations, revealing how these changes contribute to the evolutionary success of pests in agricultural ecosystems[42]. Several mechanisms for chromosomal modifications have been discussed, including nonallelic homologous recombination (NAHR)[43], nonhomologous end-joining (NHEJ)[44], microhomology-mediated break-induced replication (MMBIR)[45], and fork stalling and template switching (FoSTeS)[45].
Numerous studies have highlighted the crucial role of population genomics in insect management strategies. Researchers have managed to analyze the SNP, SV, and INDEL data to decipher the genetic structure of different agricultural insects associated with different geographical regions. For instance, recent studies have highlighted the role of SVs in shaping gene expression and local adaptation in insect pests. Expression quantitative trait locus (eQTL) analysis has provided insights into how SVs influence gene regulation, with quantitative trait locus (QTL) mapping identifying FTZ-F1 as a candidate gene associated with larval development rate in Ostrinia furnacalis[29]. Through the population genomics approach, some studies have shown that the Z chromosome of Spodoptera frugiperda drives strain divergence, with high linkage observed across this chromosome. Furthermore, a region containing a circadian clock gene linked to allochronic reproductive isolation is under strain-specific selection, pinpointing its role in maintaining reproductive barriers and ecological differentiation[46]. Genomic analysis of Halyomorpha halys (Hemiptera: Pentatomidae) revealed spatial structure but high admixture among introduced populations, leading to comparable genomic diversity between native and introduced ranges[47]. These patterns suggest a complex invasion history with multiple bridgehead events, complicating source attribution using reduced-representation genomic data. Quantifying the patterns of genomic change in Helicoverpa zea through genomic scans showed that the genomic architecture of the field-evolved Cry1Ab resistance was polygenic, which was hypothesized to arise from standing genetic variation[48]. Furthermore, researchers have explored the migration dynamics of several agricultural insects (Fig. 6). Among others, the Indochinese Peninsula was indicated as a major source of brown planthoppers in their migration to the temperate regions of China[49]. Population genomics has also enabled the elucidation of the invasive dynamics of insect species; to this end, some researchers have employed model-based diffusion approximations to get insight into lag phases in biological invasions associated with fall armyworm populations[50]. In regards to the migration dynamics of insect species, this fascinating field has enabled the identification of the Casein kinase I gene in fall armyworm, which is reported to regulate circadian rhythms and has 11 selective loci in the regulatory region that has been hypothesized to contribute to the migration behavior[51]. Gene flow dynamics, including adaptive introgression, hybridization, and horizontal gene transfer at the population genomics level, play a crucial role in driving rapid pest adaptation[52]. In a nutshell, population genomics involves inferences regarding random mating, gene flow, effective population sizes, disequilibrium, and relatedness among populations, which are based on patterns of variation at many loci.
