









-
Saline-alkali stress, characterized by high salt concentration and elevated pH, is one of the most severe abiotic constraints worldwide, significantly limiting crop growth, productivity, and quality[1]. Large areas of marginal land are affected by saline-alkali soils. Therefore, developing salt-tolerant crops is essential for ensuring food and energy security[2]. Juncao technology represents a suite of biotransformation and resource-utilization strategies that employ Juncao grasses for mushroom cultivation, livestock feed production, biomass material development, bioenergy generation, and ecological restoration[3]. Pioneered in China, it has achieved remarkable success domestically and has been extended to 87 countries, being implemented in 386 counties across 31 provinces and municipalities. Giant Juncao (Pennisetum giganteum Zhan X. Lin, syn. Cenchrus fungigraminus Zhan X. Lin) is a perennial C4 grass in the genus Cenchrus[4], widely cultivated as an important forage grass and multipurpose biomass crop. It is valued for its rapid growth, substantial biomass yield, and strong environmental adaptability. As a versatile species, Giant Juncao shares several agronomic advantages with pearl millet, including strong stress tolerance and the capacity to improve soil structure and fertility[5,6]. Recent pan-genomic studies in pearl millet have revealed structural variations associated with heat tolerance, providing evolutionary insights into stress adaptation mechanisms in related species[7]. Its extensive root system contributes significantly to soil stabilization, erosion control, and ecosystem restoration[8]. Furthermore, Giant Juncao serves as an important resource for soil-and-water conservation, forage utilization in livestock production, and biomass-based energy production.
Plants have evolved diverse adaptive mechanisms to cope with saline-alkali stress[9]. Single-cell transcriptomic studies in pearl millet further highlight the spatiotemporal dynamics of stress responses, particularly in vascular tissues[10]. Among these, transcription factors (TFs) play pivotal regulatory roles, with members of the WRKY family being particularly prominent in mediating stress-responsive gene expression. WRKY TFs have been extensively characterized in major cereal crops such as rice, wheat, sorghum, and maize, where they modulate salt tolerance through abscisic acid (ABA)- and mitogen-activated protein kinase (MAPK)-mediated signaling, reactive oxygen species (ROS) detoxification, Na+/K+ homeostasis, and osmotic adjustment[11−14]. WRKY proteins are defined by a highly conserved WRKY domain and a zinc-finger motif that binds W-box cis-elements, enabling them to regulate downstream stress-responsive genes and coordinate hormonal signaling, ROS scavenging, and ion transport under abiotic stress[15,16]. In pearl millet, genome-wide studies have revealed extensive WRKY activation and repression under salt and drought treatments, highlighting their essential role in abiotic stress responsive transcriptional networks[17,18]. As one of the largest plant-specific TF families[19], WRKY proteins also participate in plant growth and development through synergistic interactions with multiple genes and other TFs[20−22]. Recent studies also showed that non-CG DNA methylation can influence the binding specificity of WRKY transcription factors, adding an additional layer to their regulatory diversity[23,24].
Although Giant Juncao possesses substantial agronomic and ecological significance as a forage and biomass grass, a comprehensive genome-wide characterization of the WRKY gene family in this species remains lacking. Understanding the WRKY repertoire and its regulatory landscape in this polyploid perennial grass is important for elucidating mechanisms of stress adaptation and for supporting the improvement of saline-alkali-tolerant forage germplasm. Therefore, this study aims to perform a genome-wide investigation of WRKY genes in Giant Juncao, including analyses of their evolutionary relationships and regulatory features, as well as examinations of their expression patterns under salt and drought-related conditions.
-
The reference genome sequence and gene annotation files of C. fungigraminus (Giant Juncao) were retrieved from the Energy Grass Database (EGDB,
http://engrass.juncaodb.cn ) and the National Genomics Data Center under the genome accession number GWHBWDX00000000.1 (NGDC,https://ngdc.cncb.ac.cn )[25]. To identify putative WRKY transcription factors, we applied a two-step approach. First, the Hidden Markov Model (HMM) profile of the WRKY domain (PF03106) was obtained from the InterPro database (www.ebi.ac.uk/interpro , Pfam entry)[26] and used as a query to search the C. fungigraminus protein dataset with HMMER (v3.4) under an E-value threshold of 1e−5[27]. After removing redundant sequences, candidate proteins were further validated by confirming the presence of complete WRKY domains using SMART (http://smart.embl-heidelberg.de/ )[28] and InterPro[26]. Only proteins containing at least one complete WRKY domain were retained for further analyses. In addition, the basic physical and chemical properties of the identified CfWRKY proteins, including amino acid length, molecular weight (MW), and theoretical isoelectric point (pI), were calculated using the ProtParam tool available on the ExPASy server (www.expasy.org )[29]. To facilitate further analysis, the identified CfWRKY genes were sequentially renamed as CfWRKY1 to CfWRKY182 according to their physical positions on the chromosomes.Phylogenetic analysis of WRKY genes
-
To investigate the evolutionary relationships of WRKY proteins, phylogenetic analyses were conducted using the collected WRKY genes from Chlamydomonas reinhardtii, Oryza sativa, Cenchrus americanus, and C. fungigraminus. The single WRKY from Chlamydomonas reinhardtii was included as an outgroup. WRKY protein sequences were retrieved from the Plant Transcription Factor Database (PlantTFDB) and Milletdb[30,31]. Multiple sequence alignment of WRKY proteins was performed using MAFFT (v7.490) with the G-INS-i strategy and the parameters '--maxiterate 1000 --globalpair'[32]. Phylogenetic trees were constructed using the maximum likelihood (ML) method implemented in IQ-TREE2, with the best-fit substitution model automatically selected based on the Bayesian Information Criterion (BIC)[33]. Branch support values were evaluated using 1,000 ultrafast bootstrap replicates. Based on the established classification of O. sativa WRKY genes, the CfWRKY genes were assigned to the corresponding groups (I, IIa–IIe, and III)[34]. Phylogenetic trees were visualized and annotated using iTOL (Interactive Tree of Life, v6)[35].
Chromosomal distribution and gene structure analysis in C. fungigraminus
-
The chromosomal positions of CfWRKY genes were obtained from the genome annotation (GFF3) file. The identified CfWRKY genes were mapped to the 14 chromosomes (Chr01A–Chr07B) of C. fungigraminus using TBtools (v2.210)[36], and their physical locations were visualized with gene labels.
The exon–intron structures of CfWRKY genes were extracted from the genome annotation file and visualized using the Gene Structure Display Server (GSDS 2.0;
http://gsds.cbi.pku.edu.cn/ )[37] and TBtools (v2.210). Exon numbers and intron lengths were compared among WRKY subgroups to evaluate structural diversity.To explore the evolutionary mechanisms underlying family expansion, both tandem and segmental duplication events were investigated. Tandemly duplicated CfWRKY genes were defined as family members located within a 200-kb region on the same chromosome, while segmental duplications and syntenic relationships within C. fungigraminus and between C. fungigraminus and other species (A. thaliana, O. sativa, Zea mays, and C. americanus) were identified using the MCScanX toolkit[38] implemented in TBtools (v2.210). The syntenic relationships were visualized with Advanced Circos and Dual Synteny Plot modules in TBtools (v2.210). To evaluate the selective pressure on duplicated gene pairs, nonsynonymous substitution rate (Ka) and synonymous substitution rate (Ks) values were calculated using KaKs_Calculator (v2.0, Nei–Gojobori model)[39] integrated in TBtools (v2.210). Gene pairs with Ka/Ks < 1 were considered to be under purifying selection, those with Ka/Ks = 1 under neutral selection, and those with Ka/Ks > 1 under positive selection.
Conserved motifs and domain analysis of CfWRKY proteins
-
The conserved motifs of CfWRKY proteins were identified using the Multiple Em for Motif Elicitation (MEME) suite v5.5.3 (
http://meme-suite.org/ ) with default parameters[40]. The identified motifs were subsequently visualized using TBtools (v2.210).To further confirm the conserved WRKY domains, all CfWRKY protein sequences were analyzed using the Pfam database (
http://pfam.xfam.org/ ) and InterProScan (www.ebi.ac.uk/interpro ). The presence of the characteristic WRKYGQK heptapeptide and the zinc-finger motifs (C2H2 or C2HC) was examined to classify CfWRKY proteins into different subgroups. Redundant or truncated sequences lacking intact WRKY domains were excluded from downstream analyses.Cis-acting element analysis of CfWRKY genes
-
The promoter sequences of CfWRKY genes were defined as the 2,000 bp upstream regions from the translation start site (ATG) and extracted from the reference genome using TBtools (v2.210). The extracted promoter sequences were analyzed using the PlantCARE database (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ )[41] to identify cis-acting regulatory elements. The identified elements were subsequently classified into five functional categories: (1) hormone-responsive elements (e.g., ABRE, TGA-element, TCA-element, CGTCA-motif, P-box); (2) stress-responsive elements (e.g., MBS, LTR, WUN-motif, ARE); (3) growth and development-related elements (e.g., CAT-box, RY-element, O2-site); (4) core promoter elements (TATA-box, CAAT-box); and (5) other elements. The distribution of cis-acting elements was visualized using TBtools (v2.210).Plant materials, stress treatments, and RNA-seq data
-
For drought-rehydration experiments, previously published RNA-seq datasets of C. fungigraminus seedlings were obtained from our previously published study[8], where plants were subjected to progressive drought followed by rehydration.
