








-
In the era of rapid globalization and industrialization, the world is confronted with unprecedented challenges in the domains of energy and environment. Therefore, the escalating energy demand, coupled with the detrimental effects of environmental pollution, necessitates the exploration of sustainable solutions[1,2]. Among the various alternatives, sustainable carbon materials (SCMs, here referring to carbon materials derived from renewable or waste sources and synthesized through low-energy, low-emission processes), such as zero-dimensional (0D) fullerenes, one-dimensional (1D) carbon nanotubes (CNTs), two-dimensional (2D) graphene, and three-dimensional (3D) activated carbon (AC) and carbon aerogels (CAs), have emerged as promising candidates due to their exceptional physicochemical attributes and multifaceted applications in environmental remediation and energy conversion systems[3−7].
In the context of environmental pollution, organic pollutants, metal ions, and radionuclides pose severe threats to ecosystems and human health[8−12]. However, SCMs can effectively capture and eliminate pollutants in water or air through mechanisms such as physical adsorption, chemical sorption, chemical reduction, chemical complexation, etc., providing a practical and feasible solution. For instance, AC has long been utilized for water purification, while graphene-based materials have shown great potential in removing heavy metal ions and organic pollutants with high efficiency[13−15]. The ability to tailor the surface chemistry and pore structure of SCMs allows for the development of highly selective and efficient adsorbents, which can significantly improve the effectiveness of environmental remediation processes[16−18].
In the energy sector, the transition towards clean and renewable energy sources is imperative to reduce reliance on fossil fuels and mitigate climate change. SCMs play a crucial role in this transition, particularly in the CO2 reduction reaction (CO2RR), where SCMs serve as catalysts to convert carbon dioxide, a major greenhouse gas, into valuable chemicals and fuels such as methane, methanol, and syngas[19−21]. This not only helps reduce the concentration of greenhouse gases in the atmosphere but also provides a sustainable pathway for energy production. Moreover, oxygen evolution reaction (OER), and hydrogen evolution reaction (HER) are key processes in water electrolysis for hydrogen production and in metal-air batteries. Accordingly, carbon-based electrocatalysts, with their low cost, high activity, and stability, have become attractive alternatives to precious metal catalysts, thereby driving the advancement of these energy technologies[22−24].
Furthermore, the integration of artificial intelligence (AI) techniques with carbon material research has opened new avenues for the design, synthesis, and optimization of SCMs. AI algorithms can analyze vast amounts of data to predict the structure-property relationships of SCMs, guide experimental research directions, and accelerate the discovery of novel materials with tailored properties[25−27]. This synergistic approach of combining AI with materials science can significantly enhance the development of SCMs for energy and environmental applications[28].
In this context, SCMs hold immense promise for addressing the pressing challenges in the energy and environmental fields. Their unique properties, coupled with the potential of AI technologies, make them key players in the pursuit of sustainable development. As shown in Fig. 1, this work aims to provide a comprehensive overview of the construction, toxicity, and detection, as well as the structure and application of SCMs in environmental pollution control and energy conversion, and it also explores the role of AI in advancing the field of SCMs, highlighting the current state of research, challenges, and future perspectives. By delving into these aspects, it is believed that this review seeks to contribute to the ongoing efforts towards the development and implementation of SCMs for a greener and more sustainable future.
-
Carbon materials (CMs) can be classified into 0D carbon (nanometer-sized fullerenes and carbon quantum dots [CQDs], etc.), 1D carbon (CNTs, carbon nanofibers [CNFs], etc.), 2D carbon (graphene, graphdiyne, etc.), and 3D carbon. Correspondingly, various classical strategies have been utilized for synthesizing CMs, mainly including bottom-up and top-down approaches. Bottom-up synthesis constructs CMs atom-by-atom or molecule-by-molecule from precursors (e.g., hydrocarbons) to achieve precise atomic control, mainly including carbonization (or pyrolysis), chemical vapor deposition (CVD), the templating methods, etc. Top-down synthesis refers to deconstructing bulk carbon sources (e.g., graphite, biomass) into smaller nanostructures to introduce defects, pores, or reduce size, mainly involving exfoliation and etching. Besides, some newly green synthetic technologies have also emerged in the synthesis of CMs very recently, such as biomass-based hydrothermal carbonization, CO2 utilization, Flash Joule heating (FJH), and so on. Each synthesis method exhibits advantages and limitations in fabricating CMs across dimensionalities (Table 1). In this section, the principles of various synthesis strategies for CMs and cutting-edge advances since 2021 are systematically introduced, followed by structural engineering strategies (e.g., pore, heteroatom doping) to enhance their performance in energy storage (e.g., CO2RR, OER, HER, etc.) and environmental remediation (e.g., pollutant removal).