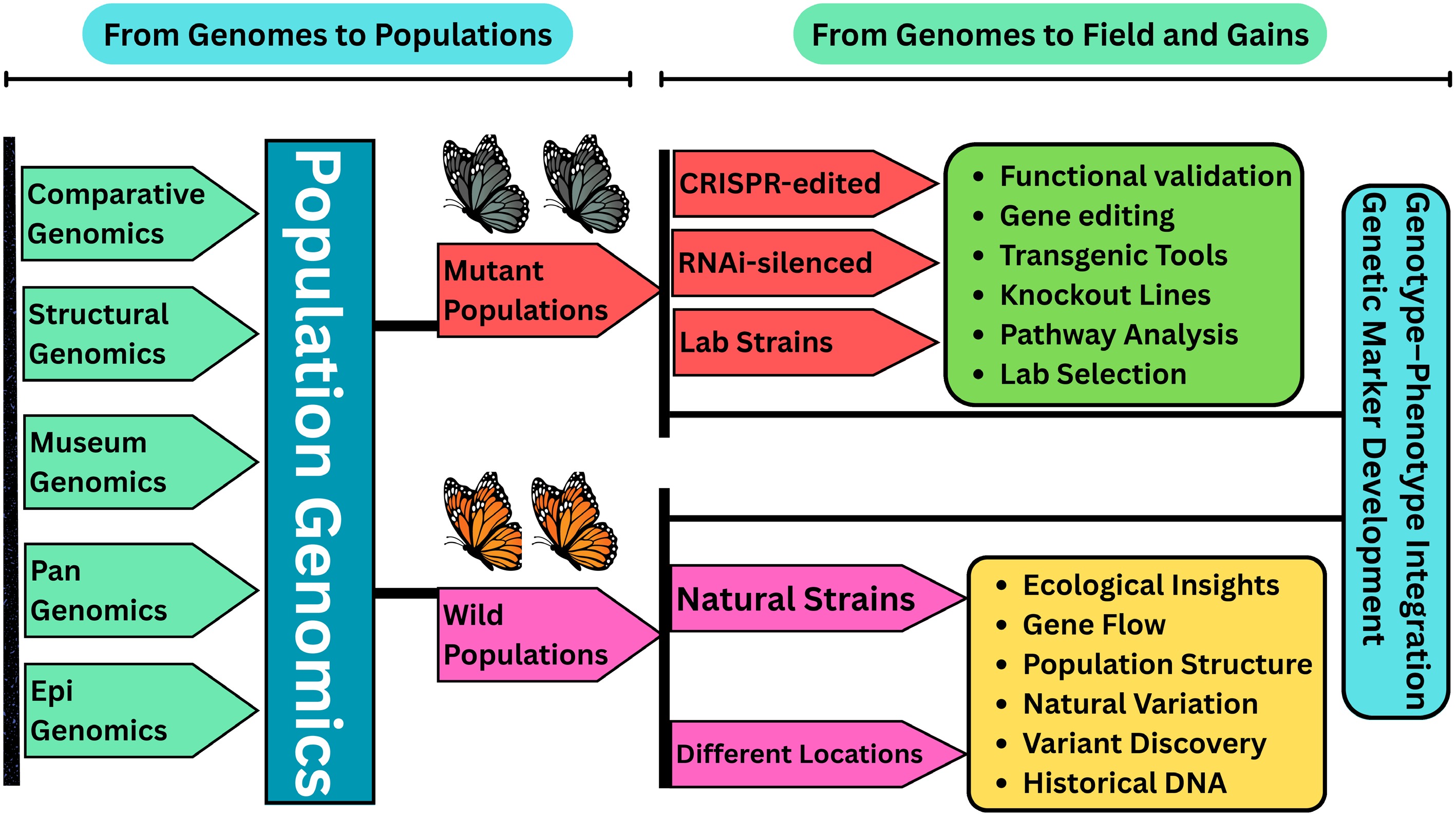
Figure 6.
Conceptual tree depicting the progression from foundational genomics to applied outcomes in agricultural insect research. The base of the tree represents genomic disciplines such as comparative genomics, structural genomics, museum genomics, pangenomics, and epigenomics that form the foundation of insect genomic knowledge. These roots converge into the central trunk of population genomics, which captures genetic variation within and between populations. From this trunk emerge two primary branches representing insights from wild-type and mutant lineages, symbolizing the evolutionary and functional pathways explored through genomic tools. Wild-type and mutant strains are then applied in the real-world 'blossoms', ranging from improved pest control strategies such as RNAi and resistance monitoring to targeted trait development and sustainable agricultural practices. The figure underscores how integrative genomic approaches are translating molecular data into actionable solutions for agricultural pest management.
Pangenomics: understanding intra-species genetic variation
-
In order to capture the intra-species genomic information from one area, pangenomics has been proposed. The pangenome can be defined as a collection of genomic information for a particular group of organisms which are related. Two types of pangenomes have been proposed, the first one being the 'presence–absence variation' (PAV) pangenome, which comprises core and accessory genomes[53]. The second type is known as the representative sequence pangenome, which comprises carefully selected genomic sequences from a particular group of species[53]. Here, core sequences are present in all individuals, while accessory sequences are dispensable across individuals[54]. Pangenomes are further classified as open (continuously gaining new genes with more sequencing, high diversity, or frequent horizontal transfer) or closed (stable gene set, minimal new acquisition)[55]; the former is very common in insects and other species. One of the key aspects in pangenomics is to identify essential genes for an organism's survival which may have direct associations with mutations found within breeding populations[53]. The pangenome is the composite reference of multiple genomes sampled from across a wider range of variability within a population or species. Agricultural insect studies, similar to other organisms, have also greatly benefitted from the fascinating field of pangenomics. The emergence of pangenomes has reduced the bias of single reference genomes, which may fail to adequately represent the genetic diversity within a population or species. Additionally, pangenomics has shown to improve the accuracy of mapping, variant calling, and genotyping in resequencing studies[56].