For salt stress experiments, uniform C. fungigraminus seedlings (~15 cm in height) were propagated from stem nodes collected from the germplasm nursery at the National Engineering Research Center of Juncao Technology, Fujian Agriculture and Forestry University. After one week of nursery growth in plug trays, seedlings with consistent vigor were transplanted individually into plastic pots (16 cm top diameter × 12.5 cm bottom diameter × 17 cm height) containing 1.5 kg of sterilized soil : sand (2:1, v/v) mixture. The substrate consisted of peat soil (organic matter > 70%, humic acid > 40%, total NPK > 3%, pH 5.0–6.5) mixed with river sand, both sieved through a 2 mm mesh and sterilized twice by autoclaving (121 °C, 0.11 MPa, 2 h, interval 1 h). Pots were pretreated with 0.1% potassium permanganate for 24 h and air-dried before use. Plants were maintained in a greenhouse under natural light with day/night temperatures of 25–35 °C and relative humidity of 50%–75%. During the growth period, seedlings were irrigated every 3 d with 100 mL distilled water and supplemented weekly with 50 mL of one-tenth strength Hoagland's nutrient solution.
After eight weeks of growth, salt treatments were applied by irrigating each seedling with 100 mL NaCl solution at concentrations of 0 (CK), 50, 100, 150, and 200 mM every 3 d for a total of 30 d. At the end of the treatments, root tissues were carefully harvested, rinsed with deionized water, cut into fragments (< 1 cm), immediately frozen in liquid nitrogen, and stored at –80 °C. Three independent biological replicates were collected per treatment. Total RNA extraction, library construction, and sequencing were performed by Novogene (Beijing, China) using the Illumina HiSeq 4000 platform following the manufacturer's protocols. The newly generated RNA-seq data from salt stress experiments were deposited in the National Genomics Data Center (NGDC) under BioProject accession number PRJCA051202.
Expression profiling and co-expression network analysis of CfWRKY genes
-
RNA-seq data from C. fungigraminus seedlings treated with 0, 50, 100, 150, and 200 mM NaCl was used to analyze the expression patterns of CfWRKY genes. Expression levels were normalized to transcripts per million (TPM), log2-transformed and visualized as heatmaps using the R package pheatmap v1.0.12 (
https://CRAN.R-project.org/package=pheatmap ). Pairwise differential expression analyses among the salt treatments were conducted using DESeq2[42], and differentially expressed genes were defined with thresholds of |log2FC| ≥ 1 and adjusted p-value < 0.05. The numbers of significantly upregulated and downregulated genes for each comparison were summarized using bar plots generated with the R package ggplot2[43]. Overlaps of upregulated genes were visualized using the VennDiagram package[44], and global intersection patterns of all DEGs were illustrated with UpSetR[45] to assess shared and treatment-specific responses across multiple contrasts.Co-expression network analysis of CfWRKY genes under drought–rehydration
-
Weighted gene coexpression network analysis (WGCNA) was conducted using the WGCNA package (v1.73)[46] in R to explore the regulatory roles of CfWRKY genes under drought and rehydration conditions. Prior to network construction, low-expression genes were filtered, and TPM values were log2-transformed. A soft-thresholding power (β = 8) was selected based on the scale-free topology criterion. Gene modules were identified using dynamic tree cutting with a minimum module size of 50 genes, and grey modules representing unassigned genes were excluded from downstream analysis. Module eigengenes were correlated with drought and rehydration treatments to identify stress-associated modules. Hub genes within significant modules were determined based on eigengene connectivity (kME). Coexpression networks were visualized using Cytoscape (v3.7.0)[47]. Gene Ontology (GO) functional annotation for cyan-module genes was obtained using the eggNOG-mapper server (
http://eggnog-mapper.embl.de/ ) (v2.1.1)[48], and GO enrichment analysis was performed using the R package clusterProfiler (v4.0). Visualization was conducted in clusterProfiler[49] and ggplot2[43]. -
A total of 182 non-redundant WRKY genes were identified in the C. fungigraminus genome based on conserved-domain screening. These genes were designated CfWRKY1–CfWRKY182 according to their physical chromosomal positions (Fig.1a), and detailed genomic information including gene ID, chromosome, start and end positions, and strand orientation, is provided in Supplementary Table S1.
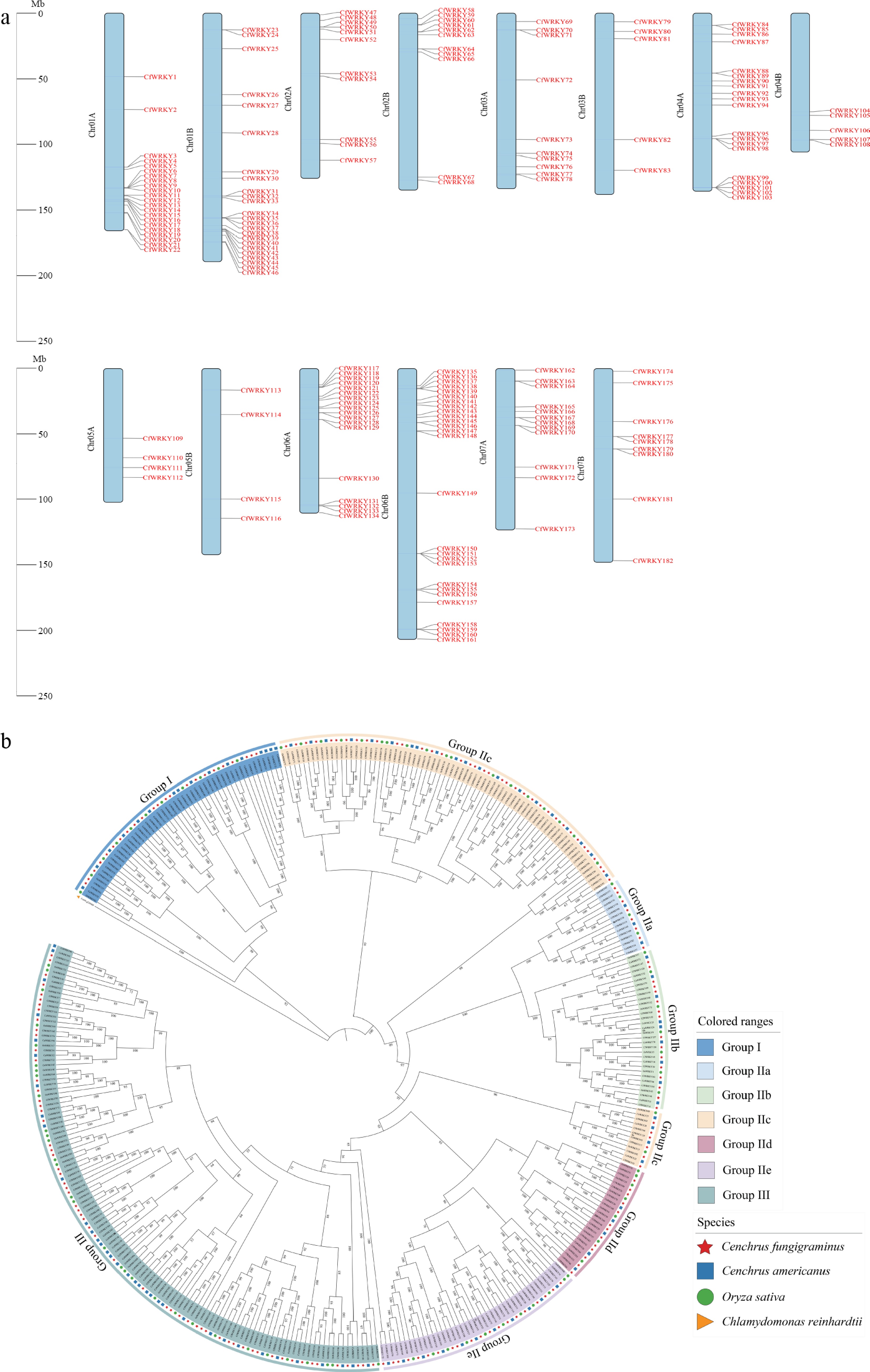
Figure 1.