Table 1. Comparison of various synthesis strategies for CMs
Methods Year developed Advantages Limitations Products Refs. Carbonization (pyrolysis) 1980s Low cost, scalable, tunable porosity via precursors (e.g., biomass, polymers) High energy input (> 600 °C), Limited crystallinity (ID/IG > 1.5), amorphous morphology AC, biochar, carbon NPs, carbon foam [42,729] Hard templating 1990s Precise pore size control (2–50 nm), ordered architectures, high surface area Toxic template removal (HF/NaOH), low yield, high cost Ordered mesoporous carbon, CNTs, 3D carbon frameworks, CAs [489,730] Soft templating 1990s Template-free, self-assembly, tunable mesopores (2–10 nm) Poor thermal stability (< 400 °C), surfactant residue contamination Mesoporous carbon spheres, CNFs [731,732] CVD 1960s
(modern: 2004)High crystallinity (ID/IG < 0.1), atomic-level thickness control High cost, slow growth rate, substrate limitations Graphene, CNTs, carbon fibers (CFs), fullerenes, graphdiyne [53,733] Mechanical exfoliation 2004 Simple (e.g., Scotch tape), minimal chemical defects, preserves intrinsic properties Low yield (< 5%), non-uniform flake sizes, labor-intensive Graphene nanosheets, few-layer graphdiyne [734] Chemical exfoliation 2008 High yield, solution-processable, scalable Defect generation (ID/IG >1.0), requires harsh oxidants (H2SO4/KMnO4), residual functional groups Graphene oxide, CQDs [735] Electrochemical exfoliation 2012 Mild conditions (room temp), tunable surface groups, high purity (> 95%) Limited to conductive precursors, low throughput, solvent dependency Few-layer graphene, fluorinated CNTs [62,736] Chemical etching 2010 Defect engineering (edge sites), hierarchical porosity, enhanced surface reactivity Over-etching risks, corrosive reagents (HNO3/KOH) Holey graphene, porous CNFs [63,65] Physical etching 2015 No chemical waste, plasma/ion beam precision (nm-scale), high reproducibility High equipment cost, slow processing, limited to thin films Patterned graphene, nanoperforated carbon membranes [66,737] HTC 2001 Eco-friendly (aqueous, < 200 °C), spherical morphology control Low graphitization (ID/IG > 1.5), limited surface area (< 500 m²/g) Carbon microspheres, hydrochar from biomass, CQDs [68,69] CO2 utilization 2015 Carbon-negative process, converts waste CO2 Energy-intensive (600–1,000 °C), low efficiency (< 30% conversion) CNTs, graphene, fullerenes, PC [70] FJH 2019 Ultrafast (< 1 s), energy-efficient (~0.1 kWh/kg), upcycles waste precursors Limited to conductive precursors, non-uniform heating in bulk samples Turbostratic graphene, 3D PC, CQDs [64] Synthesis strategies
Bottom-up synthesis
Carbonization (or pyrolysis)
-
The carbonization technique, as one of the most widely used ways to produce AC, involves carbonizing carbon-rich organic precursors at high temperatures (500–1,500 °C) in an inert atmosphere. During the carbonization process, volatile molecules (e.g., CO, CO2, and alkanes) are removed to produce porous carbon (PC) structures. Many studies show that various carbon precursors (like organic polymers, biomass, metal organic frameworks (MOFs), etc.) and carbonization conditions can regulate the morphology and structure of CMs (such as porosity, doping, and nanostructure). For example, Zhang et al. fabricated a sodium polyacrylate-based PC by direct carbonization (Fig. 2a)[29]. The results showed that at the carbonization temperature of 500 °C under an Ar atmosphere, the pores of the PC exhibited irregular morphologies with many large macropores (> 1 µm), while at 600 °C, the pores became denser, with some being smaller than 500 nm (Fig. 2b, c). At the carbonization temperature above 800 °C, the pores in PC were mainly composed of large diameter pores (> 2 μm) (Fig. 2d). Additionally, they also found that compared to carbonized in Ar, the pore structure of PC in an air atmosphere exhibited more severe structural collapse (Fig. 2e). Zhu et al. synthesized a nitrogen-rich polymer and used it as a precursor[42]. After being activated with KOH, and doped with melamine and urea, the polymer precursor could produce nine carbonized derivatives by adjusting the carbonization temperature. Thanks to the synergistic effect of nitrogen- and oxygen-doped functional groups in its carbonized framework, the CMs (carbonized at 600 °C) exhibited a high adsorption capacity of 2,782 mg/g for gaseous iodine, and 1,854 and 867.55 mg/g for bromine and iodine in solution, respectively. Mo et al. synthesized nitrogen-doped porous carbon (NPC) via pyrolysis of biomass-derived carbon precursors co-treated with urea and ZnCl2 under N2 atmosphere[43]. As the carbonization temperature increased from 800 to 1,000 °C, the NPC-1000 samples exhibited significantly more medium-sized pores (~1 to 10 nm) and fine pores (< 1 nm) in microflake structures compared to NPC-800 and NPC-900. These pore characteristics induced a spatial confinement effect that favored a four-electron reduction pathway in the oxygen reduction reaction (ORR). Consequently, despite having the lowest contents of pyridinic nitrogen and graphitic nitrogen, the NPC-1000 sample demonstrated superior ORR activity, outperforming commercial Pt/C catalysts.
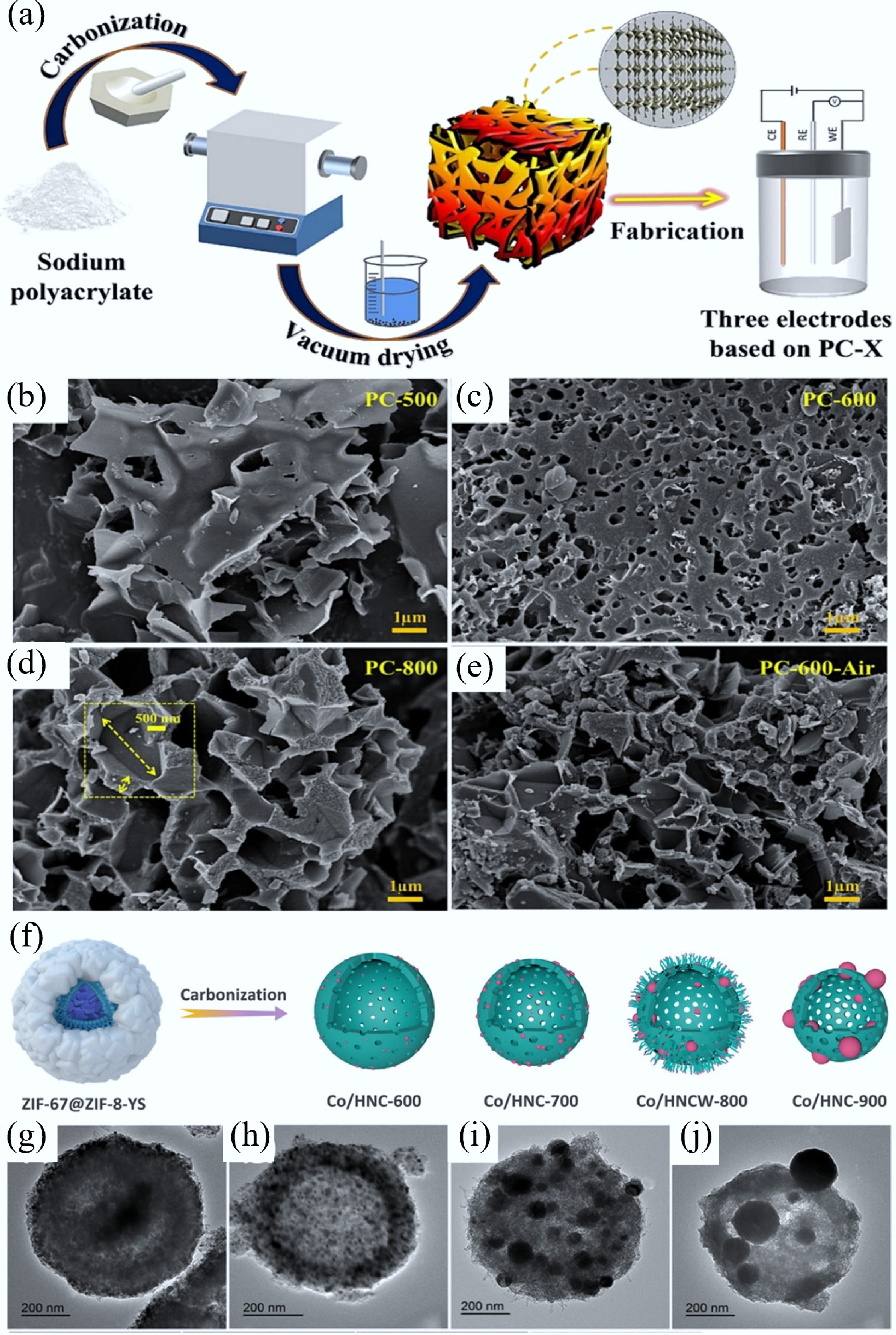
Figure 2.
