









-
Cis-acting elements, also known as cis-regulatory elements (CREs), are non-coding DNA regions that regulate the expression of nearby genes[1]. They play a crucial role in controlling gene expression related to plant growth, development, yield, and stress resistance[2−4]. In recent years, significant progress has been made in this field. For instance, the Basenji2-long deep learning model has been employed to predict and identify numerous candidate CREs across the maize genome, with experimental validation confirming the enhancer activity of some[5]. Additionally, a short sequence-expression prediction model has been developed to precisely identify promoter CREs, along with the innovative introduction of the 'editing plasticity' concept to quantify the regulatory potential of gene promoter editing on expression[5].
In terms of identification methods, the sea-ATI technology has been established to systematically characterize all known and unknown transcription factor-binding CREs with physiological and biochemical functions, further enriching the understanding of plant cis-regulatory grammar[6]. Despite these advancements, challenges remain, such as accurately distinguishing CRE activity states and experimentally validating their regulatory functions[7,8]. Future research will continue to focus on the precise identification and in-depth functional analysis of CREs.
Alfalfa (Medicago sativa L.) is a widely cultivated perennial legume belonging to the Medicago genus. Due to its high protein content and strong adaptability, alfalfa holds significant importance in animal husbandry[9]. To enhance biomass yield, improve winter hardiness, and boost resistance to foliar diseases—three critical agronomic challenges in alfalfa—precise modulation of gene expression is essential[10]. These traits are regulated by intricate transcriptional networks that govern photosynthetic capacity, cold-responsive carbohydrate metabolism, and defense gene induction, respectively. Identifying robust enhancer elements offers cis-regulatory tools to elevate the expression of rate-limiting genes, avoiding the positional effects and instability often linked with random transgene insertion, thus accelerating molecular breeding. Transcriptional enhancers interact with transcription factors to enhance the efficiency of transcriptional initiation, playing a central regulatory role in plant growth, development, and stress responses. Systematic screening of transcriptional enhancer motifs in the alfalfa genome will facilitate precise gene expression regulation, thereby enabling targeted cultivar improvement.
This study systematically identified enhancer-type CREs in the promoter regions of highly expressed genes (HEGs) in alfalfa using transcriptome data mining and bioinformatics analysis. Their functional activity was validated through a tobacco transient transformation system. The findings provide important theoretical support and functional element resources for elucidating gene expression regulation mechanisms and molecular breeding in alfalfa. Specifically, 20 constitutively highly expressed genes were first screened from transcriptome databases, and 11 potential CREs regulating high-level gene expression were identified through bioinformatics analysis. Based on motif length, promoter distribution characteristics, and copy number, four core CREs were selected for functional validation. By constructing LUC reporter vectors containing target CREs and employing the tobacco transient transformation system with luciferase activity assays, functional elements with significant transcriptional enhancement activity were successfully identified.
-
Nicotiana benthamiana seeds were sown in a 1:1 mixture of nutrient soil and vermiculite. Plants were grown under 16 h light/8 h dark at 22 °C for two weeks before agroinfiltration.
Identification of candidate enhancer CREs
-
The reference genome of alfalfa (Medicago sativa cv. 'Zhongmu No. 1') was obtained from the study by Shen et al.[11]. To identify genes with stable and high expression levels, the top 20 constitutively highly expressed genes (HEGs) were selected based on their Fragments Per Kilobase of transcript per Million mapped fragments (FPKM) values and low coefficient of variation (CV < 0.2) across three major alfalfa tissues (leaf, stem, and root of 15-day-old seedling). The RNA-seq dataset used for this selection was obtained from Zhang et al. The top 1, 5, 10, 15, 20, and 25 genes ranked by mean FPKM were compared to determine the optimal number of genes for the analysis (Supplementary Fig. S1). The top 20 genes were selected as they captured the majority of HEGs, ensuring a robust and unbiased dataset for subsequent cis-regulatory element (CRE) identification.
Vector construction
-
The In-Fusion primers were designed based on the candidate CRE sequences (Table 1). Using In-Fusion technology, the recombinant promoters containing CREs were inserted upstream of the LUC gene in the pGreenII0800-Luc vector to construct recombinant plasmids (e.g., MsEF1-αPro::LUC, ΔM11-MsEF1-αPro::LUC, etc.). After verifying the correctness of the recombinant plasmids by sequencing, they were transformed into Agrobacterium GV3101 via the liquid nitrogen freeze-thaw method. Positive clones were then selected on YEP solid medium supplemented with kanamycin (50 mg/mL) and rifampicin (50 mg/mL). Finally, positive bacterial strains were identified through PCR.
Table 1. Primers used for vector construction.