Chromosomal distribution and phylogenetic analysis of CfWRKY genes in Cenchrus fungigraminus. (a) A total of 182 CfWRKY genes were mapped onto the 14 chromosomes (Chr01A–Chr07B) of C. fungigraminus. Chromosomes are shown as colored bars, and gene positions are indicated in red. (b) Phylogenetic analysis of CfWRKY proteins together with WRKY members from C. americanus, O. sativa, and C. reinhardtii (outgroup). CfWRKY genes are marked with red stars, and different groups are indicated by distinct colors.
To investigate the basic characteristics of the CfWRKY family, we analyzed protein length, molecular weight (MW), theoretical isoelectric point (pI), WRKY type, zinc-finger motif, and predicted subcellular localization. These physicochemical properties of 182 CfWRKY proteins exhibited considerable variation. Protein lengths ranged from 92 to 1,345 amino acids, and MWs ranged from 10.19 to 149.21 kDa. Based on pI values, 93 proteins were classified as acidic (pI < 6.5), 29 as neutral (pI 6.5–7.5), and 60 as basic (pI > 7.5). Subcellular localization predictions showed that most CfWRKY proteins were localized to the nucleus, while a few were predicted to localize to the extracellular space, mitochondria, chloroplast, or cytoplasm (Supplementary Table S2). Secondary structure analysis revealed that random coils were the predominant structural element (70.4%), representing the major component of WRKY proteins (Supplementary Table S3). This was followed by α-helices (17.1%), extended strands (6.4%), and β-turns (2.1%).
The 182 CfWRKY genes were unevenly distributed across the 14 chromosomes. C. fungigraminus is an allotetraploid species (2n = 4x = 28) composed of two subgenomes, A and B, each containing seven chromosomes (Chr01A–Chr07A and Chr01B–Chr07B). The highest densities were observed on Chr01A, Chr01B, Chr04A, Chr06A and Chr06B, whereas Chr05A and Chr05B each contained only four WRKY genes. The number of genes on Chr02A/B, Chr03A/B and Chr07A/B were relatively comparable (Fig.1a). There were 10 genes located on Chr03A, and five on Chr03B, while 12 genes were found on Chr07A, and nine genes on Chr07B. The 182 CfWRKY genes were distributed across the A (95 genes) and B (87 genes) subgenomes with near-equal representation, indicating balanced gene retention following the allopolyploidization event.
Phylogenetic analysis, multiple sequence alignment and sequence logo of CfWRKY genes
-
To investigate the evolutionary relationships of the WRKY gene family, a maximum likelihood (ML) phylogenetic tree was constructed using 182 CfWRKY proteins together with 103 WRKY proteins from C. americanus, 105 O. sativa WRKYs, and WRKY proteins from C. reinhardtii (outgroup) (Fig. 1b). Consistent with established WRKY classification schemes, the CfWRKY proteins were grouped into three main clades (I, II, and III), with Group II further divided into five subgroups (IIa–IIe). Among these, Group III represented the largest clade, encompassing 57 CfWRKY members, followed by subgroup IIc (43) and Group I (23). The remaining subgroups contained eight (IIa), 16 (IIb), 15 (IId), and 20 (IIe) members, respectively.
Multiple sequence alignment revealed that most CfWRKY proteins contained the highly conserved WRKYGQK heptapeptide and either C2H2 or C2HC zinc-finger motifs (Fig. 2a). Members of Group I generally possessed two WRKY domains, whereas three proteins exhibited incomplete domains, including CfWRKY165 (lacking the C-terminal domain) and CfWRKY166 and CfWRKY176 (lacking the N-terminal domain). Groups II and III contained a single WRKY domain; however, several members exhibited sequence variations or deletions. In Group II, nine CfWRKY proteins (CfWRKY160, CfWRKY129, CfWRKY38, CfWRKY148, CfWRKY133, CfWRKY18, CfWRKY142, CfWRKY12, and CfWRKY123) showed a Q-to-K substitution (WRKYGKK), while CfWRKY45 displayed a Q-to-R substitution (WRKYGRK). CfWRKY44 exhibited both substitutions and deletions in the heptapeptide, and CfWRKY88 and CfWRKY89 contained multiple mutations that disrupted the integrity of the WRKY domain. In Group III, most members displayed partial deletions of the domain, often resulting in the WRKY--GQ motif, and six members showed extensive deletions or multiple mutations, leading to loss of a complete WRKY domain.

Figure 2.
Conserved domain analysis of WRKY proteins in C. fungigraminus. (a) Multiple sequence alignment of WRKY proteins. Based on phylogenetic classification, the CfWRKY proteins were grouped into I, II, and III. The conserved WRKY domain and the associated zinc-finger motifs are indicated above the aligned sequences. The number displayed on the far right of each row indicates the position of the corresponding sequence within the full-length protein. (b) Sequence logos of the WRKY domain regions from the three groups, showing the degree of amino-acid conservation; the larger the letter height, the higher the conservation at that position.
Sequence logo analysis illustrated the conservation pattern across groups (Fig. 2b). Members of Group I showed two highly conserved WRKY domains with information content values approaching the maximum (≈ 4.0), indicating that most Group I proteins retain two intact WRKY domains. In Group II, the heptapeptide motif was predominantly WRKYGQK, whereas a few members displayed substitutions such as WRKYGKK or WKKYGQK. In Group III, many CfWRKY proteins possessed incomplete WRKY domains with multiple gaps, and the C-terminal region often lacked a typical zinc-finger structure.
Gene structure and conserved motif analysis of CfWRKY genes
-
Ten highly conserved motifs were identified among CfWRKYs (Fig. 3a), with each member containing two to six motifs. Most CfWRKY proteins possessed motif 1 and motif 2, which together form the core WRKY domain. In Group II, all members contained both motifs except CfWRKY30, CfWRKY44, CfWRKY88, CfWRKY92, and CfWRKY163. Members within the same subgroup generally shared similar motif composition and arrangement. For example, most Group III proteins contained either motifs 1-2 or motifs 1-6-9, whereas many Group I proteins harbored six motifs (motifs 1, 2, 3, 4, 5, 7) arranged in a conserved order of 4-7-3-1-2-5. Such subgroup-specific motif patterns may underlie functional diversification of WRKY proteins. Intra-group differences were also observed; for instance, CfWRKY94 and CfWRKY67 in Group II lacked motif 9, whereas CfWRKY34 and CfWRKY73 possessed it, suggesting potential functional divergence even among closely related members.