(a) Synthesis process of sodium polyacrylate-based PC. SEM graphs of PC carbonized at (b) 500 °C, (c) 600 °C, and (d) 800 °C in an Ar atmosphere, and (e) PC-600 fabricated in air[29]. (f) Diagram of Co/HNC-x samples (where x denotes calcination temperatures: 600, 700, 800, and 900 °C), prepared by carbonizing the ZIF-67@ZIF-8-YS precursor for 2 h in N2, and the corresponding TEM images of (g) Co/HNC-600, (h) Co/HNC-700, (i) Co/HNCW-800, and (j) Co/HNC-900[46].
Apart from organic polymers and biomass precursors, MOFs have been recognized as superior sacrificial precursors for synthesizing CMs in environmental remediation and energy storage/conversion fields[44]. Their tunable chemical compositions and adjustable hierarchical structures enable precise control over heteroatom doping profiles (e.g., N, P, S) and porous architectures in the derived CMs, thereby providing prominent advantages for adsorption capacity, catalytic activity, and electrochemical performance[45]. Through designing the composition of MOF precursors (e.g., organic ligands, metal nodes, coordination manners, and carbonization conditions), various MOF-derived CMs, including metal-free pure carbon or heteroatom/metal/metallide-doped CMs, can be obtained. Zhu et al. constructed cobalt nanoparticles (NPs)/nitrogen-doped carbon composite materials at different carbonization temperatures using hollow ZIF-67 microspheres as precursors (Fig. 2f)[46]. With the increase in carbonization temperature, the particle size of this carbon composite decreased slightly (Fig. 2g−j). At 800 °C, a certain number of carbon nano-whiskers were anchored on the surface of the hollow sphere, forming a unique hierarchical hollow structure. After further phosphating, the cobalt metal/cobalt phosphides/nitrogen-doped carbon composites were obtained, which showed excellent OER electrocatalytic performance. Ling et al. reported a K-defect-rich PC material with 12-vacancy-type defects without N doping by direct carbonization of K+-confined MOFs at 1,100 °C[47]. Remarkably, the resulting K-defect-rich PC presented an ultrahigh CO Faradaic efficiency (FE) up to 99% at −0.45 V, much better than that of N-doped carbon for electro-catalytic CO2 reduction. Zhang et al. fabricated a hierarchical 1D/3D NPC by carbonizing a composite of Zn-MOF-74 crystals that were grown in situ on commercial melamine sponge (MS) for electrochemical CO2RR[48]. The unique spatial architecture of 1D/3D NPC enhanced CO2 adsorption capacity and facilitated electron transfer from the 3D nitrogen-doped carbon framework to the 1D carbon domains, thereby improving the reaction kinetics of CO2RR.
Generally, the carbonization method offers low-cost, scalable production of CMs with moderate to high conductivity, broadband absorption, high specific surface area (SSA, 100–3,000 m2/g), tunable porosity, and rich surface O/N groups via temperature and precursor tuning. Defects or O/N groups in CMs reduce conductivity and photoluminescence but increase active sites, enhancing catalytic activity (e.g., ORR outperforming Pt/C) and pollutant adsorption. However, this method requires high temperatures (500–1,500 °C), leading to substantial energy consumption; the products have limited crystallinity (ID/IG >1.5), limiting high conductivity. Moreover, strict dependence on inert atmospheres complicates the process, and biomass-derived carbon yields are only 47%–50%. Though CO2 conversion and other techniques mitigate environmental impact, its sustainability lags behind greener alternatives like hydrothermal methods and flash Joule heating.
Templating method
-
The template method, including hard templating and soft templating, was first adopted to obtain ordered mesoporous carbons (OMCs) by removing various pre-existing sacrificial templates in precursors. As early as 1999, OMCs were first prepared using ordered mesoporous silicates as hard templates (Fig. 3a)[30]. The carbon precursor was filled inside the mesopores, polymerized, and then carbonized. After removing the silica scaffold by etching, OMCs that replicated the silica meso-channels were produced. Following that, OMCs were also synthesized by a self-assembly approach, i.e., soft templating (Fig. 3b). Soft templates (e.g., supramolecular aggregates) and carbon precursors are co-assembled into 3D-ordered meso-structures. After eliminating the supramolecular aggregates by regulating the calcination temperature, meso-carbonaceous framework was preserved. In the past five years, CMs with various morphologies and pore structures have been synthesized for environmental and energy applications. For example, Zhu et al. reported atomically dispersed Ce sites embedded in a hierarchically macro–meso–microporous N-doped carbon catalyst (Ce SAS/HPNC) by a hard-template approach (Fig. 3c)[49]. During the synthesis process, Ce-doped ZIF-8 precursors with a SiO2 ball embedded in every face of the rhombododecahedron were first prepared. After carbonization and acid leaching, SiO2 was removed to produce a hierarchically macro–meso–microporous N-rich PC. At 1,150 °C in flowing N2, an atomically dispersed Ce SAS/HPNC catalyst was finally formed. As shown in Fig. 3d−f, the as-formed Ce SAS/HPNC catalyst exhibited a porous-rhombododecahedral single particle morphology with holes at an average of 167 nm. Benefiting from the atomic dispersion of Ce atoms and a unique 3D hierarchical ordered porous architecture, the Ce SAS/HPNC possessed dramatic catalytic activity due to enhanced mass transport and exposing more active sites. Through a simple soft template polymerization and activation strategy, Zhang et al. synthesized polypyrrole (PPy)-based N, S co-doped porous CNTs[50]. During the synthesis, rod-like soft templates were first formed by using methyl orange (MO) and FeCl3 as templating agents. Then, pyrrole monomer was polymerized on MO-FeCl3 template to form PPy nanotubes, while surfactant hexadecyl trimethylammonium bromide was added to modify the branched PPy nanotube structure. Subsequently, the activated N, S co-doped porous CNT was produced after pyrolysis using sulfur as a dopant and ZnCl2 as an activator. As a result, this N, S co-doped porous CNT electrode exhibited high energy storage capacity, excellent rate performance, and superior cyclic stability.