Primer pair name Primers sequences (5’-3’) EFM1-LUCln-F TTGATATCGAATTCCTGCAGGACCGCGCCCGGCGTGGTGCGACCTGGGCCCGGGGCTCCCCATTAGGATATTTTCGGAA EF-LUCln-R GCTCTAGAACTAGTGGATCCGTTGTCTTAAAACTGCGAT EFM2-LUCln-F TTGATATCGAATTCCTGCAGCAGGTCCCGCCCCCCCCCGCCCCCCCACCCACCCCCCTCCCCCCCCCCATTAGGATATTTTCGGAA EF-LUCln-R GCTCTAGAACTAGTGGATCCGTTGTCTTAAAACTGCGAT EFM7-LUCln-F TTGATATCGAATTCCTGCAGCCACACCGCGCTACCTGGCCTCTGGCTCCCATTAGGATATTTTCGG EF-LUCln-R GCTCTAGAACTAGTGGATCCGTTGTCTTAAAACTGCGAT EFΔM11-LUCln-F TTGATATCGAATTCCTGCAGCATTAGGATATTTTCGGAA EF-LUCln-R GCTCTAGAACTAGTGGATCCGTTGTCTTAAAACTGCGAT LEA2M1-LUCln-F TTGATATCGAATTCCTGCAGGACCGCGCCCGGCGTGGTGCGACCTGGGCCCGGGGCTCCCGAGTTTAATTGCATATTCA LEA2-LUCln-R GCTCTAGAACTAGTGGATCCGATTTATAGTGAATAACAC LEA2M2-LUCln-F TTGATATCGAATTCCTGCAGCAGGTCCCGCCCCCCCCCGCCCCCCCACCCACCCCCCTCCCCCCCCCGAGTTTAATTGCATATTCA LEA2-LUCln-R GCTCTAGAACTAGTGGATCCGATTTATAGTGAATAACAC LEA2M7-LUCln-F TTGATATCGAATTCCTGCAGCCACACCGCGCTACCTGGCCTCTGGCTCCGAGTTTAATTGCATATTCA LEA2-LUCln-R GCTCTAGAACTAGTGGATCCGATTTATAGTGAATAACAC LEA2M11-LUCln-F1 TTGATATCGAATTCCTGCAGCAATACCTTAACCTAACCTTCTTTATTTCATCCTTTACCTAA LEA2M11-LUCln-F2 TATTTCATCCTTTACCTAAAAAACTCACAAAAGCATATTCCATG LEA2M11-LUCln-F3 ACAAAAGCATATTCCATGCGCCACCACTCCCCAACGTTCAGAGTTTAATTGCATATTCA LEA2-LUCln-R GCTCTAGAACTAGTGGATCCGATTTATAGTGAATAACAC Agrobacterium-mediated transient expression
-
The GV3101 strain harboring recombinant plasmids (OD600 = 0.6) was infiltrated into N. benthamiana leaves along with the P19 helper strain. After maintaining the plants in darkness for 48 h, LUC enzyme activity and fluorescence intensity were measured in the infiltrated leaf regions.
Luciferase assay
-
Leaf sections from infected areas were treated with 10 μL of a 10% luciferase imaging solution. After two minutes, fluorescence images were captured using a LUC imaging system (three biological replicates per treatment). The activities of firefly luciferase (FLUC) and Renilla luciferase (RLUC) in the infected zones were measured using a dual-luciferase assay kit. The relative FLUC/RLUC ratio was calculated (eight biological replicates per treatment) to quantify the regulatory effects of CREs.
Statistical analysis
-
The raw data underwent standardization in Excel, followed by significance analysis using SAS software (p < 0.05 indicates significance, p < 0.01 indicates high significance). Figures were generated with Graphpad Prism 9.5.
-
Using the lab's RNA-seq database[12], sequencing reads were aligned to the reference genome via STAR/HISAT2, and transcript quantification was performed using StringTie. From this analysis, 20 constitutively highly expressed genes (HEGs) across leaf, stem, and root were identified, selected based on the highest mean FPKM and low expression variability (CV < 0.2) across all nine RNA-seq libraries. The promoter regions of these HEGs were then extracted, spanning 2,000 bp upstream to 500 bp downstream of the transcription start site (TSS). As a control, promoter sequences from an equal number of non-highly expressed genes (non-HEGs) were collected. To identify potential cis-regulatory elements (CREs), these promoter sequences were analyzed using the PlantCARE database and the FIMO tool from the MEME Suite. Only known CREs with a stringent significance threshold (p < 1e-4) were retained. Through Fisher's exact test (adjusted p < 0.05), 10 candidate enhancer-like motifs significantly enriched in HEG promoters were identified. Figure 1 illustrates the distribution of these ten candidate motifs across the promoters of the 20 HEGs. These elements were designated as M1 to M10 (Fig. 1, Table 2).

Figure 1.
Sequence logos and promoter distributions of ten candidate motifs in 20 high-expression genes (HEGs). (a) Sequence logos for the ten candidate motifs. (b) Color key for motif identities. (c) Promoter-localized motif occurrences across the 20 HEGs.
Table 2. The 11 candidate enhancer motifs founded in alfalfa highest-expressed genes’ pormoters.
Motif ID Alt ID Width Frequency Best possible match 1 GAYSRCNCNYSSCGWDGTKYGNCCTGRRCMCGKGGYTSMM MEME-1 40 0.35 GACCGCGCCCGGCGTGGTGCGACCTGGGCCCGGGGCTCCC 2 CARSYCMHDMHNHMMHYYDYHCYYNNWCMMWCMCYYYTCYCYCCCCY MEME-2 47 0.90 CAGGTCCCGCCCCCCCCCGCCCCCCCACCCACCCCCCTCCCCCCCCC 3 AARAAAAAMAARAAA MEME-3 15 3.25 AAGAAAAACAAGAAA 4 CCCCMSAGASRSTS MEME-4 14 0.45 CCCCCGAGACGCTC 5 SVTCSBTGGCAMACGMAHGAHGGGYAVABGCATYKCNTCTHGMC MEME-5 44 0.35 GCTCGGTGGCACACGCACGAAGGGCACACGCATCGCATCTAGCC 6 GAAGMGAGAAA MEME-6 11 1.60 GAAGCGAGAAA 7 CCMMWVCRCGYTACSTKKNCTVTGKNTSC MEME-7 29 0.55 CCACACCGCGCTACCTGGCCTCTGGCTCC 8 KRGHACMVCTNGCSCNKCCSYVACARVCADTBCTWTNCC MEME-8 39 0.40 GGGCACCGCTAGCCCAGCCGCCACAGCCAATCCTTTACC 9 TCABAMRKAYTYCMYCYRTCCCWTA MEME-9 25 0.70 TCACACGTACTCCCTCCGTCCCTTA 10 TTWTKAWKTTTTTTTTTTSAA MEME-10 21 3.15 TTTTGAAGTTTTTTTTTTCAA 11 MEME-11 89 no CAATACCTTAACCTAACCTTCTTTATTTCATCCTTTACCTAAAAAACTCACAAAAGCATATTCCATGCGCCACCACTCCCCAACGTTCA ALT ID, alternative identifier; Frequency, occurrence frequency (no. per promoter).Frequency was calculated as the total number of motif detections across the 20 highest-expressed gene promoters divided by 20. In addition to the 10 motifs (M1–M10), core promoter-proximal elements within the promoters of highly expressed genes were also examined. An 89 bp fragment was identified upstream of the constitutively highly expressed gene MsEF1-α[13], spanning from –1,128 to –1,216 bp. This region contains a typical CAAT-box (CAAT, located at –1,173 bp). The fragment is situated approximately 600 bp upstream of the TATA-box, consistent with the 'long-range regulation' characteristic of enhancers[14] (Kyrchanova & Georgiev). This segment exhibits enhancer-like cis-regulatory element (CRE) characteristics and has been designated as M11 for further functional characterization (Table 2).