Pangenomic analysis in silkworms enabled the identification of the BmE2F1 gene, which is involved in cell cycle progression. It harbors one deletion and three insertions in its cis-regulatory region. Further, CRISPR-cas9 mediated knockout of BmE2F1 reduced the number of silk gland cells by 7.68% and silk yield by 22%[57]. Furthermore, analysis of chromosome 11 in the pnd strain of B. mori identified 10 genes with exonic variations, highlighting BmTret1-like (KWMTBOMO06872), a key sugar transporter for diapause. A 747 bp 3′-untranslated region (UTR) deletion was achieved in pnd homozygotes, while heterozygotes retained both copies[57]. Leveraging pangenomics, Huang et al. recently generated a pangenome for fall armyworm. In their findings, they brought in new insights regarding this insect pest. For instance, they identified 17,616 cores genes present all the samples they employed, representing 60.20% of all annotated genes. Additionally, the ABC gene family had the highest proportion of core genes, contributing 81% of the detoxification-related genes in the pangenome. This may demonstrate the critical role of this gene family in regard to detoxification. Furthermore, they identified 19 horizontally acquired genes, with responses across 16 insecticide treatments. Notably, three (SFR02618, SFR05248, and SFR05249) co-expressed with heat shock proteins and were induced by avermectin and cypermethrin[58]. Not only that, another butterfly pangenome study revealed that structural variation shapes chromatin accessibility. Assay for transposase-accessible chromatin using sequencing (ATAC-seq) of caterpillar head tissue identified 31,066 accessibility differences, with 30.4% in lineage-specific SVs and 9.4% linked to TE insertions[59]. A pangenome constructed from three geographic populations revealed structural variation and gene family evolution in Ostrinia furnacalis. Ortholog analysis identified 15,214 gene families, 96.72% of which were shared across genomes. Ka/Ks analysis found 982 positively selected genes, enriched in developmental and signaling functions. Structural variation analysis at the pangenomic level revealed 76,059 biallelic and 52,535 multi-allelic SVs, with over 50% derived from transposable elements, with 78.43% impacting genes[29]. All these breakthroughs in pangenomic research cement the critical significance of this field in insect pest management. Building on these core genomic approaches, temporal genomics extends our insights across evolutionary time by leveraging historical and preserved specimens to track long-term genetic changes and adaptation trajectories.
-
Museum genomics, which leverages genomic research on traditional museum specimens and cryogenic collections, has significantly advanced studies in ecology, evolutionary biology, extinct species, and the effects of human activities on biodiversity[60]. Paleogenomics focuses on using museum specimens to reconstruct the genomes and evolutionary history of extinct species[61]. Historic genetic information can reveal the genetic diversity and structure of a species before the reduction in population numbers through human activities[62]. A museum genomics study uncovered significant shifts in allele frequencies over time in a butterfly species, suggesting strong genetic drift or cycles of extinction and recolonization[63]. Analysis of heterozygosity in samples from 1998–1999 indicated evidence of a historical population bottleneck, aligning with a documented decline in the late 1970s that affected the studied population and several others across the UK[63]. Museum genomics can also aid molecular identification when a species thought to be extinct is rediscovered[64]. By integrating modern sequencing technologies with historical specimens, this field provides unique opportunities to explore genetic changes over time, reconstruct evolutionary histories, and assess the impact of environmental as well as anthropogenic pressures on species diversity. Although this is a fascinating field for studying the evolutionary patterns of insect species, two critical related bottlenecks cannot be overlooked. These bottlenecks include the fragmentation of DNA molecules over time, which may affect the genetic analyses, and restrictions in accessing museum specimens[65]. Therefore, it should be noted that conducting genetic analyses using specimen samples is more complicated than using fresh samples because of specimen-specific issues related to DNA degradation.