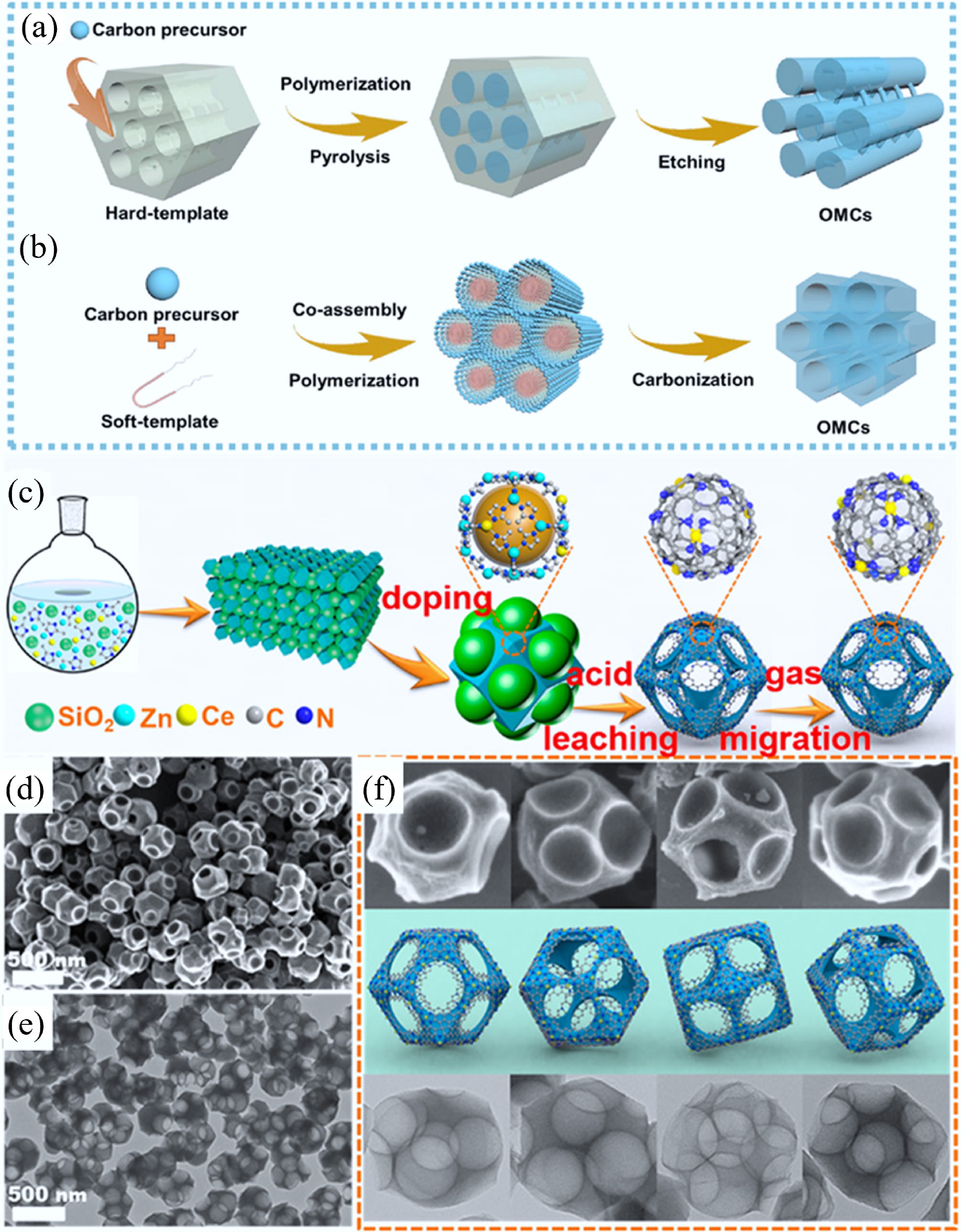
Figure 3.
Scheme for the synthesis of OMCs by using (a) ordered mesoporous silicates as hard-templates and (b) supramolecular aggregates as soft-templates[30]. (c) Fabrication procedure of a hierarchically macro–meso–microporous N-doped carbon (Ce SAS/HPNC) catalyst by using SiO2 as hard-templates, (d) SEM, (e) TEM, and (f) both images of Ce SAS/HPNC corresponding to the models from different angles[49].
The hard templating approach generally enables precise pore size control and ordered architectures, while the soft templating approach creates tunable mesopores, enabling hierarchical porosity. Both templating approaches exhibit relatively low conductivity owing to their amorphous or partially graphitic structures. Compared to carbonization-derived CMs, the ordered porosity and low defect density of templated CMs reduce light scattering, thus improving optical absorption efficiency. Templated CMs typically possess high SSA and tunable surface chemistry, with soft templating allowing heteroatom doping (e.g., N, P) during synthesis to enhance surface reactivity. The ordered porosity and high SSA of templated CMs facilitate mass transfer and atomic-level active site exposure, promoting pollutant capture (e.g., heavy metals, dyes), and enhancing CO2RR electrocatalytic activity. However, the low conductivity of templated CMs limits their suitability for HER. Meanwhile, multi-step synthesis processes, high costs, high chemical waste generation, and energy-intensive procedures limit their scalability and sustainability.