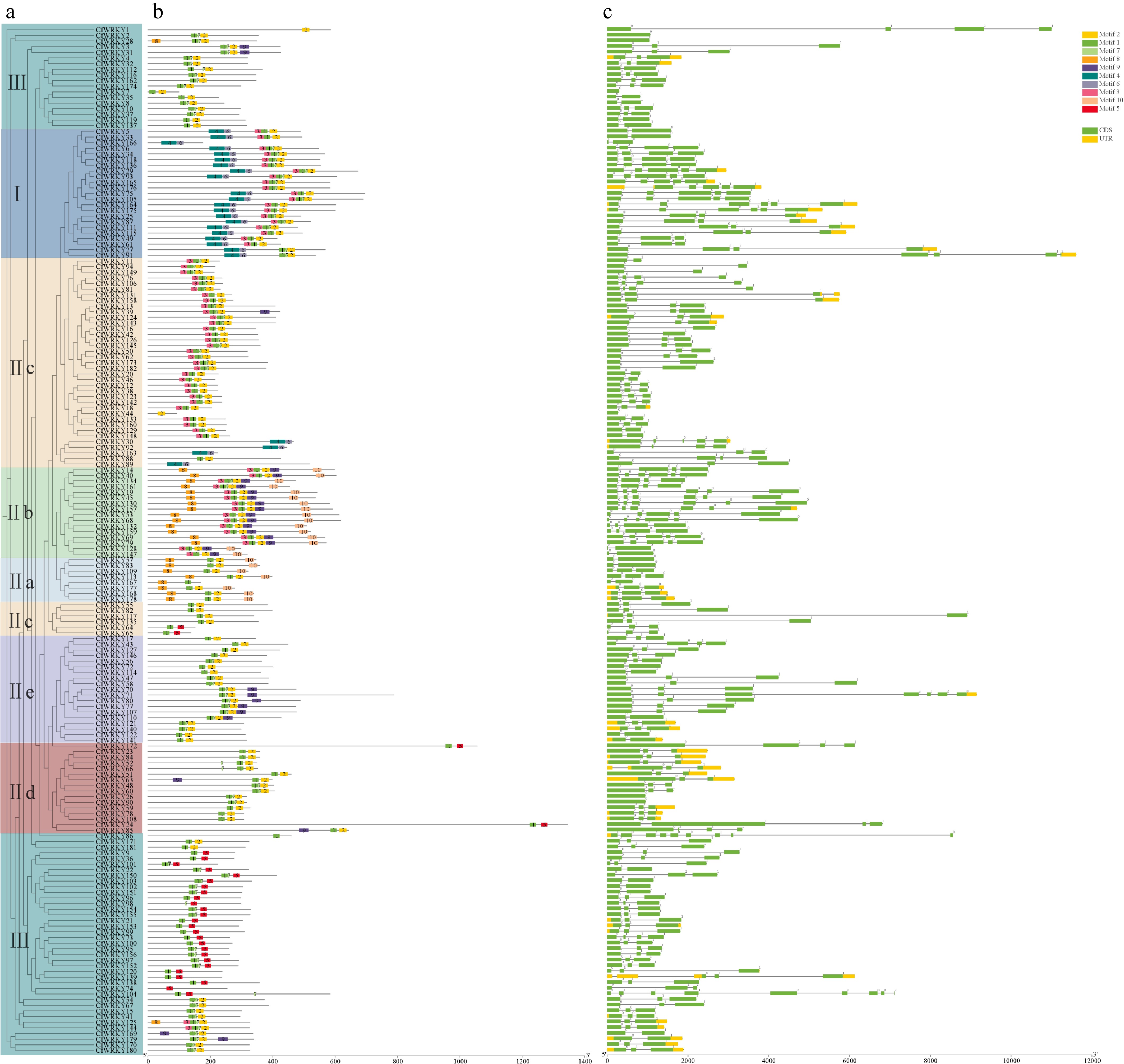
Figure 3.
Conserved motif architecture and exon–intron structures of CfWRKY genes. (a) MEME-derived motifs (motifs 1–10) along CfWRKY protein sequences, grouped by phylogenetic clades (I, II, III). (b) Exon–intron organizations of CfWRKY genes. Members within the same subgroup generally share similar motif composition/arrangement and comparable gene structures.
Exon–intron structure analysis (Fig. 3b) revealed that CfWRKY genes contained two to nine exons, with most genes harboring four exons. For example, CfWRKY182, CfWRKY173, and CfWRKY148 contained only two exons; CfWRKY130 and CfWRKY157 had six exons; and CfWRKY104 and CfWRKY86 had nine exons. The variation in exon numbers may contribute to their functional diversity. Genes within the same subgroup generally exhibited similar exon numbers and gene lengths; for instance, CfWRKY177 and CfWRKY178 in Group II both had three exons and two introns and showed comparable gene lengths.
Genomic positioning and duplication driven expansion of the CfWRKY family
-
Members of the WRKY gene family are typically classified into three major groups (I, II, and III) based on the number of WRKY domains and the type of zinc-finger motif. To explore the origin and evolutionary expansion of the CfWRKY gene family, we first examined whole-genome duplication (WGD) signals. The high-quality, near-complete genome of C. fungigraminus revealed that this species is an allotetraploid, containing two highly syntenic subgenomes (A and B), which form clear homeologous chromosome pairs such as Chr01A/Chr01B and Chr06A/Chr06B. This genomic architecture demonstrates that WGD acted as a major force driving the initial doubling of ancestral WRKY gene content in Giant Juncao.
Following the WGD event, the CfWRKY family underwent substantial lineage-specific expansion through segmental duplication. We identified 136 segmentally duplicated CfWRKY gene pairs across the genome, whereas no tandem duplication events were detected. These duplicated pairs were predominantly enriched on Chr01A, Chr01B, Chr06A, and Chr06B, corresponding to large collinear blocks preserved between homeologous chromosomes. This enrichment pattern indicates that segmental duplication, rather than tandem duplication, represents the major force driving the recent expansion of the CfWRKY repertoire in C. fungigraminus (Supplementary Table S2). Most duplicated pairs were retained within the same WRKY subgroup, reflecting strong structural and functional conservation. For example, CfWRKY5–CfWRKY33 form a Group I duplicated pair, while CfWRKY2–CfWRKY28 represent a duplicated pair within Group III. Such paralogs likely contribute to improved responsiveness to biotic and abiotic stresses due to their functional redundancy and high sequence conservation. Ka/Ks analysis further supported this interpretation (Supplementary Table S4). Except for the CfWRKY120–CfWRKY152 pair, which showed an undefined Ks value, all duplicated pairs exhibited Ka/Ks < 1, indicating that strong purifying selection has acted to maintain their functional integrity during evolution.
A genome-wide intraspecific synteny analysis of CfWRKY genes further supported these duplication patterns (Fig. 4a). Extensive collinearity was observed across all 14 chromosomes of C. fungigraminus, and numerous CfWRKY gene pairs were linked by strong syntenic connections—especially between chromosome pairs such as Chr01A/Chr01B and Chr06A/Chr06B. The retention of these large ancestral collinear blocks highlight the allotetraploid origin of Giant Juncao and illustrates how WGD combined with subsequent segmental duplication has shaped the expansion and redistribution of WRKY genes within the genome.
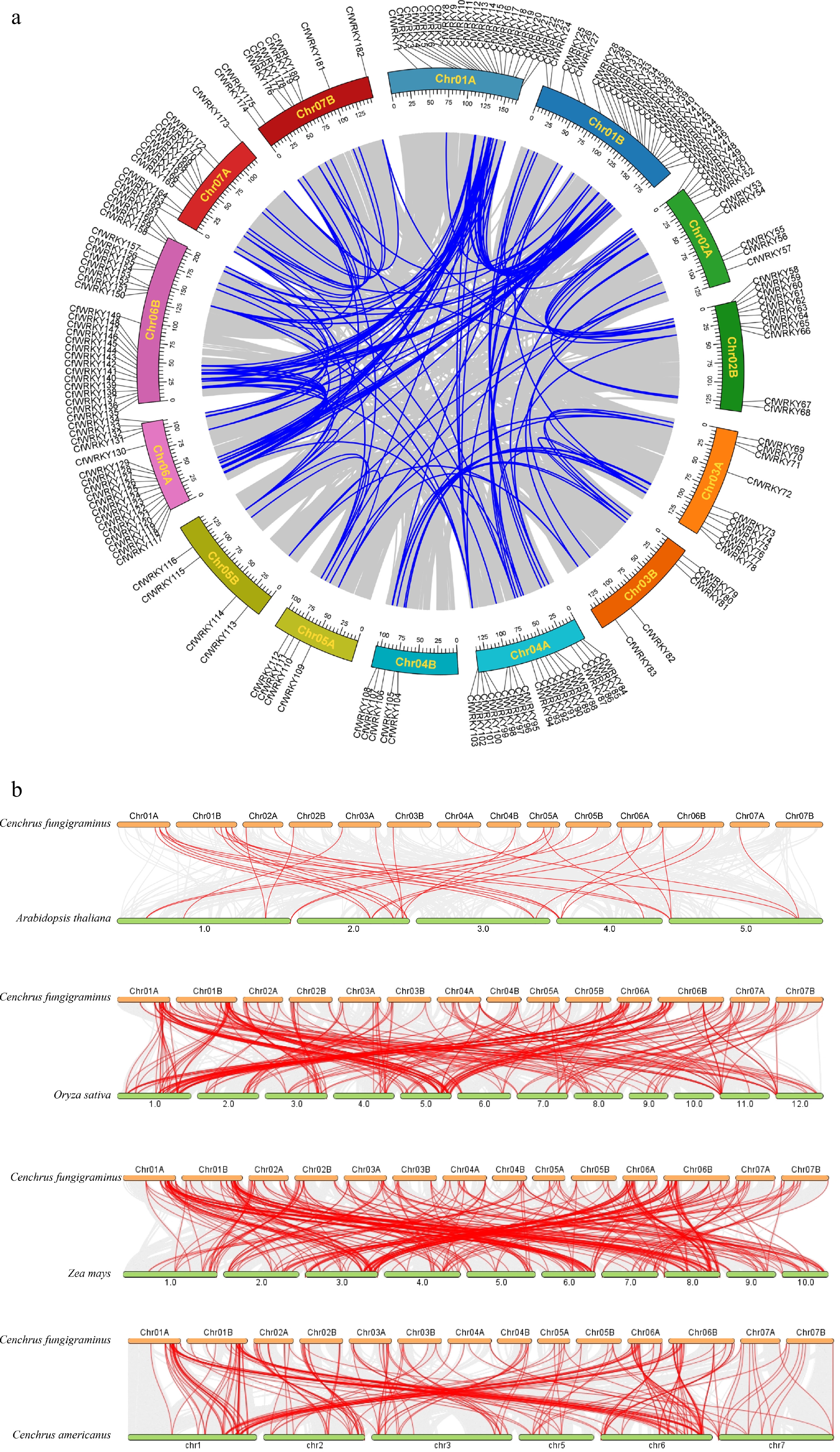
Figure 4.