Vector construction and identification
-
Analysis of the distribution and abundance of motifs M1–M10 in the promoters of highly expressed genes revealed that M1, M2, and M7 were the most prevalent in the promoters of the top ten highest-expressed genes (Fig. 1c). Based on these findings, M1, M2, M7, and M11 were selected for further functional characterization. To investigate the roles of these motifs, primers were designed to insert M1, M2, M7, and M11 individually into the promoter regions of MsEF1-α or MsLEA2, approximately 600 bp upstream of the TATA box. These resulting constructs were then integrated into the pGreenII0800-Luc vector via homologous recombination, generating the corresponding recombinant plasmids.
The recombinant plasmids constructed based on the MsEF1-α promoter were designated as follows: MsEF1-αPro::LUC (containing M11), ΔM11-MsEF1-αPro::LUC(with M11 deleted), M1-ΔM11-MsEF1-αPro::LUC (containing M1), M2-ΔM11- MsEF1-αPro::LUC (containing M2), M7-ΔM11- MsEF1-αPro::LUC (containing M7).
The recombinant plasmids constructed based on the MsLEA2 promoter were designated as follows: MsLEA2Pro::LUC (containing the native MsLEA2 promoter), M1-MsLEA2Pro::LUC (with M1 variant), M2-MsLEA2Pro::LUC (with M2 variant), M7-MsLEA2Pro::LUC (with M7 variant), M11-MsLEA2Pro::LUC (with M11 variant).
All recombinant plasmids were verified by sequencing to confirm their correct construction (Fig. 2).
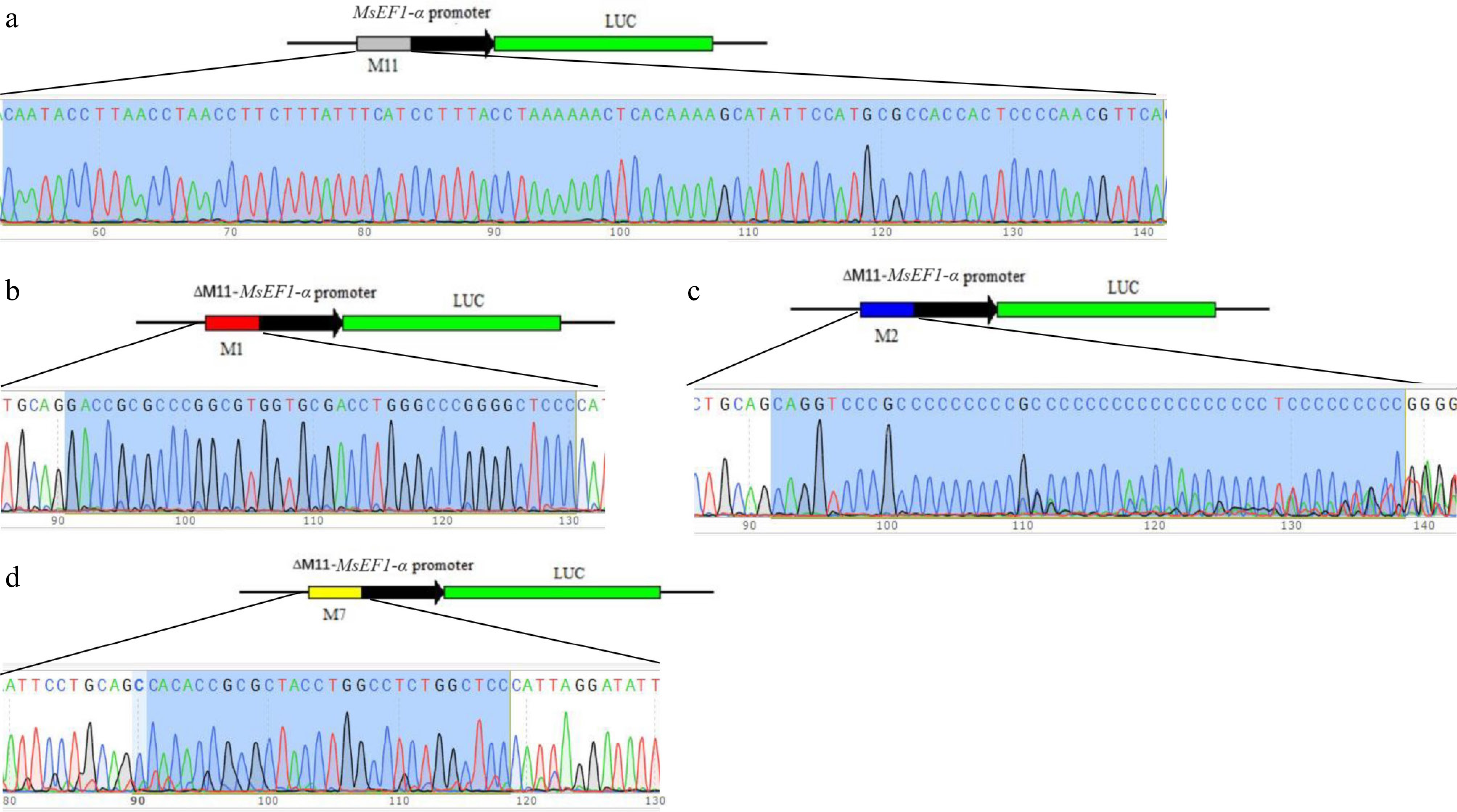
Figure 2.