CVD
-
The principle of the CVD technique to synthesize CMs is that carbon source gases (i.e., methane and acetylene) decompose on the surface of catalysts (such as metal NPs) at high temperatures (700–1,200 °C), and carbon atoms grow and assemble on the substrate to form nanoscale CMs. This technique was originally developed in the 1960s and 1970s and used in the production of carbon fibers (CFs) and carbon nanofibres[51,52]. In 1996 and 2008, CVD was separately reported as a potential method for large-scale synthesis and production of CNTs[53] and graphene. Studies have shown that different carbon sources, catalyst substrates, and reaction conditions would greatly influence the types, morphology, and structure of CMs. Wang's group employed ring-rich polystyrene (PS) vapor and chain-rich polyethylene (PE) vapor as carbon precursors to study the growth mechanisms of carbon nanomaterials (CNMs) on biochar substrates[54,55]. When PS vapor was used as a carbon source, three types of CNMs, i.e., bulk amorphous carbon, monolayer amorphous carbon, and CNFs, were produced. Conversely, only bulk amorphous carbon and CNFs were found by using PE vapor. Meanwhile, they found that the substantial carbon deposition on biochar surfaces was attributed to stable C-C bonds formed between PS/PE, and biochar. The presence of aromatic rings in PS vapor resulted in the monolayer amorphous carbon growth, while the presence of sp2 carbon in PE vapor leads to CNFs growth. Chen et al. developed a space-confined-CVD method to prepare hard CMs with graphite-like carbon domains filled into the micropores of AC[55]. During the synthesis process, benzene carbon sources tended to be adsorbed onto the pore wall inside AC due to the van der Waals force between π orbital on the carbon planes and the electronic density in the benzene. At 700 °C, benzene was pyrolyzed into a flat graphene layer. Through changing the space confined-CVD residence time and the temperature of post-heat treatment, the interlayer spacing and size of graphitic carbon were facilely adjusted. Zhu et al. investigated the wrinkling/folding process of graphene by using ethylene as a carbon precursor and single-crystal Cu–Ni(111) foils as substrates[56]. They found that when the growth temperature was above 1,030 K, the folds would form during the subsequent cooling process. Through rationally controlling the growth temperature between 1,000 and 1,030 K, they successfully synthesized large-area, fold-free, single-crystal single-layer graphene films. Jia et al. fabricated a mixed-dimensional CF/CNTs@Fe3C@Fe3O4 heterostructure by a continuous process including carbonization of melamine foam (MF), synthesis of carbon foam (CF)/FeOOH, and the in-situ growth of CNTs in a C2H2 atmosphere by the catalytic chemical vapor decomposition (Fig. 4a)[57]. The SEM images showed that the as-formed material exhibited good 3D interconnected networks without the collapse of skeleton (Fig. 4b). And the closer SEM results suggested that this material possessed a rough surface and many flocculent CNTs growing around the entire skeleton (Fig. 4c–d). Especially, the content of CNTs in the CF/CNTs@Fe3C@Fe3O4 samples could be effectively enhanced by increasing the pyrolysis time of the C2H2 carbon source. It is worth noting that although CVD yields high-quality graphene/CNTs, the impacts of spatially non-uniform reactions, catalyst instability, and energy-intensive precision control on the mass production of graphene/CNTs merit serious consideration.
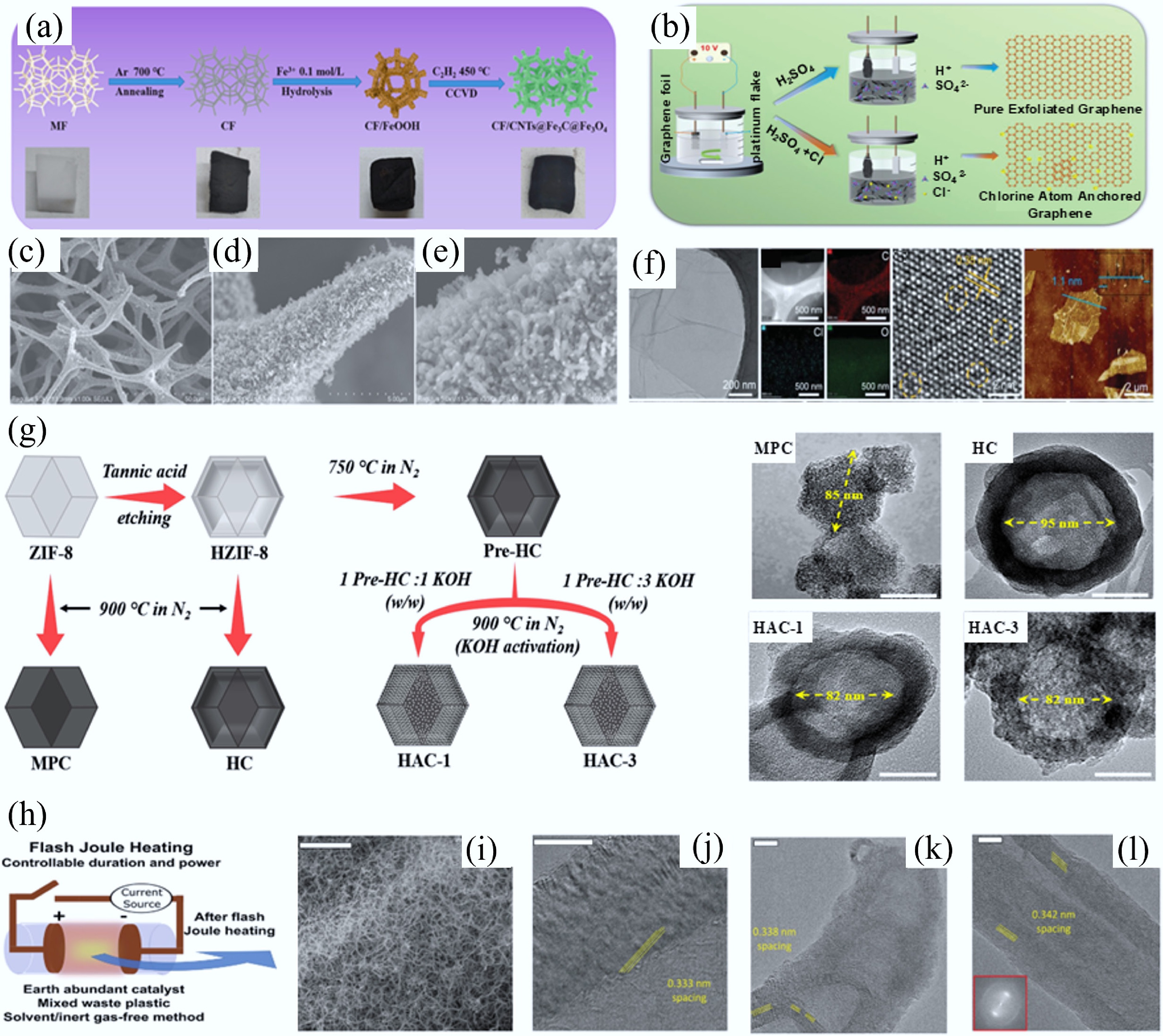
Figure 4.