A recent analysis of historical Helicoverpa armigera museum specimens spanning over a century across Australia revealed minimal changes in the spatial and temporal population structure. However, researchers detected a moderate decline in genetic diversity between 1960 and 1970. Additionally, temporal genome scans provided strong evidence of selective pressures following the application of insecticide[66]. Museum genomics also uncovered the rapid expansion of Leptinotarsa decemlineata in the 19th century, which reduced genetic diversity and erased the population structure. Resequencing of 78 historical specimens showed that selection predominantly leveraged standing genetic variation, not new mutations. Temporal genomic scans linked insecticide resistance genes to modern field populations in Wisconsin and New York[67]. These findings validate modern selective sweeps, demonstrating museum genomics' power to decode adaptive processes in agricultural pests, including the evolution of resistance. It is worth mentioning that museum genomics, integrated with paleogenomics and field studies, can offer a powerful approach to reconstructing the genomes of extinct agricultural insect pests. While temporal genomics reveals how insect genomes evolve across time, integrative genomics bridges multiple molecular layers, linking nuclear and mitochondrial data, transcriptomics, and epigenetics to resolve complex traits and deepen our understanding of adaptive biology.
-
Structural genomics, a field dedicated to mapping the 3D organization and functional elements of genomes, has emerged as a critical resource for investigating agricultural insect pests. This is especially true as advancements in sequencing technologies enable broader access to precise, high-resolution genomic data. Structural genomics projects aim to solve the experimental structures of all possible protein folds[68]. The resolution of these new structures has driven large-scale data science and infrastructure initiatives, whose integration with breakthroughs in deep learning such as AlphaFold 3[69] have revolutionized computational molecular biology. Three-dimensional structures provide essential information on protein folding and function, and are important elements of drug and insecticide discovery. Through molecular docking, molecular dynamics (MD) simulations, and mutagenesis, structural genomics has enabled researchers to comprehend protein–ligand, protein–protein, and protein–membrane interactions facilitating in the development of precision insecticides. By resolving molecular structures at atomic resolution, researchers gain insights into the mechanisms underlying pests' adaptations, insecticide resistance, and host–pathogen interactions[70]. For instance, structural analyses of cytochromes P450s (CYPs) have identified a conserved active site motif for substrate carboxylate binding in peroxygenases. Additionally, mechanistic and transient kinetic studies have confirmed the formation of reactive iron-oxo species, which drive the hydroxylation and decarboxylation of fatty acids[71].
Leveraging structural genomics, researchers have discovered the key residues of Sip1Aa protein associated with insecticidal activity against Colaphellus bowringi, an important agricultural insect. This protein is produced by Bacillus thuringiensis (Bacillales: Bacillaceae) and was reported to be highly associated with insecticidal activities[72]. Structural genomics has also led to the discovery of ionic channel domains, such as the ryanodine receptors (RyRs) NTD, RyR Repeat34, and RyR SPRY2 in Plutella xylostella (Lepidoptera: Plutellidae)[73]; TRPV-Nan and TRPV-Iav in Nilaparvata lugens (Hemiptera: Delphacidae)[74]; and TRPN-NompC and RDL-GABAR in Drosophila melanogaster[75], which serve as insecticide targets. Moreover, a homology model of the diamondback moth's RyRs revealed that the key residues responsible for ryanodine binding are fully conserved in insect RyRs compared with their mammalian counterparts[76]. Structural genomics identified a plant-derived protein variant, resolved at 1.98 Å, that mimics the Cry toxins of Bacillus thuringiensis (Bt) but lacks their essential C-terminal domain. Notably, this truncated fern protein effectively targets fall armyworm strains that are resistant to Bt Cry1Fa and Cry2A toxins[77]. In another study, through docking simulations, the ryanodine receptor protein (RyR) of fall armyworm demonstrated comparable binding affinity between limonoid biopesticides and chlorantraniliprole, a commercial insecticide[78]. Furthermore, characterizing the PBAN-receptor (PBAN-R or PR) active binding domains using chimeric G protein-coupled receptors (GPCRs) and proposed that extracellular loop 3 is critical for ligand selection in Helicoverpa zea[79]. Another analysis of the Bacillus thuringiensis Vip3Aa toxin, combining homology modeling and molecular docking, pinpointed critical amino acid residues within its C-terminal domain. Functional validation via site-directed mutagenesis demonstrated that the Y619A mutation boosted insecticidal potency against Helicoverpa armigera, a major crop pest. Conversely, W552A and E627A substitutions severely impaired toxicity in both Helicoverpa armigera and Spodoptera exigua[80]. These findings also cement the importance of structural genomics in the development of targeted pesticides.