Sanger sequencing validation of candidate motifs in the recombinant MsEF1-α promoter-LUC plasmid. (a) M11 motif (gray box) in MsEF1-αPro::LUC. (b) M1 motif (red box) in M1-ΔM11-MsEF1-αPro::LUC. (c) M2 motif (blue box) in M2-ΔM11-MsEF1-αPro::LUC. (d) M7 motif (yellow box) in M7-ΔM11-MsEF1-αPro::LUC.
Functional validation of enhancer CREs
-
By transiently transforming tobacco with plasmid constructs and performing LUC imaging analysis, it was observed that the deletion of the M11 region from the full-length MsEF1-α promoter significantly reduced its transcriptional activity, indicating that M11 plays an enhancer role in the MsEF1-α promoter (Fig. 3). When the M11-deleted MsEF1-α promoter was individually fused with the M1, M2, or M7 CREs, none of these combinations restored the promoter's activity. To investigate whether M11's enhancer function is transferable to other promoters, the MsLEA2 promoter is used as a backbone and constructed chimeric promoters by incorporating M1, M2, M7, or M11. Subsequent transient transformation assays in tobacco and LUC imaging analysis further confirmed M11's enhancer capability (Fig. 4). Among M1, M2, and M7, M2 exhibited a modest enhancer effect, though its activity appeared highly sensitive to changes in transcriptional context (Fig. 5).
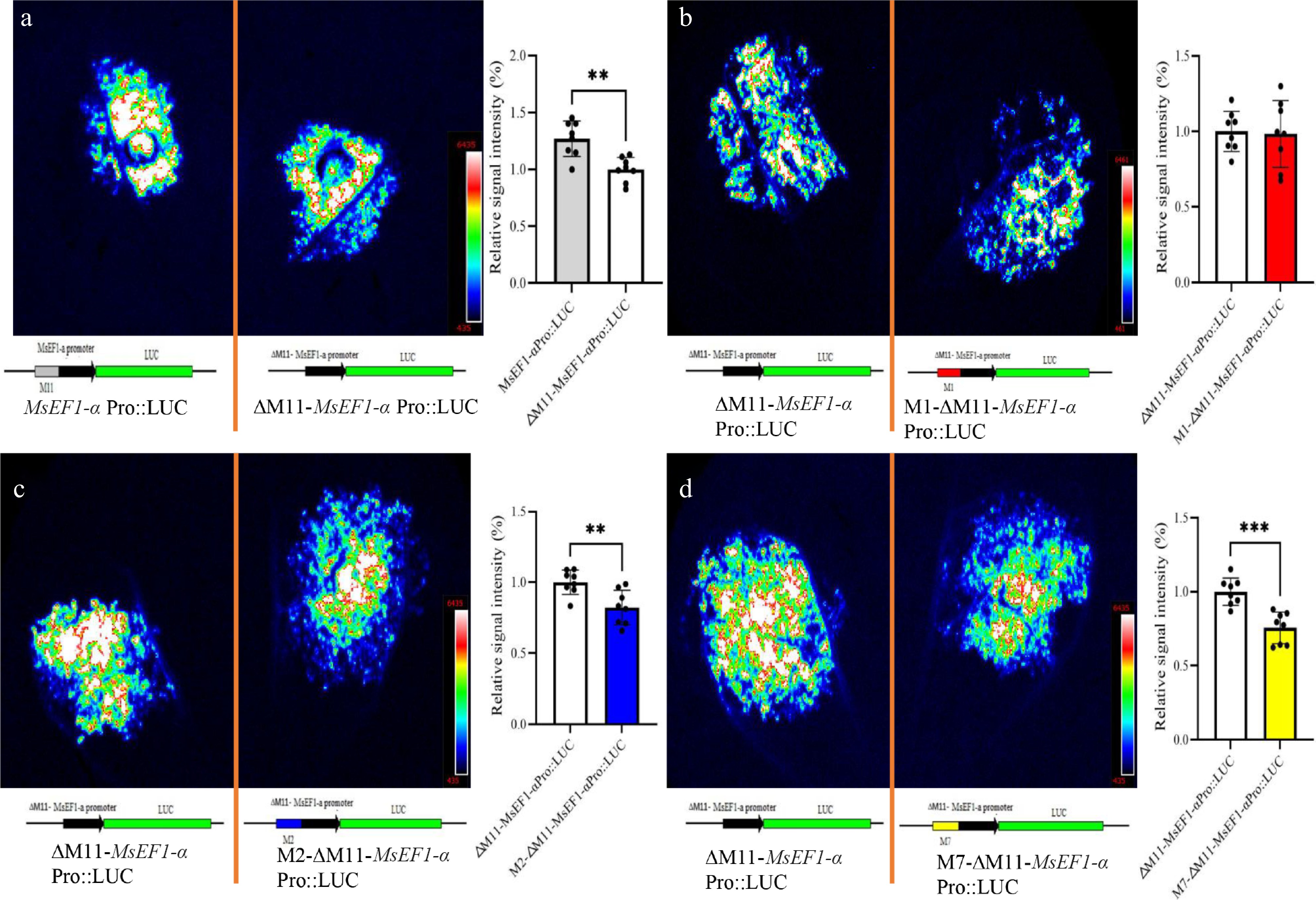
Figure 3.