(a) Synthesis of CF/CNTs@Fe3C@Fe3O4 via in-situ growth of CNTs via catalytic CVD of C2H2 at 450 °C. (b)–(d) SEM images of CF/CNTs@Fe3C@Fe3O4 with different magnifications[57]. (e) Preparation process of chlorine-doped graphene (working electrode: graphite flake; counter electrode: platinum foil; electrolyte: sulfuric acid with chloride salt; applied potential: +10 V) and (f) morphology and structure characterizations[62]. (g) Synthetic pathways and corresponding morphologies of MPC (microporous carbon from ZIF-8 carbonization), HC (hollow carbon from tannic acid-etched ZIF-8 carbonization), HAC-1 and HAC-3 (hollow AC from HC activated with KOH/pre-HC weight ratios of 1 and 3, respectively, at 900 °C in N2)[63]. (h) Preparation of flash 1D materials via FJH using waste polymer as starting material (~3,000 K temperatures generated in 0.05–3 s) and (i)−(l) different morphologies of flash 1D materials[64].
Unlike templated or etched CMs that have high-porosity CMs (SSA > 1,000 m2/g), the CVD CMs typically have non-porous, dense structures (e.g., graphene, CNTs) with relatively low SSA (< 500 m2/g). Notably, CVD CMs exhibit exceptional conductivity (> 104 S/m) due to high crystallinity, outperforming carbonized or templated CMs. Meanwhile, CVD CMs, with tunable band gaps (e.g., graphene quantum dots) and high transparency (e.g., atomically thin CVD graphene), are ideal for optoelectronic applications. Different from etched or templated CMs, which are featured with abundant defects and edge sites, the CVD CMs have smooth, pristine conductive surfaces with few active sites, which is beneficial for their regeneration when used as electrode materials. The high conductivity of CVD CMs makes them perfect candidates for HER. Engineered defects or heteroatom doping can effectively enhance their CO2RR/ORR/OER catalytic activity. However, the CVD method requires high temperatures (500–1,500 °C), metal catalysts, and toxic precursors (e.g., methane), restricting its scalability and sustainability.
Top-down synthesis
Exfoliation
-
Exfoliation techniques are mainly used to synthesize graphene and its derivatives. Generally, exfoliation begins with the bulk graphite. With the help of external forces, the van der Waals forces between graphite layers can be broken, thus allowing graphene to be stripped from the graphite[58]. Based on the external force, exfoliation can be categorized into mechanical exfoliation, chemical exfoliation, and electrochemical exfoliation, etc. Mechanical exfoliation was first reported to synthesize graphene in 2004 by Novoselov and Geim, and they were awarded the Nobel Prize in Physics in 2010[59]. In their work, the few-layer graphene sheets were separated from graphitic flakes by repeatedly peeling bulk graphite crystals with Scotch Tape. This method is simple and reliable, but the yield is low and not suitable for reproducibility or large-scale production of graphene. Chemical exfoliation obtains graphene by using chemical reagents (such as strong acids, strong oxidants, intercalating agents, etc.) to break the interlayer bonding forces in graphite. A representative technique of chemical exfoliation is the Hummers method[58]. During the preparation process, acids and oxidizing agents intercalate into graphite to expand the interlayer spacing and form graphene oxide (GO). Under ultrasonication or mechanical stirring, GO is exfoliated into the single- or few-layer graphene. Finally, graphene is produced through the chemical reduction of GO using a reductant (e.g., hydrazine). This method is simple, convenient, and scalable for the mass production of graphene, but it may introduce structural defects and degrade its electronic properties for certain applications. Electrochemical exfoliation is another fast, cost-effective, environmentally benign, and scalable way to prepare graphene. By applying voltage to a graphite electrode, ions in the electrolyte (e.g., (NH4)2SO4) are driven to insert and expand the graphite layers. Meanwhile, the generated gas (H2/O2) further impairs interlayer bonds, thereby mechanically exfoliating graphite into few-layer graphene with low defects[60]. In 2008, Liu et al. adopted this method to obtain exfoliated product with an average length of ~700 nm, a width of 500 nm, and a thickness of 1.1 nm, demonstrating complete exfoliation from graphite to graphene nanosheets[61]. In the past five years, exfoliation techniques have been adopted to synthesize various doped graphene. For example, Liu et al. prepared the high-quality and solution-processible chlorine-doped graphene nanosheets by the electrochemical exfoliation technique[62]. In the exfoliation process, a graphite flake was used as the working electrode with dilute sulfuric acid containing chloride salt as the electrolyte (Fig. 4e). By applying a +10 V potential for 10 min, ultrathin and rugged 2D chlorine-doped graphene nanosheets with a large lateral size of ~10 µm have been synthesized. Morphological and structural characterization results indicated that chlorine-doped graphene nanosheets exhibited high structural uniformity, homogeneous distributions of carbon, chlorine, and oxygen elements, a high degree of crystallinity, abundant defects, and a topographic thickness of 1.1 nm (Fig. 4f).
Graphene prepared by the three exfoliation methods shows different structural and performance features. Mechanically exfoliated graphene has fewer pores, excellent conductivity (retaining its pristine sp2 structure), and superior optical properties from layer-dependent absorption, but lacks surface functionality. Chemically exfoliated graphene forms pores via sheet restacking, experiences significant conductivity loss due to oxidative defects, exhibits tunable optical transitions, and contains abundant surface O groups. In contrast, electrochemically exfoliated graphene exhibits moderate conductivity and porosity, fewer surface O groups, and retains decent optical absorption. For environmental remediation, chemical exfoliation outstrips mechanical exfoliation (inert surfaces) owing to rich O groups and porosity, while electrochemical exfoliation balances adsorption capacity and stability. For catalysis, mechanically exfoliated graphene with pristine basal planes excels in HER, while chemically/electrochemically exfoliated graphene, with defect-mediated activity, performs better in CO2RR/ORR/OER. Owing to aqueous processing, moderate yield, low chemical usage, and energy efficiency, electrochemical exfoliation outperforms the other two in scalability and sustainability. Overall, while superior to carbonization/templating in targeted graphene synthesis, these methods face inherent efficiency-performance tradeoffs.