Integration of nuclear and mitochondrial genomics
-
Mitochondrial genomes are the most extensively studied genomic systems in insects, outnumbering insect nuclear genomes. In the past few decades, mitochondrial genomic data have sharply increased in various genomic databases. As is the case with nuclear genomes, mitochondrial genomes, as the cellular powerhouse[81], have also benefited from NGS technologies. However, despite these advancements, mitogenome annotation software has lagged behind improvements in assembly methods, leading to gaps in functional analysis. In agricultural pests, mitochondrial genomes have proven useful in resolving cryptic species complexes, tracking pest invasions, and understanding pesticide resistance mechanisms[82]. Mitochondrial markers, particularly cytochrome oxidase I (COI), serve as cornerstone tools for DNA barcoding, enabling precise species identification, phylogeographic reconstruction, and detection of cryptic pest species. These markers are integral to global pest surveillance programs, facilitating rapid tracking of invasive species and informing quarantine protocols to mitigate biosecurity risks[83].
Despite their central role in energy metabolism, mitochondria may influence evolutionary trajectories in agricultural insect pests through nuclear mitochondrial DNA sequences (NUMTs), which are fragments of mitochondrial DNA (mtDNA) that have been integrated into the nuclear genome; in other words, NUMTs are mitochondrial DNA segments transferred to the nuclear genome that are found in various insect groups[84]. These NUMTs have been suggest to reflect key evolutionary processes such as gene flow and mitochondrial lineage diversification in study of 32 Chrysomelidae beetle species[84]. NUMTs are pervasive in insect genomes, including high-impact pests like Bemisia tabaci (Hemiptera: Aleyrodidae) (silverleaf fly) and Helicoverpa armigera, where they reported to contribute to mito-nuclear discordance and complicate phylogenetic inference[85]. NUMTs also pose challenges for DNA barcoding, a cornerstone of pest surveillance. In Spodoptera frugiperda, NUMTs have been linked to global invasions[86]. Critically, NUMTs may interact with transposable elements, destabilizing genomes or creating novel regulatory elements that shape traits like pesticide resistance[87]. As agricultural systems increasingly rely on genomic tools for pest management, integrating mitochondrial and nuclear datasets will be prime to avoid analytical debacles and uncover hidden drivers of pest diversification. This synergy is exemplified by CRISPR-based strategies targeting mitochondrial genes such as COI to induce sterile insect technique (SIT) programs, where NUMT interference must be minimized for precision[88]. By resolving NUMT dynamics, researchers can refine insect pest phylogenetics, monitoring resistance, and biocontrol designs, ultimately fostering sustainable agriculture.