Synteny analysis of the CfWRKY gene family. (a) Intraspecific synteny and chromosomal distribution of CfWRKY genes in C. fungigraminus. The outer track indicates the positions of 182 CfWRKY genes on 14 chromosomes (Chr01A–Chr07B). Blue lines inside the circle represent segmentally duplicated CfWRKY gene pairs. Chromosomes are drawn to scale (Mb). (b) Comparative interspecific synteny of WRKY genes between C. fungigraminus and A. thaliana, O. sativa, Z. mays, and C. americanus, illustrating conserved orthologous gene pairs and their evolutionary relationships.
Interspecific synteny analysis was conducted between C. fungigraminus and four representative plants (A. thaliana, O. sativa, Z. mays, and C. americanus) (Fig. 4b). We identified 314 WRKY orthologous gene pairs between Z. mays and C. fungigraminus, 251 between O. sativa and C. fungigraminus, 209 between C. americanus and C. fungigraminus, and 35 between A. thaliana and C. fungigraminus. Grass–Cf syntenic blocks were widely distributed across all chromosomes, indicating that the macroscopic chromosomal structure and gene order of WRKY loci have been largely conserved during species divergence.
Cis-acting element analysis of CfWRKY genes
-
To further elucidate the regulatory landscape of CfWRKY, we systematically analyzed the cis-acting elements present in their promoter regions. In total, 52 distinct types of cis-elements were identified, indicating a highly complex promoter architecture associated with various biological processes (Supplementary Fig. S1). These motifs were predominantly related to light responsiveness, hormone signaling, responses to biotic and abiotic stresses, and plant growth and development. Light-responsive elements constituted the largest category (22 types), followed by hormone-responsive (11 types), and growth/development-related motifs (11 types), while stress-responsive elements were comparatively fewer (six types). Box 4, G-Box, and the GT1-motif were consistently detected across all three WRKY groups, suggesting key roles in light-regulated functions. Among stress-related elements, ARE, CCAAT-box, GC-motif, LTR, and MBS were prevalent, and among hormone-responsive elements, ABRE (ABA), TGA-element (auxin), and CGTCA/TGACG-motifs (JA) were enriched, indicating that ABA, auxin, and JA signaling may predominantly regulate CfWRKY expression. Growth- and development-related motifs such as A-box, AT-rich elements, GCN4_motif, HD-Zip 1, MSA-like, O2-site, AACA_motif, and RY-element were also widely distributed, implying diverse developmental roles of the CfWRKY family. Consistently, ABRE sites were detected in 165 promoters, MBS elements in 108, whereas TC-rich repeats were present in 47 genes, supporting the predominant integration of CfWRKYs into ABA- and drought-associated regulatory circuits and a comparatively selective engagement in biotic-stress pathways.
Salt-responsive expression dynamics of CfWRKY family members
-
To examine the transcriptional plasticity of WRKY genes under salinity stress, RNA-seq analysis was performed on C. fungigraminus seedlings subjected to a gradient of NaCl concentrations (CK and 50, 100, 150, 200 mM). Gene expression exhibited marked differences across salt concentrations. Among all comparisons, S4 (200 mM vs CK), S8 (150 vs 100 mM), and S9 (200 vs 100 mM) showed the highest numbers of differentially expressed genes, indicating the strongest transcriptional responses (Fig. 5a, b). In contrast, S6 (150 vs 50 mM) displayed almost no differentially expressed genes, suggesting minimal transcriptional changes between these treatments (Fig. 5c). Overall, salt stress induced distinct expression patterns across different concentration comparisons, with S9 likely representing the most pronounced response stage (Fig. 5d).
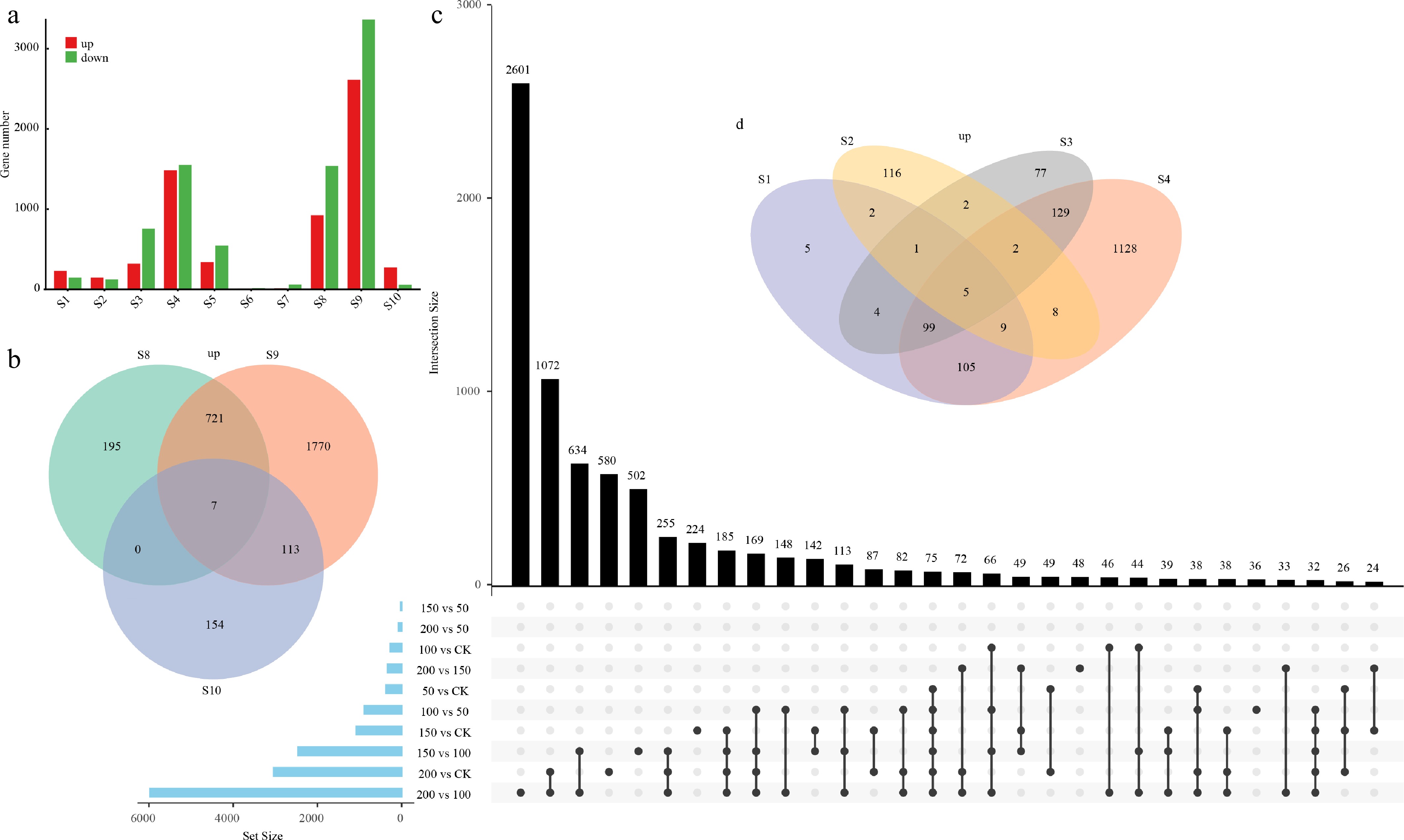
Figure 5.