Etching
-
The etching strategy, involving chemical and physical etching, mainly relies on selectively removing certain substances from the CMs, thereby regulating their microstructures (such as pore size, defects, morphology, etc.) to obtain specific properties. Chemical etching primarily employs strong acids, strong bases, or oxidants to react with CMs, selectively removing amorphous carbon or introducing pores. For example, Kim and his colleagues reported hollow ACs with a hollow nanoarchitecture and high SSA via a chemical etching strategy[63]. They first adopted tannic acid as a selective etching agent to create a hollow cavity in the center of zeolite imidazolate framework-8 (ZIF-8) (Fig. 4g). Then, hollow carbon was obtained after a carbonization process. Furthermore, through the reaction between KOH and carbon atoms (6KOH + 2C ↔ 2K + 3H2 + 2K2CO3), hollow AC-x (where x = 1 or 3 corresponded to the KOH/pre-hydrochar weight ratio) with more nanopores and an increased SSA was generated. These well-designed hollow activated CMs, featuring a hollow nanoarchitecture and high BET SSA, show great potential in the energy storage field. Instead of the usage of traditional strong acids, Zhang et al. utilized a selenic-acid-assisted etching strategy to synthesize Co0.85Se1−x@C electrocatalysts toward OER[65]. The selenic acid, with weak acidity, can only partially etch the ZIF-67 framework and serves as a selenium source. After subsequent calcination, a carbon layer-coated Co0.85Se1−x with abundant Se vacancies was obtained under an inert atmosphere. The unique structure of Co0.85Se1−x@C improved the catalytic activity for OER through increasing the number of active sites, enhancing the conductivity, and reducing reaction barriers for the formation of intermediates.
The physical etching method utilizes high-energy particle bombardment (i.e., plasma) or gas-phase (i.e., CO2) reactions to obtain porous CMs. Liu et al. utilized the N2 plasma-etching strategy to regulate defects and N species in CNTs. By controlling the plasma-etching time, the vacancy defects, C-O, pyrrolic N, and graphitic N could be rationally designed[66]. The ID/IG (from 0.56 to 0.94) and C–O contents (from 0.07% to 0.44%) of N-CNTs rose with increasing etching time. Zheng et al. developed a mild CO2-etching and carbonization strategy to generate abundant closed pores in starch-derived hard carbon[67]. During the CO2-etching process, abundant open micropores were created in the AC matrix via the reaction (CO2 + C → 2CO). Due to CO2 etching, the pore diameter in hard carbon microspheres increased from 3.82 to 4.86 nm, with corresponding pore volume rising from 0.045 to 0.117 cm3/g.
Both chemical and physical etching generate hierarchical porosity via selective atomic removal, but chemically etched CMs typically have larger SSA than physically etched ones. Etching-induced defects reduce conductivity, yet optimized defect density accelerates charge transfer kinetics in electrocatalysis. Enhanced light scattering and absorption from increased defects and porosity in etched CMs benefit their photothermal applications. Chemical etching enriches surfaces with O/N groups and enables precise doping, while physical etching creates reactive edges with few heteroatoms. Generally, chemically etched CMs show strong affinity for organic contaminants and excel in CO2RR (O-mediated active centers) and ORR (defect sites) but underperform in HER due to low conductivity; physically etched ones show moderate ORR/HER activity via edge exposure. Both lack scalability and sustainability: chemical etching uses corrosive reagents (H2SO4/KOH), risking harm; physical etching faces energy/cost barriers. Etching excels at tailoring surface sites for specific CM catalysis but lags behind templating in pore uniformity and CVD in electrical properties.
Emerging sustainable synthetic technologies
-
Very recently, some green synthetic techniques have also emerged to synthesize CMs, involving biomass-based hydrothermal carbonization (HTC) method[5,68,69], CO2 utilization[70], FJH[64], etc. Particularly, biomass-based HTC has become a research hotspot over the past decade for synthesizing CMs from types of biomass waste due to its eco-friendliness, non-toxicity, and energy-efficiency. Considering that glucose is the most abundant sugar in biomass and the main product of lignocellulose acid hydrolysis, Ischia et al. systematically investigated the fundamentals of the HTC conversion over several operating conditions (180–270 °C and 0–8 h) by using a 1.1 M glucose solution as a carbon precursor[69]. Results revealed that hydrochar changed notably with time only at 180 °C, and the solid yield stabilized at 47%–50% with the carbon content of 67%–70% above 180 °C. Meanwhile, hydrochars were found to comprise distinctive nano/microspheres whose size distribution correlated with operating conditions. These materials mainly exhibited an amorphous structure and gradually evolved toward graphitization with intensified HTC severity. Hessien[68] adopted a microwave-assisted HTC method to prepare hydrochar from pomegranate peel waste at 200 °C for 1 h with a peel to water mass ratio of 1:10. The results confirmed that the as-formed amorphous hydrochar exhibited a porous structure, showing potential as an adsorbent for methylene blue (MB) dye removal.
HTC generates CMs with moderate porosity and amorphous nature, leading to relatively low conductivity and broad optical absorption. HTC-derived CMs inherit rich O-groups from biomass precursors, which is beneficial for polar pollutant adsorption and facilitating CO2RR via improved CO2 adsorption, but impede ORR/HER due to poor conductivity and insufficient graphitic active sites. The scalability/sustainability of HTC for CMs fabrication lies in its mild conditions (< 250 °C), use of biomass feedstocks, and minimal energy/chemical usage.
CO2 utilization has emerged as a sustainable strategy for synthesizing CMs via CO2 conversion, offering a dual benefit of resource utilization and CO2 emission mitigation. Yuan et al. reported the transformation of CO2 and ethane into CNTs using earth-abundant metals (Fe, Co, Ni) as catalysts at 750 °C[70]. Through regulating the H2/CO ratios, this strategy not only generated a rapid rate of CNTs' production, but also yielded valuable syngas. In the absence of CO2, direct pyrolysis of ethane underwent rapid deactivation, while the participation of CO2 contributed to 30% of CNT formation. Meanwhile, CNTs generated from Co- and Ni-based catalysts had a diameter of 20 nm with a micrometer length, whereas those using Fe-based catalysts were bamboo-like. This study establishes a breakthrough CO2-to-CNT conversion platform, demonstrating dual environmental and economic advantages through carbon-negative synthesis of high-performance nanotubes for energy storage applications.