Epigenomics: beyond the DNA sequence
-
Epigenetic information guides the development of distinct cellular and organismal phenotypes from a common genome[89]. Epigenetics particularly considers heritable changes to gene regulation that occur in response to intercellular and extracellular environmental stressors. Insects adapt to environmental stressors through epigenetic regulation, without altering their DNA sequence. Epigenetic information acts as a molecular intermediary, converting environmental signals into changes in gene expression. This enables the cell, and ultimately the organism, to produce a phenotype that is better adapted to its environment[90]. DNA methylation, the modification of histone proteins, and the action of noncoding RNAs are the key mechanisms involved in epigenetics. Genomic patterns of DNA methylation in insects are preferentially directed towards genes that are widely expressed across different tissues and organismal morphs. Although DNA methylation in insects is relatively low, constituting 0%–3% of cytosine followed by guanine (CpG)[91], its involvement in the phenotypic plasticity of insects cannot be ruled out. In regards to histone modifications, it has been linked to transcriptional variations associated with behavioral transitions and stage-specific variations associated with head development[92]. Histone modifications might also be linked to effective response to developmental cues and environmental stressors[93]. Noncoding RNAs have been reported to play a role in regulating cellular processes[94]. Four types of noncoding RNAs (ncRNAs) have been identified as potentially influencing epigenetic regulation: PIWI-interacting RNAs (piRNAs), microRNAs (miRNAs), small interfering RNAs (siRNAs), and long noncoding RNAs (lncRNAs)[92]. The evidence documenting the involvement of noncoding RNA in epigenetic regulations in insects has been well captured[95]; on the other hand, general epigenetics in insects has been thoroughly examined by Glastad et al.[92]. In addition, recent studies have suggest that rice crops respond to fall armyworm infestations through H3K9ac epigenetic regulation in the jasmonic acid (JA) signaling and phenolamide biosynthesis pathways[96]. These findings again provide room for more studies related to such mechanisms. Building on the synergy of integrative genomics, translational genomics emerges as the conduit through which genomic insights are transformed into tangible agricultural innovations.
DNA methylation, as a key mechanism of epigenetics, has been reported to vary across insect species. For example, insects like aphids and planthoppers display significant methylation, whereas ants, bees, wasps, and sawflies exhibit only minimal levels[97]. Some researchers identified a gene duplication in maintenance DNA methyltransferase 1 (Dnmt1) in some Hymenoptera, with paralogs undergoing divergent, non-neutral evolution[98]. The key function of Dnmt1 was also shown in tomato leaf miner, Tuta absoluta (Lepidoptera: Gelechiidae), where scientists demonstrated that this gene is key in response to changing environments[99]. It is important to note that DNA methylation is thought to occur through the action of the DNA methyltransferase (Dnmt) gene family[90]. Recent studies have shown the role of DNA methylation in host–pathogen interactions. For example, exposure to pathogens induces changes in the methylation patterns of Drosophila melanogaster[100], suggesting that epigenetics might be associated with innate immunity. Additionally, a study identified N6-methyladenine (m6A), a DNA methylation modification occurring at the sixth position of adenine nucleotides, as a critical epigenetic regulator necessary for survival in Bombyx mori[101]. Yoon et al. discovered an important gene duplication in Dnmt3, which was demonstrated to play a crucial role in the early development of a pea aphid[102]. It should also mentioned that insecticides influence DNA methylation in a highly random manner, with varying exposure levels altering distinct methylation sites, as demonstrate in a Colorado potato beetle study[103]. Moreover, stress can impact small noncoding RNA types and post-translational modifications of histone, ultimately affecting DNA's accessibility for transcription[103]. Similarly, changes in histone modifications may contribute to insecticide resistance by increasing chromatin'saccessibility and activating genes involved in enzymatic detoxification[90].
-
In the context of insect studies, translational genomics applies gene editing and RNAi silencing techniques based on genomic datasets to improve insect pest management. Translational genomics in agriculture applies genomic breakthroughs to real-world agricultural challenges, facilitating the development of pest-resistant crops, targeted pest control strategies, and sustainable management practices (Fig. 6)[104]. Advances in genome sequencing, functional genomics, and gene editing technologies have enabled researchers to identify key genetic determinants of host adaptation, insecticide resistance, and metabolic detoxification pathways in agricultural pests. For instance, RNAi-based pest control strategies leverage species-specific gene silencing to suppress essential genes[105]. This has enabled the development of dsRNA spray targets that reduce some insect populations[106,107]. Studies have unraveled the mechanisms by which silencing of the function of a particular gene occurs in the red flour beetle, providing critical insights into gene regulation in pest insects[108]. CRISPR-based functional validation approaches have been applied to disrupt the mating potential in Spodoptera frugiperda by knocking out the major sex pheromone gene[109]. Additionally, gene editing of Sfabd-B in Spodoptera frugiperda using CRISPR-Cas9 uncovered its regulatory role in body segmentation and reproductive organ development, providing a foundation for engineered sterility-based pest management[110]. In another study, CRISPR/Cas9 gene editing of HzABCA2 in Helicoverpa zea confirmed its role in Bt Cry2Ab resistance, generating resistant strains with > 200-fold increased tolerance. These findings established HzABCA2 as a key molecular marker for monitoring resistance to Cry2Ab resistance in field populations[111]; as such, molecular diagnostic kits have been developed to detect ABCC2 mutations and forecast resistance outbreaks. On the other hand, GWASs have refined our understanding of how genetic variation underlies pests' resistance mechanisms, aiding the design of novel pest management strategies[112].