Distribution and overlap of differentially expressed genes (DEGs) under various treatment conditions. (a) Numbers of upregulated and downregulated DEGs identified in each comparison group (S1–S10). The comparison groups are defined as follows: S1, 50 vs CK; S2, 100 vs CK; S3, 150 vs CK; S4, 200 vs CK; S5, 100 vs 50; S6, 150 vs 50; S7, 200 vs 50; S8, 150 vs 100; S9, 200 vs 100; and S10, 200 vs 150. The variation in DEG numbers across comparisons reflects the transcriptional responses under different stress intensities and contrasts. (b) Venn diagram showing the shared and unique upregulated DEGs among S8 (150 vs 100), S9 (200 vs 100), and S10 (200 vs 150). (c) UpSet plot illustrating the intersection patterns and set sizes of DEGs across all comparison groups (S1–S10), revealing both common and distinct transcriptional responses across the stress gradient. (d) Venn diagram showing the overlap of upregulated DEGs among S1 (50 vs CK), S2 (100 vs CK), S3 (150 vs CK), and S4 (200 vs CK), highlighting the core stress-responsive genes under increasing treatment intensities relative to the control.
Among those differentially expressed genes, 51 CfWRKYs showed significant changes in their expression levels, indicating broad integration of the WRKY family into the salinity-responsive regulatory network (Fig. 6). Among those CfWRKY responses to salt stress, we found several genes, such as CfWRKY57, CfWRKY63, CfWRKY14, CfWRKY159, and CfWRKY152 exhibited progressively increased expression with rising salt concentrations, showing strong induction at high salinity (150–200 mM NaCl). These genes may function in high-salt–activated defense and signaling pathways. In contrast, genes including CfWRKY11 and CfWRKY182 showed slightly reduced expression under high-salt treatment compared with the control, suggesting potential roles in maintaining basal transcriptional states under normal growth conditions. Additionally, CfWRKY17, CfWRKY127, CfWRKY138, and CfWRKY147 reached peak expression at moderate salinity levels (50–100 mM NaCl), indicating involvement in early salt-stress perception and response. Meanwhile, genes such as CfWRKY5, CfWRKY78, and CfWRKY140 displayed relatively stable expression across treatments, likely serving housekeeping or constitutive regulatory functions. Collectively, these results demonstrate that the CfWRKY family participates broadly in salt-stress adaptation, exhibiting multi-layered and dynamic regulatory behaviors during salinity exposure. For example, CfWRKY57, CfWRKY63, CfWRKY14, CfWRKY159, and CfWRKY152 were strongly induced under high salinity (150–200 mM), whereas CfWRKY17, CfWRKY127, CfWRKY138, and CfWRKY147 peaked at moderate salinity (50–100 mM). In contrast, CfWRKY11, CfWRKY182, and CfWRKY138 showed slight repression under high salt, while CfWRKY5, CfWRKY78, and CfWRKY140 maintained relatively stable expression levels.
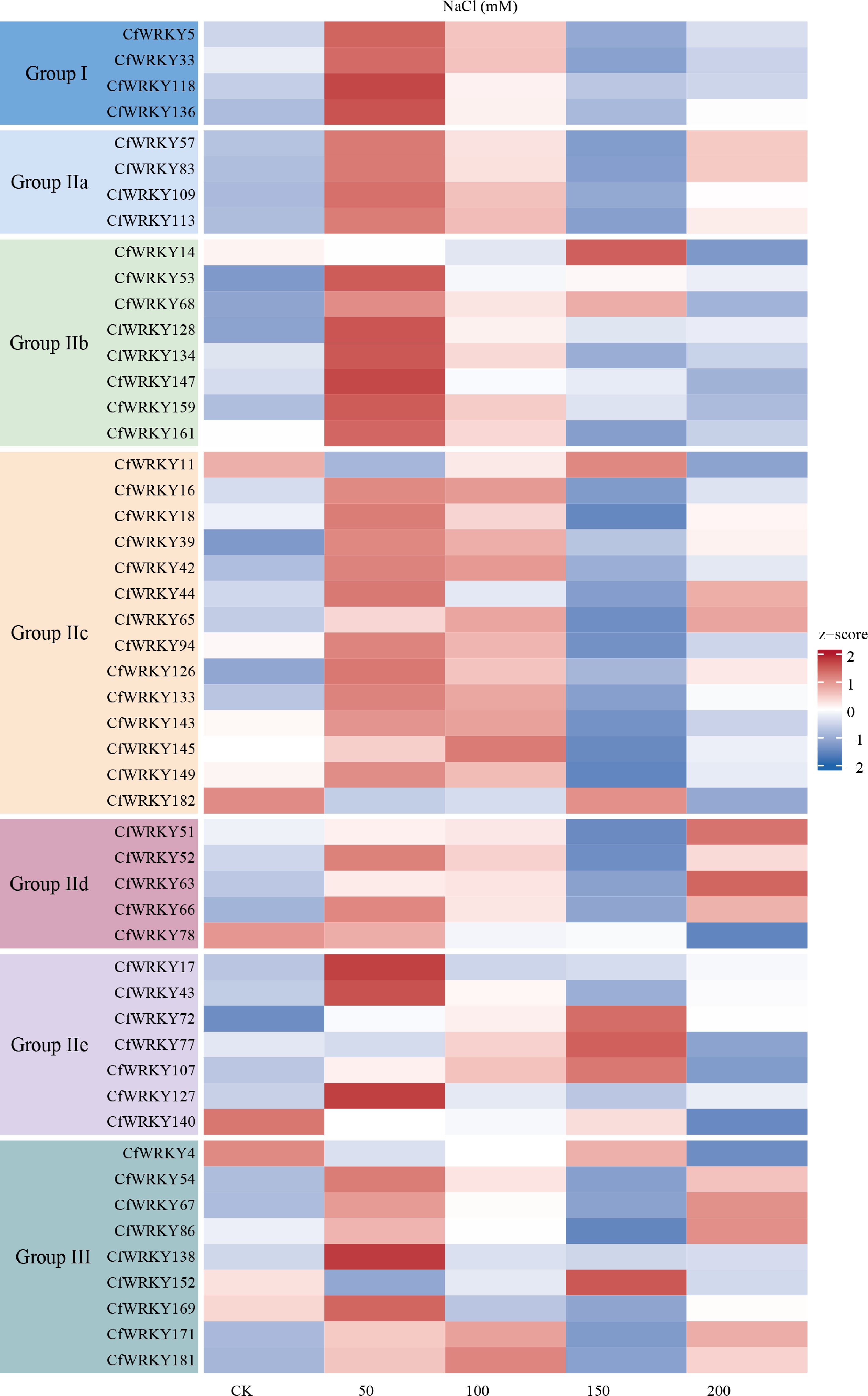
Figure 6.
Heatmap of WRKY transcription factor (TF) gene expression in C. fungigraminus under salt stress. Gene expression levels were derived from RNA-seq data and transformed as log2(TPM + 1) for visualization. The x-axis shows control (CK) and NaCl treatments at 50, 100, 150, and 200 mM with biological replicates; the y-axis lists all 51 WRKY family members. The color scale represents relative expression from low (blue) to high (red), and genes are organized by phylogenetic group, as indicated by colored blocks on the left.
Drought rehydration responsive co expression modules involving CfWRKY genes
-
To complement the salt-induced expression patterns of CfWRKY, we further examined their regulatory roles under drought stress and subsequent rehydration using a coexpression network framework. A total of 123 coexpression modules were identified. Module–trait association analysis revealed that 31 modules were significantly correlated with drought and/or rehydration stages (|r| ≥ 0.5 and FDR < 0.05; Fig. 7a), indicating extensive transcriptional reprogramming during water-deficit stress and recovery. The module–trait association heatmap showed that most drought-responsive modules were strongly positively correlated with drought treatments, indicating that these modules were markedly induced under water-deficit conditions. In contrast, their expression levels rapidly decreased after rehydration, suggesting a potential involvement in activating metabolic and repair-related pathways during recovery. Across all drought-associated modules, 9,774 genes were identified, including 27 WRKY transcription factors. Hierarchical clustering and module assignment confirmed clear modular organization of the transcriptome (Fig. 7b).