LUC activity in N. benthamiana leaves expressing MsEF1-α promoter motif variants. (a) Wild-type M11 motif. (b) M1 motif in ΔM11 background. (c) M2 motif in ΔM11 background. (d) M7 motif in ΔM11 background. The MsEF1-α promoter with M11 deletion served as the control. Fluorescence intensity was quantified by selecting the eight brightest spots from N. benthamiana leaves and calculating their ratio to the mean fluorescence intensity of the control. Data represent mean ± SE (n = 8); ** p < 0.01, *** p < 0.001 (one-way ANOVA).
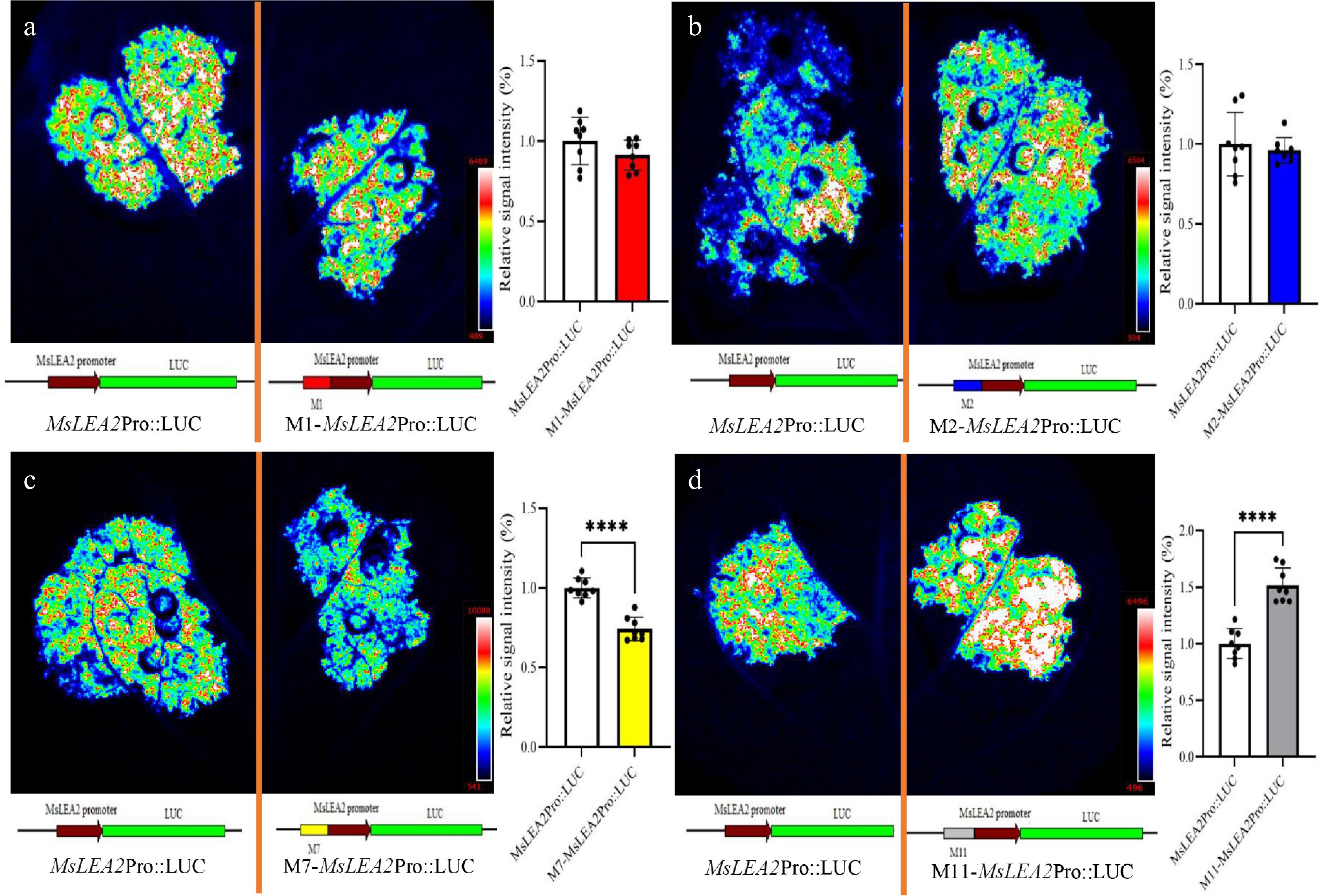
Figure 4.
LUC activity in N. benthamiana leaves expressing MsLEA2 promoter motif variants. (a) Construct with M1 motif. (b) Construct with M2 motif. (c) Construct with M7 motif. (d) Construct with M11 motif. The empty MsLEA2 promoter served as a control. Fluorescence intensity quantification methods followed those described in Fig. 3. Data are presented as mean ± SE (n = 8); **** p < 0.0001 (one-way ANOVA).