Beyond pest control, translational genomics also enhances integrated pest management (IPM) by informing the breeding of pest-resistant crops and developing predictive models for pest outbreaks. For example, genomic surveillance of Helicoverpa armigera and Helicoverpa zea hybrids in Brazil identified 8,511 ancestry-informative markers, confirming hybridization. These results emphasize genomic datasets' power to detect introgression, with critical implications for managing pesticide resistance and host adaptation in these pests[113]. Furthermore, genomic insights into the gut microbiota composition in agricultural insect pests have opened new avenues for microbial-based pest control interventions[114]. Emerging tools like gene drives and microbiome engineering further exemplify the integration of genomics into ethical, species-specific pest suppression[115]. Additionally, comprehensive analyses of odorant receptor genes have enabled the development of synthetic lures mimicking host volatiles that improve pheromone traps' efficiency. As sequencing technologies become more accessible, translational genomics will continue to bridge the gap between laboratory research and practical applications. It should be pointed out the field of translational genomics has not been adequately explored in the agricultural systems compared with medical research. Nevertheless, the integration of Bacillus thuringiensis (Bt) toxins in insect pest management has the been the most applicable approach associated with translational genomics in agricultural systems. Xiao and Wu have extensively described the scope of Bt toxins in insect pest management[1].
-
Given the abovementioned points, we propose 13 questions that we think and believe are of critical importance as far as genomic research of agricultural insects is concerned.
i. Optimization of gene regulation: Can intron-mediated enhancement (IME) be leveraged to fine-tune gene expression in transgenic crops or RNAi-based pest control systems, improving both efficacy and target specificity?
ii. Noncoding regulatory networks: What functions do small RNAs, long noncoding RNAs, and other noncoding genomic elements play in regulating key adaptive traits such as diapause and host–plant specialization in agricultural insects?
iii. Genetic innovation and adaptation: To what extent do horizontal gene transfer (HGT) and gene duplication events contribute to the rapid acquisition of adaptive traits, including insecticide resistance and detoxification across insect pest populations?
iv. Microbiome–pest dynamics: How do symbiotic microbial communities shape pests' virulence, nutrient acquisition, and resistance evolution, and can these interactions be disrupted through microbiome engineering?
v. Cross-resistance risk assessment: How can genomic datasets – including resistance allele frequencies, epigenetic modifications, and structural variants – be integrated to predict and mitigate cross-resistance risks across different insecticide classes?
vi. Artificial intelligence-driven pest surveillance: Can machine learning models integrate genomic, transcriptomic, and field data to forecast emerging resistance mechanisms or invasive pest threats before they destabilize agroecosystems?
vii. Biocontrol genomics: Is this another emerging genomic field? What genomic traits such as host-seeking behavior and detoxification capacity underpin the efficacy of natural predators or parasitoids, and can these traits be enhanced through selective breeding or gene editing?
viii. Pangenomics for targeted insecticide development: Can pangenomics capturing global pest diversity identify conserved molecular targets for next-generation, species-wide insecticides?
ix. Historical adaptation insights: How can museum genomics and paleogenomics clarify historical adaptation patterns (e.g., before and after the insecticide era) to improve predictive models of pest evolution?
x. Climate-driven invasions: Can genomic markers, including adaptive SNPs and stress-response genes, help predict the range expansion, host shifts, and invasive potential of agricultural pests under changing climate conditions?