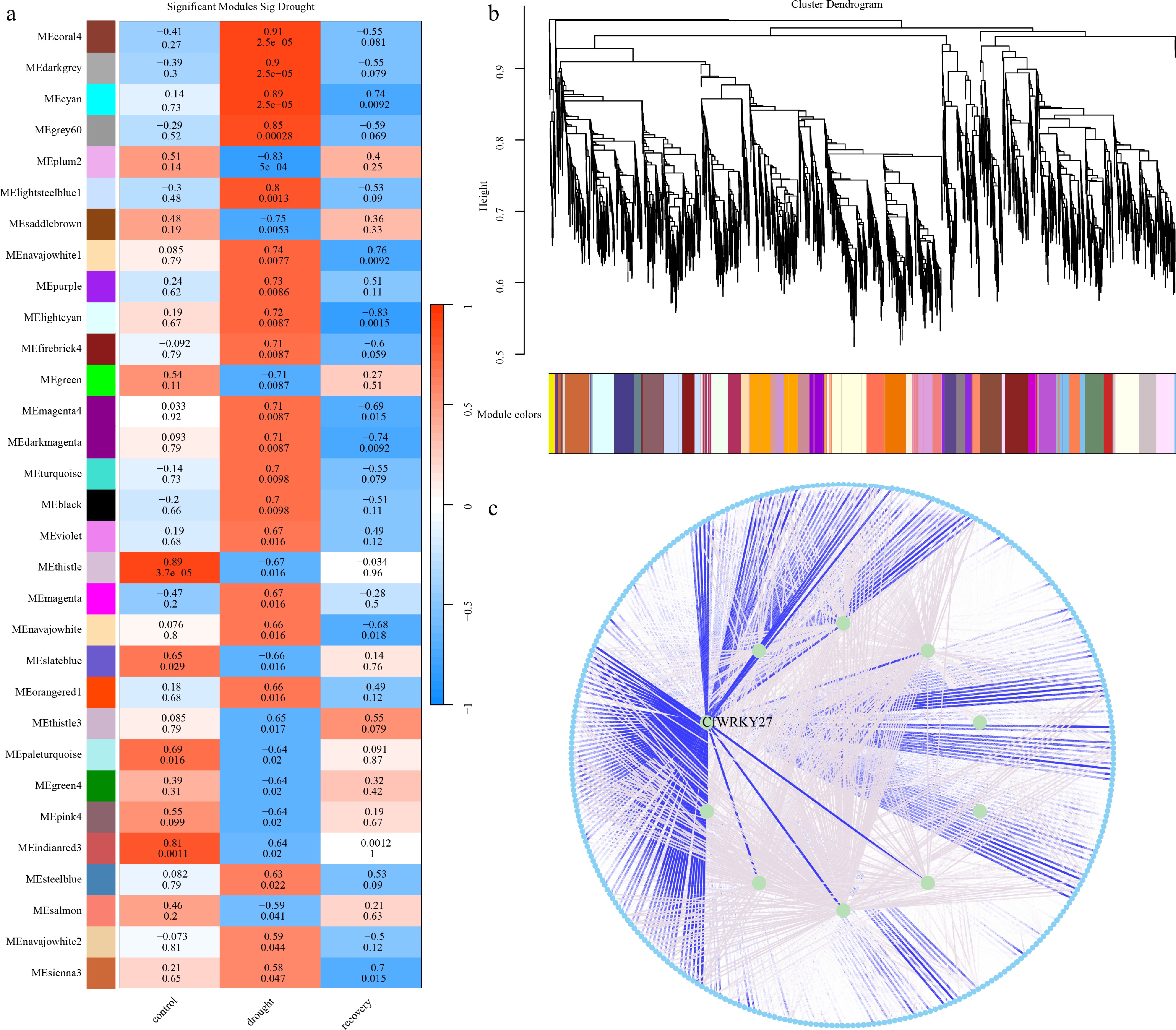
Figure 7.
WRKY-associated drought modules and hub network in C. fungigraminus. (a) Module–trait correlations under control, drought, and recovery conditions, showing drought-responsive modules. (b) Gene clustering dendrogram and WGCNA module assignment. (c) Drought-responsive coexpression network highlighting the hub transcription factor CfWRKY27, which displays strong connectivity with stress-related genes.
Among the drought-responsive modules, the cyan module comprising 344 genes showed a characteristic induction under drought stress followed by a gradual return towards baseline after rewatering. Notably, this module contains a WRKY transcription factor, CfWRKY27, which exhibited a high module eigengene connectivity (kME = 0.942), ranking among the top genes within the module. CfWRKY27 also displays extensive coexpression with stress- and metabolism-related genes (Fig. 7c), suggesting a central regulatory role in drought–rehydration responses. Functional enrichment analysis demonstrated that cyan-module genes are significantly enriched in pathways related to phosphate metabolic regulation, ubiquitin-mediated protein degradation, and autophagy, highlighting their involvement in cellular homeostasis and stress response (Supplementary Fig. S2). Collectively, these findings suggested that CfWRKY27 acts as a potential hub regulator mediating drought adaptation and post-stress recovery in C. fungigraminus.
-
In this study, we identified 182 WRKY genes in C. fungigraminus, a number substantially higher than that reported in sugarcane (53)[50], another Poaceae species but with an autopolyploid origin. To facilitate downstream analyses, CfWRKY genes were designated as CfWRKY1–CfWRKY182 according to their chromosomal positions. Compared with other allotetraploid crops, this number is lower than that in Brassica napus (287)[51] and tetraploid cotton (Gossypium hirsutum, 239; Gossypium barbadense, 213)[52], reflecting distinct gene family expansion patterns among Poaceae, Brassicaceae, and Malvaceae. These results suggest that the markedly larger WRKY repertoire in C. fungigraminus is likely associated with its allotetraploid genomic architecture, which retains duplicated homeologs more effectively than autopolyploid genomes.
Although polyploidization shaped the genome of C. fungigraminus, the WRKY gene family exhibited nearly balanced retention between the two subgenomes (A: 95 genes; B: 87 genes), suggesting that both subgenomes have preserved similar functional capacity for stress response regulation. Despite this balance at the subgenome level, these members were unevenly distributed across the 14 chromosomes, with Chr06B harboring the highest number of members, whereas Chr05A and Chr05B contained the fewest. Such non-uniform distribution in other plant species likely reflects lineage-specific chromosomal rearrangements and local chromatin accessibility that influences gene retention patterns[53]. In C. fungigraminus, a total of 136 duplicated WRKY pairs were identified, all of which originated from segmental duplication, with no evidence of tandem duplication. Similar expansion patterns have been reported in C. purpureus[20] and barley[54], indicating that polyploidization and large-scale chromosomal duplication rather than proximal duplications were the potential primary forces driving WRKY expansion in Poaceae. Given that C. fungigraminus is an allotetraploid species[4], the prevalence of segmental duplication likely reflects the legacy of ancient WGD events that shaped its genome. Nearly all CfWRKY paralogs exhibited Ka/Ks ratios below 1, indicating strong purifying selection[55], suggesting that most WRKY family members were maintained to preserve essential regulatory functions during evolution, which might be associated with stress adaptation, metabolism, and developmental regulation.
Furthermore, synteny analysis showed extensive one-to-one and one-to-many orthology between CfWRKYs and WRKYs in maize and pearl millet, with 75% of CfWRKYs maintaining syntenic anchors with ZmWRKYs[56] and 77% with CaWRKYs[18]. In contrast, only four CfWRKYs showed exclusive syntenic relationships with Arabidopsis, whereas 29 CfWRKYs were syntenic across Arabidopsis, rice, maize, and pearl millet[18]. Notably, the proportion of group III WRKYs in C. fungigraminus was 31.87% (58 members), which is lower than the 42% reported in Vaccinium[57], indicating lineage-specific evolutionary strategies in the WRKY subgroup expansion among monocots and dicots. These findings support the hypothesis that WRKY genes originated prior to monocot–dicot divergence and that core WRKY gene modules have been evolutionarily conserved across major angiosperm lineages. However, lineage-specific losses and expansions still occurred, reflecting adaptive pressures and genome innovation during grass evolution.
Phylogenetic analysis classified the CfWRKY family into three major groups and five subgroups, a pattern consistent with previous studies in pea, asparagus, cucumber, and maize[54,58,59]. Group III contained the largest number of CfWRKY members (57; 31.32%), followed by subgroup IIc (43) and Group I (23), consistent with our phylogeny. Variations in exon–intron organization were also evident, ranging from two to nine exons, suggesting structural plasticity and possible alternative splicing diversity, similar to patterns observed in Vaccinium[57] and other dicot lineages.
Motif analysis revealed conserved WRKY DNA-binding elements (motifs 1 and 2) in nearly all CfWRKY proteins, reinforcing core functional conservation. Subgroup-specific motif architectures were also conserved; for example, IId and IIe WRKYs in C. fungigraminus shared characteristic motif combinations similar to those reported in peanut, suggesting functional homogeneity within these subgroups. Meanwhile, the presence of WRKYGKK variants in several CfWRKYs, consistent with reports in cucumber and sorghum suggests that adaptive amino acid replacement may fine-tune DNA-binding specificity. A few CfWRKYs (CfWRKY89 and CfWRKY167) lacked a complete C2H2 zinc-finger motif, a feature also reported in sugarcane WRKYs, suggesting possible pseudogenization or annotation uncertainty[50]. The Q→K substitution introduces a positively charged residue, which may enhance electrostatic interaction with the DNA phosphate backbone or alter local WRKY-domain conformation, leading to subtle yet biologically meaningful shifts in target recognition and binding affinity. Previous reports demonstrated that WRKYGKK variants may increase DNA-binding affinity toward distinct cis-elements[59,60], implying that such structural diversification may drive functional divergence in stress-responsive pathways.