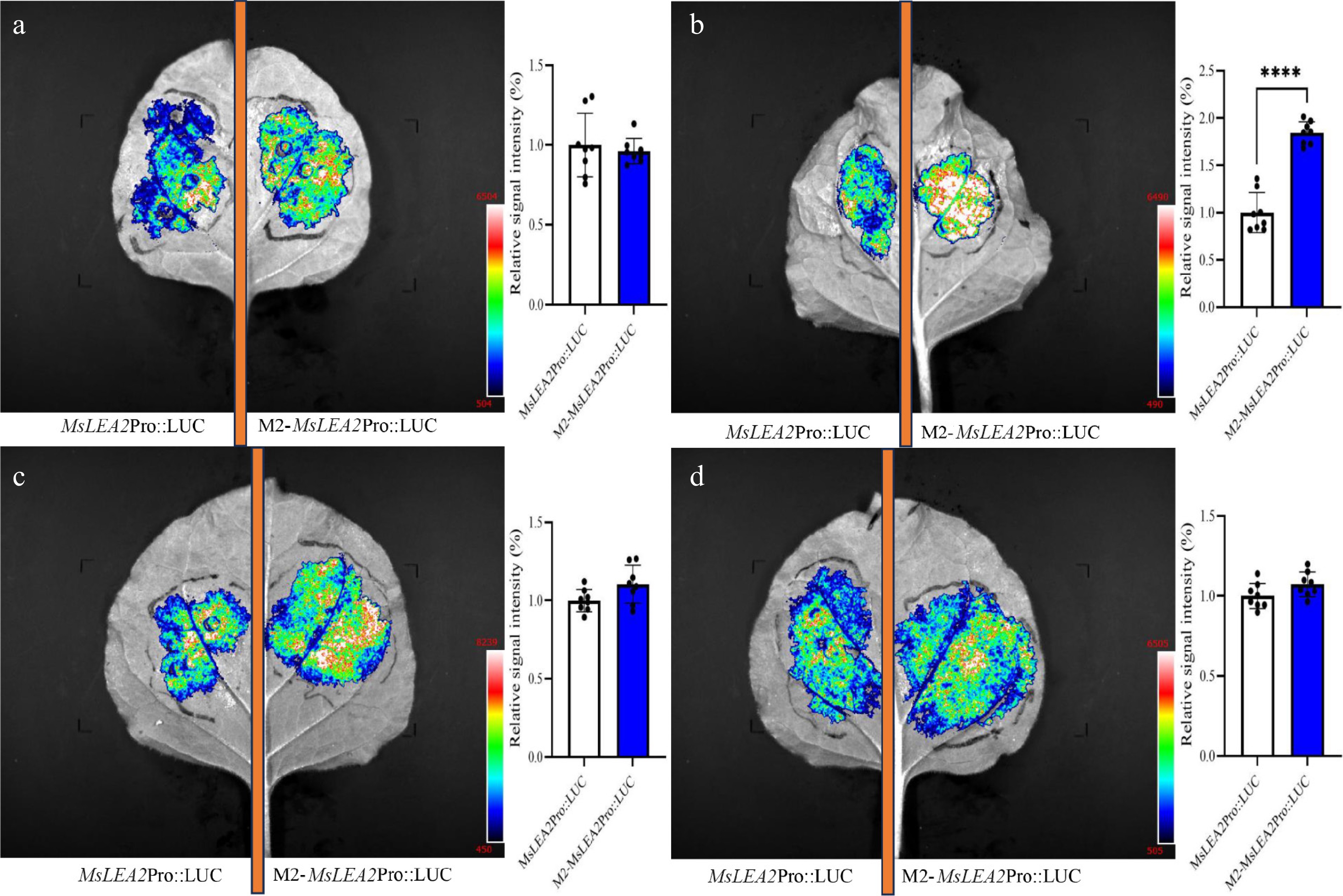
Figure 5.
LUC activity in N. benthamiana leaves after infiltration with M2-MsLEA2Pro::LUC. Representative images from four independent biological replicates (a)–(d) illustrate variable enhancement by motif M2. The empty MsLEA2 promoter served as a control. Fluorescence intensity quantification methods followed those described in Fig. 3. Data are presented as mean ± SE (n = 8); **** p < 0.0001 (one-way ANOVA).
By measuring LUC enzyme activity in transiently transformed tobacco leaves, it was observed that the deletion of the M11 element from the MsEF1-α promoter led to an approximately 55% reduction in activity. Conversely, when the M11 element was inserted into the MsLEA2 promoter, its activity increased by about 183% (Fig. 6). These findings further demonstrate that the M11 element possesses significant enhancer properties.