xi. Democratization of genomic data: What technical, financial, and educational barriers hinder the integration of genomic tools into agricultural extension systems, and how can these be addressed to benefit smallholder farmers and developing regions?
xii. Global data standardization: How can international collaborations standardize the collection, sharing, and utilization of genomic data to improve its applicability across diverse agricultural systems?
xiii. Gene editing ethics: What ecological and ethical safeguards should be established to responsibly deploy CRISPR-edited pests or gene-drive technologies in open ecosystems, considering risks such as off-target effects, genetic introgression, and unintended ecological disruptions?
-
In summary, genomic technologies have metamorphosed the study of agricultural insects, providing profound insights into their evolutionary dynamics, adaptive resilience, and sustainable management. WGS provides the backbone for high-quality genome assemblies, enabling precise gene annotation and functional studies. Comparative and population genomics decrypt species' divergence and track adaptive evolution over time, elucidating the pathways behind pesticide resistance and host–plant specialization. Emergent tools like pangenomics capture intra-species diversity, while epigenomics uncovers the regulatory landscapes governing traits such as diapause and virulence. Structural genomics identifies molecular targets for next-generation insecticides, whereas museum genomics reconstructs historical adaptation patterns, contextualizing modern pest invasions. Translational genomics bridges these discoveries to the field, driving innovations like RNAi-based biopesticides and CRISPR-edited crops that are resistant to key pests. However, critical gaps persist, from the dynamics of horizontal gene transfer to the ethical risks of gene drives, demanding interdisciplinary collaboration to fully leverage genomic tools. Addressing these challenges will require the democratization of genomic resources, refining predictive models for climate-driven invasions, and ensuring equitable access to technologies, ultimately securing global food systems against escalating pest threats.
This work was financially supported by the National Natural Science Foundation of China (32302352), the Shenzhen Science and Technology Program (Grant No. RCBS20231211090649086), the STI 2030–Major Projects (Grant No. 2022ZD04021), and the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (Grant No. CAASZDRW202412).
-
The authors confirm their contributions to the paper as follows: conceptualization and project lead: Xiao Y; manuscript drafting and writing: Yesaya A; manuscript drafting and revision: Zhang L, Peng Y. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are publicly available in the NCBI repository.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yesaya A, Zhang L, Peng Y, Xiao Y. 2025. Surges and insights from the genomics of insect pests. Genomics Communications 2: e017 doi: 10.48130/gcomm-0025-0017 

Surges and insights from the genomics of insect pests
- Received: 22 May 2025
- Revised: 15 July 2025
- Accepted: 18 July 2025
- Published online: 20 August 2025
Abstract: Genomics has revolutionized our understanding of agricultural insects by revealing the genetic basis of their adaptation, pesticide resistance, and ecological success, while also driving the development of sustainable pest management innovations. Breakthroughs across sequencing technologies, comparative genomics, and population genomics have mapped genomes, identified resistance-linked genes such as P450s, monitored invasive lineages, and tracked alleles' spread in real time. While pangenomics captures the intra-species diversity critical for resilience, epigenomics uncovers heritable noncoding mechanisms shaping phenotypic plasticity under environmental stress. Structural genomics elucidates the protein targets for novel insecticides, and museum genomics reconstructs historical adaptations by employing archival DNA. Mitochondrial genomics allows species identification and dispersal tracking. Translational genomics bridges discoveries to field applications, such as CRISPR-edited sterile insects or RNA interference (RNAi)-based methods. Nevertheless, key gaps persist: the role of noncoding regions, horizontal gene transfer in trait acquisition, and intron–exon dynamics in adaptive evolution remain underexplored. Integrating multi-omics with artificial intelligence (AI)-driven predictive approaches could forecast climate-induced pest shifts and resistance trajectories. Ethical frameworks must parallel technical advances to address gene-driven risks and equitable access. By merging cutting-edge genomics with cross-disciplinary collaboration, this field holds unparalleled potential to combat pesticide resistance, pre-empt emerging threats, and redefine pest management in an era of agricultural uncertainty.