Cis-regulatory element enrichment further demonstrated the presence of ABRE, MBS, MeJA, and SA responsive sites in CfWRKY promoters, similar to findings in Forsythia[61], maize[56], peanut[62], and tobacco[63], revealing deep integration of WRKYs into ABA- and JA-mediated signaling cascades. Interestingly, ABRE sites were dominant, suggesting the crucial role of CfWRKYs in ABA-dependent stress responses. A relatively low frequency of TC-rich repeats indicated that CfWRKYs may prioritize abiotic stress defense vs biotic stress responses, consistent with the adaptive history of grasses in fluctuating drought and soil salinity environments. Taken together, these results indicate that while CfWRKYs maintain the hallmark WRKY structural identity, motif diversification, intron variation, and cis-element evolution jointly contributed to functional specialization, allowing this transcription factor family to respond efficiently to environmental fluctuations and developmental demands.
Salt stress is a major abiotic factor limiting plant growth and productivity. Transcriptome profiling revealed that the majority of CfWRKY genes responded to NaCl treatment, highlighting their central role in salinity tolerance. Interestingly, among the salt-responsive CfWRKYs, 35 genes responded robustly to moderate salt stress (50–100 mM), while eight genes showed peak expression only at high salinity (150–200 mM). Importantly, the two groups of salt-responsive WRKYs were largely non-overlapping, suggesting a division of labor between WRKY modules responsible for moderate-stress acclimation vs acute high-salt defense. A similar stepwise WRKY activation pattern was previously observed in asparagus[64], supporting the hypothesis that WRKY-mediated salt tolerance operates through tiered transcriptional networks. In addition to salt stress, WGCNA analysis revealed that CfWRKY27 acts as a potential hub gene in drought–rehydration responses, displaying strong connectivity with stress- and metabolism-related genes. This suggests that CfWRKY27 may coordinate stress recovery and homeostasis pathways, consistent with WRKY-mediated drought rescue mechanisms reported in other cereal crops. Taken together, salt- and drought-responsive WRKY modules indicate that CfWRKYs act in both acute stress defense and post-stress restoration, supporting their role as master regulators in stress resilience of polyploid C4 grasses.
These findings also echo results from maize and rice, where specific WRKYs function as hubs to orchestrate salinity signal transduction, ion homeostasis, ROS detoxification, and osmoprotectant biosynthesis pathways[56]. CfWRKYs enriched in ABRE and MBS motifs may participate in ABA-dependent stress alleviation, while JA-responsive WRKYs may regulate cross-talk between growth and defense pathways. These patterns underscore an evolutionarily conserved WRKY strategy of orchestrating hormone signaling and transcriptional modulation to achieve stress tolerance.
Genes showing highly induced expression under severe salt stress represent promising candidates for functional characterization and breeding. Future studies could employ CRISPR knockout, promoter dissection, yeast one-hybrid assays, and DAP-seq/ChIP-seq to identify direct WRKY target genes and illuminate chromatin-level regulatory mechanisms. Ultimately, these WRKY regulators hold promise for engineering salinity-tolerant C4 biomass grasses and improving resilience in high-salinity ecological restoration systems.
-
We present the first comprehensive WRKY atlas in C. fungigraminus, integrating genome-wide identification, phylogeny, motif and gene-structure analyses, duplication history, cis-regulatory profiling, and stress-responsive expression. The CfWRKY repertoire comprises 182 members with uneven chromosomal distribution and expansion primarily driven by segmental duplication under purifying selection. Conserved WRKY motifs coexist with subgroup-specific variants (e.g., WRKYGKK), suggesting fine-tuning of DNA-binding properties. Promoter enrichment of ABRE, MBS, and JA-related elements link CfWRKYs to hormone-mediated stress signaling. Salt-gradient RNA-seq uncovers a tiered, largely non-overlapping activation of WRKY modules at moderate vs high salinity, while WGCNA highlights CfWRKY27 as a drought–rehydration hub. These findings provide valuable omics-based insights into WRKY-mediated stress tolerance in forage grasses and offer promising candidate genes for improving salinity and drought resistance in Giant Juncao used for forage production and ecological restoration.
-
The authors confirm contributions to the paper as follows: study conception and design: Luo L, Lu G, Xiao J, Lin D, Zhu F; data collection: Chen Y, Liu H, Zhang Y, Tan F, Ke S, Yi C; analysis and interpretation of results: Luo L, Chen Y; draft manuscript preparation: Luo L; supervision and project administration: Zhu F, Lin Z, Gu L, Lu G, Xiao J, Lin D. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are available in the National Genomics Data Center (NGDC) under the BioProject accession number. These data were derived from the following resources available in the public domain: https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA051202. The processed data are also available on the EGDB website (https://engrass.juncaodb.cn/download.html). The code used in this study is publicly available on GitHub (https://github.com/2201494814/Giantjuncao_wrky).
-
This research was funded by the Major Science and Technology Project of the Xinjiang Uygur Autonomous Region "Research and Development of Key Technologies for High-Yield and High-Quality Juncao Cultivation and Comprehensive Utilization in Arid and Barren Lands" (2024A03009-5), the Science and Technology Program of the Xizang Autonomous Region "Evaluation and Demonstration of Juncao Varieties in High-Altitude Cold and Arid Regions" (XZ202501ZY0021), the National Key Research and Development Program of China (2024YFC3407200), and the National Natural Science Foundation of China (32570656, 32370582). We thank the Functional Chemical Genomics Core Facility of Fujian Agriculture and Forestry University Metabolomics Center for automation assistance. We also thank Associate Researcher Jing Zhou for providing the drought transcriptome data of Giant Juncao.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/grares-0026-0002.
- Supplementary Table S1 Genomic features of 182 CfWRKY genes in C. fungigraminus.
- Supplementary Table S2 Protein features and chromosomal distribution of the identified 182 CfWRKYs.
- Supplementary Table S3 Information on the predicted secondary structure of CfWRKY proteins.
- Supplementary Table S4 Ka/Ks analysis of CfWRKY collinearity gene pairs.
- Supplementary Fig. S1 Distribution of cis-acting regulatory elements in the promoters of CfWRKY genes.
- Supplementary Fig. S2 GO enrichment analysis of WRKY family genes in Cenchrus fungigraminus.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Luo L, Chen Y, Zhang Y, Liu H, Tan F, et al. 2026. Genome-wide identification of WRKY transcription factors in Giant Juncao (Cenchrus fungigraminus) and their transcriptional responses to salinity and drought stresses. Grass Research 6: e009 doi: 10.48130/grares-0026-0002 

Genome-wide identification of WRKY transcription factors in Giant Juncao (Cenchrus fungigraminus) and their transcriptional responses to salinity and drought stresses
- Received: 16 December 2025
- Revised: 27 January 2026
- Accepted: 09 February 2026
- Published online: 14 April 2026
Abstract: Giant Juncao (Cenchrus fungigraminus) is a perennial C4 forage and bioenergy grass with high biomass productivity and strong tolerance to adverse environments. However, the transcriptional regulatory mechanisms underlying its stress adaptation remain poorly understood. WRKY transcription factors are key regulators of plant stress responses, but their genomic features and functional potential in this species have not been systematically investigated. Here, we identified 182 CfWRKY genes and classified them into three major groups based on phylogenetic relationships. Comprehensive analyses of their chromosomal distribution, gene structures, conserved motifs, cis-regulatory elements, and duplication patterns revealed that segmental duplication, alongside whole-genome duplication, was the primary force driving WRKY family expansion. Transcriptome analysis under salt treatments showed diverse and dynamic expression responses, with several CfWRKY genes exhibiting strong induction and early responsiveness. Weighted gene co-expression network analysis under drought and rehydration conditions further identified 31 stress-related modules and 27 CfWRKY candidates potentially involved in water-deficit adaptation. This study presents the first genome-wide characterization of the WRKY family in Giant Juncao and provides integrative insights into their evolutionary patterns and stress-responsive regulatory roles. Our findings offer valuable candidate genes and a molecular basis for functional genomics and breeding of stress-resilient forage and energy grasses.