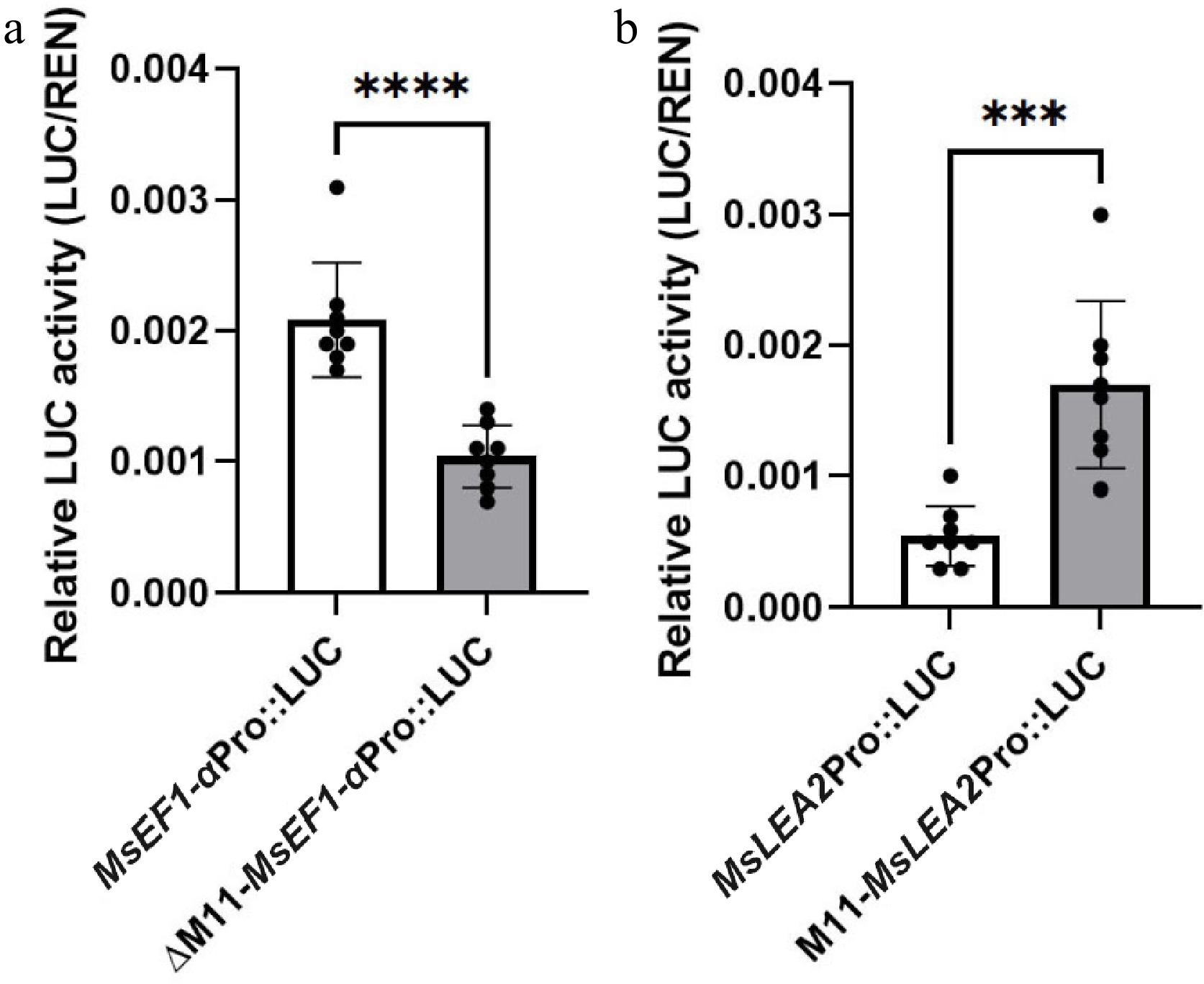
Figure 6.
Normalized luciferase activity (LUC/REN) of M11-containing promoter variants in N. benthamiana. (a) MsEF1-α promoter with (+M11) vs. without (ΔM11) the motif. (b) MsLEA2 promoter with (+M11) vs. empty promoter control. Data are mean ± SE (n = 8); *** p < 0.001, **** p < 0.0001 by one-way ANOVA.
-
This study employed bioinformatics analysis and transient tobacco transformation assays to identify enhancer-type cis-regulatory elements (CREs) within the promoters of highly expressed genes (HEGs) in Medicago sativa. Among these, the M11 element demonstrated robust and stable transcriptional enhancement activity, serving as a key functional component for research on gene expression regulation and molecular breeding in alfalfa. Building upon existing research, the biological significance, potential mechanisms, and limitations of these findings are further discussed.
Identification and functional validation of enhanced CREs: the potent activity of M11 as a key finding
-
The key finding of this study is the identification of an 89 bp fragment (M11) from the housekeeping gene MsEF1-α promoter, which exhibits significant enhancer activity. Experimental results demonstrated that deletion of M11 from the MsEF1-α promoter reduced LUC activity by approximately 55% (Fig. 5). Conversely, inserting M11 into the MsLEA2 promoter increased its activity by about 183% compared to the original promoter (Fig. 5). These findings conclusively demonstrate that M11 functions as a strong CRE in alfalfa. Importantly, its enhancer activity is not dependent on specific promoter contexts (e.g., housekeeping or stress-responsive gene promoters), highlighting its broad applicability in genetic engineering.
The robust activity of M11 is closely associated with its sequence characteristics: (1) It contains a CAAT box—a common enhancer element in plant promoters—that binds to NF-Y family transcription factors to facilitate RNA polymerase II recruitment[15,16]; (2) It is located approximately 600 bp upstream of the TATA box in the MsEF1-α promoter, consistent with the 'long-distance regulation' positional feature of enhancers[17]; (3) Its sequence comprises an AT-rich region (upper segment) and a GC-rich region (lower segment). The AT-rich region promotes chromatin unwinding, while the GC-rich region maintains structural stability, synergistically enhancing regulatory efficiency[17,18]. These features explain why M11 maintains strong enhancer activity across diverse promoter contexts.
The activity of M1, M2, and M7
-
In addition to M11, this study also examined the functions of M1, M2, and M7. The results revealed that neither M1 nor M7 exhibited a significant promoter-enhancing effect when inserted into either the MsEF1-α or MsLEA2 promoter, indicating that at least single copies of these elements lack intrinsic enhancer activity. The functional expression of M1 and M7 may require cooperation with other CREs (e.g., pairing with M11 or unidentified components) or depend on specific transcription factors (e.g., those exclusively expressed in certain tissues or under stress conditions)[19−21]. Based on these findings, it is concluded that M1 and M7 do not possess constitutive enhancer properties and are therefore unsuitable for standalone application in subsequent modern molecular predictions.
The M2 element appears to possess certain enhancer functions, particularly when inserted into the MsLEA2 promoter, where its effects become more pronounced. However, these results were not consistently stable (Fig. 6). This observation indicates that M2's enhancer activity is substantially influenced by transcriptional environmental factors, such as the plant's growth status and the availability of transcription factors[22,23]. Alternatively, the heterologous tobacco system may lack alfalfa-specific transcription factors required for stable M2 functionality. These findings highlight that functional validation of CREs should be conducted in homologous systems (e.g., stable transformation in alfalfa) to more accurately reflect their native biological roles. Moreover, because the current reference genome is a haploidized assembly, allele-specific expression between sub-genomes could not be quantified; potential allele-specific expression (ASE) bias in the selected HEGs will need to be addressed once phased tetraploid assemblies are available.
This research aims to provide key genetic components for molecular breeding in alfalfa
-
Alfalfa, known as the 'King of Forage', requires precise regulation of stress-resistant and quality-related genes for effective variety improvement. With alfalfa breeding now entering the 4.0 era, the introduction of gene-editing technology has significantly accelerated progress in this field. Using gene-editing techniques, enhancer elements can be precisely delivered to specific gene promoters in the alfalfa genome, boosting the expression of stress-resistant and quality-related genes, ultimately leading to the development of superior new varieties. In this study, the M11 element, identified in alfalfa, demonstrated a remarkable ability to enhance reporter gene expression—whether inserted into the MsEF1-α promoter or the MsLEA2 promoter—making it a highly effective enhancer. By employing a gene-editing delivery system, the M11 element can be accurately integrated into target gene promoters at precise locations, promoting the high-efficiency expression of desirable genes and greatly expediting alfalfa breeding efforts[24]. Thus, the confirmation of M11's gene expression enhancement function holds significant practical value for modern molecular breeding in alfalfa.
-
This study identified a highly potent cis-regulatory element, M11, in alfalfa, which exhibits strong enhancer activity independent of specific promoter contexts. This discovery provides a crucial genetic component for research on gene expression regulation and molecular breeding in alfalfa. The robust activity of M11 is closely associated with its sequence characteristics, including the presence of a CAAT box, its genomic position, and AT/GC content balance. Moving forward, employing advanced molecular techniques such as gene editing to precisely integrate M11 into targeted genomic loci could enhance the expression of desirable genes, thereby increasing its practical utility and accelerating the improvement of alfalfa cultivars.
This work was supported by the National Key R & D Program of China (Grant No. 2022ZD04012), and Special Fund for Scientific Research of Shanghai Landscaping & City Appearance Administrative Bureau (Grant No. G242422).
-
The authors confirm their contributions to the paper as follows: study conception and design: Zhou P, Shen Z; experimental work: Shen Z; experimental discussion and guidance: Liu M; LUC imaging system guidance and transcriptome/bioinformation data analysis: Liu M, Wen W; LUC imaging system use and data analysis and material preparation: Song Y; experimental support: Xing Q; manuscript revision: An Y; draft manuscript preparation: Zhou P, Shen Z. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are available upon request from the corresponding author.
-
The authors declare that they have no conflict of interest.
- Supplementary Fig. S1 The average FPKM (Fragments Per Kilobase of transcript per Million mapped fragments) and pooled Coefficient of Variation (CV) for different numbers of gene sets (top 1, top 5, top 10, top 15, top 20 and top 25), (table on the top). The following figure depicts the average FPKM trend curve for varying numbers of gene sets.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Shen Z, Liu M, Wen W, Song Y, Xing Q, et al. 2026. Screening and functional validation of transcriptional enhancer cis-elements in Medicago sativa L. Grass Research 6: e006 doi: 10.48130/grares-0025-0036 

Screening and functional validation of transcriptional enhancer cis-elements in Medicago sativa L.
- Received: 27 September 2025
- Revised: 12 November 2025
- Accepted: 26 November 2025
- Published online: 10 March 2026
Abstract: Alfalfa (Medicago sativa L.) is a globally important forage crop. Identification of enhancer cis-regulatory elements (CREs) in high-expression gene promoters is essential for precise gene regulation and molecular breeding. This study aimed to systematically screen and functionally validate enhancer CREs in alfalfa. Twenty constitutively highly expressed genes (coefficient of variation < 0.2) were identified from leaf, stem, and root tissues of 15-day-old alfalfa seedlings (nine RNA-seq libraries, n = 3 per tissue). Promoter regions (2,000 bp upstream to 500 bp downstream of the transcription start site) were extracted and analyzed using PlantCARE, FIMO, and Fisher's exact test (adjusted p < 0.05). Ten candidate enhancer CREs (M1–M10) were identified, and an 89-bp fragment containing a CAAT box from the MsEF1-α promoter, named M11, was selected for functional validation. Recombinant promoter-LUC vectors containing M1, M2, M7, and M11 were constructed and transiently expressed in Nicotiana benthamiana. Luciferase (LUC) activity was quantified to assess enhancer function. Deletion of M11 from the MsEF1-α promoter reduced LUC activity by approximately 55%, whereas insertion of M11 into the MsLEA2 promoter increased activity by approximately 183%. M1 and M7 exhibited no significant enhancer activity, while M2 showed unstable enhancing effects. M11 is a robust and promoter-independent enhancer CRE in alfalfa, providing a valuable tool for gene regulation and molecular breeding.
-
Key words:
- Medicago sativa /
- Cis-regulatory elements /
- Enhancer /
- Gene expression /
- LUC assay