CO2 utilization-derived CMs exhibit tunable porous structures via CO2 activation and moderate conductivity attributed to defect-rich frameworks. Their optical absorption is broad but less tunable due to inherent defects and porosity. Surface groups derived from CO2, such as carbonate species, enhance CO2 affinity, conferring superiority in CO2RR through a direct carbon-negative pathway and tailored active sites. However, limited graphitic domains result in modest ORR/HER activity. While this method achieves net carbon negativity by directly sequestering CO2, outperforming others in emissions mitigation, its high energy demands (600–1,000 °C) and low conversion efficiency constrain scalability compared to HTC or FJH.
Recently, FJH has emerged as a rapid, sustainable, and scalable method for solvent-free carbon production. This technique uses electricity and resistance to generate extreme temperatures (~ 3,000 K) in milliseconds (0.05–3 s), converting low-value waste into high-value products without solvents. Using the FJH technique, Wyss et al. successfully demonstrated the conversion of waste plastic into flash 1D materials and hybrid graphitic 1D/2D materials with controllable morphologies through adopting a variety of earth-abundant catalysts (Fig. 4h)[64]. Through FJH parameter tuning, fiber-like products were formed (Fig. 4i). TEM images showed that morphologies of flash 1D materials included ribbon-type and bamboo-like nanofibers, as well as multi-walled nanotubes (Fig. 4j–l). The as-produced flash 1D materials outperformed commercial CNTs in many properties.
Compared to other methods, FJH-derived CMs integrate ultrahigh processing efficiency with superior performance. Explosive degassing during synthesis generates ultrahigh SSA and hierarchical pores (micro/meso/macro), surpassing templated carbons in pore complexity. By rapidly repairing defects within the sp2 framework, FJH-derived CMs achieve near-metallic conductivity—far outperforming pyrolysis, HTC, and exfoliation methods while rivaling CVD-derived carbons. Simultaneously, tunable optical absorption/emission is enabled through in situ heteroatom doping (N, P, and S). Beyond superior pollutant capture via rapid adsorption kinetics enabled by hierarchical pores, these FJH CMs also surpass counterparts in ORR/OER/HER catalysis through balanced graphitic domains and atomic-scale active sites. Doped variants further demonstrate competitive CO2RR performance. Though templated CMs offer finer pore uniformity and CVD higher crystallinity, FJH dominates in harmonizing high SSA, extremely high conductivity, and millisecond processing—establishing it as the most scalable and energy-sustainable strategy for high-performance carbons.
Structure engineering
-
To advance the application of CMs in environmental protection and energy sectors, recent efforts have focused on modifying their physicochemical and structural properties through strategies such as pore structure and morphology optimization, heteroatom doping, surface modification, and composite formation.
Pore structure
-
The large SSA and hierarchical porosity of CMs serve as fundamental determinants for their deployment in sustainable energy and environmental remediation. Pore structures are classified into three categories based on IUPAC standards: micropores (< 2 nm) that enable molecular sieving effects and maximize active sites for small-molecule adsorption/charge storage, mesopores (2–50 nm) that accelerate ion diffusion kinetics, and macropores (> 50 nm) that act as mass transfer highways to minimize diffusion resistance[71]. Owing to the synergistic effects of multiscale porosity, hierarchically porous carbons have been established as a strategic material platform for sustainable energy and environmental remediation, thereby driving cutting-edge research in targeted pore engineering. For example, Wang & Mu constructed a hierarchically porous waste soybean meal AC via three steps of pre-curing, carbonization, and alkali activation[72]. A honeycomb-like skeleton structure was first obtained via hydrogen bonding crosslinking by pre-curing. Then, macropores were maintained by carbonizing the porous skeleton, while mesopores and micropores were created by alkali activation. The resulting material possessed an ultra-high SSA (3,536.95 m2/g), as well as a hierarchical macro–meso–microporous structure. Consequently, this hierarchical porous structure exhibited extremely high adsorption capacities for MB (3,015.59 mg/g), MO (6,486.30 mg/g), and mixed dyes (8,475.09 mg/g), and fast adsorption kinetics for MB and MO (~30 min). Its highly efficient adsorption performance for dyes was mainly ascribed to its excellent hierarchically porous structure and the synergy of multiple adsorption mechanisms containing π–π stacking, pore-filling, electrostatic interaction, and hydrogen bond interaction.
Heteroatom doping
-
Heteroatom doping can adjust the energy band structure of CMs owing to the differences in the size and the electronegativity of carbon and heteroatoms (N, S, B, P, etc.), thereby enhancing the electron transfer ability[73]. Meanwhile, the introduction of heteroatoms can provide adsorption or catalytic active sites while enhancing hydrophilicity through polar functional groups, thus optimizing the surface chemical characteristics of CMs[74]. Hence, heteroatom-doping has been reported as a versatile modification strategy that remarkably improves the functional capabilities of carbon-based materials, particularly in environmental remediation, chemical transformation processes, and sustainable energy technologies. For example, Chang et al. reported a 3D N/P/S-tri-doped nanoflower with highly branched CNTs bifunctional catalyst by a simple self-assembly pyrolysis method (Fig. 5a)[34]. Density functional theory (DFT) calculations revealed that the synergistic effects between the heterogeneous active sites play a vital role in improving the catalytic activity of the catalyst. The simultaneous introduction of N/P/S can facilitate the redistribution of electron density at the catalyst interface. Meanwhile, the appropriate P-doping not only improves the electronic conductivity of the substrate but also promotes the charge transfer in the OER/ORR process. The accelerated durability experiments displayed that there was only a 9 mV decay in E1/2 after 5,000 cyclic voltammetry (CV) cycles for ORR and only a tiny potential change of 8 mV after 5,000 CV cycles for OER. The Co/SP-NC cathode assembled zinc-air batteries exhibited excellent cycling performance (> 280 h). Co/SP-NC showed no X-ray diffraction (XRD) peak change post ORR/OER stability tests, confirming good structural stability. Notably, strong C–Co bonds enhanced structural integrity via dopant anchoring, but higher P-doping reduced stability and cyclic performance. Recently, the enhancement of catalytic activity of CMs via the synergistic effect of heteroatom-doping and transition metal centers has been widely explored for energy and environmental applications. Specifically, Yang et al. synthesized hollow nitrogen-doped carbon capsules supporting iron single-atom sites, which were functionalized with amidoxime groups[35]. Due to the synergistic effect of amidoxime groups and site-isolated FeNx centers, this electrode material can decrease the uranium concentration in seawater to below 0.5 ppb from ~3.5 ppb with an extraction capacity of ~1.2 mg/g within 24 h.
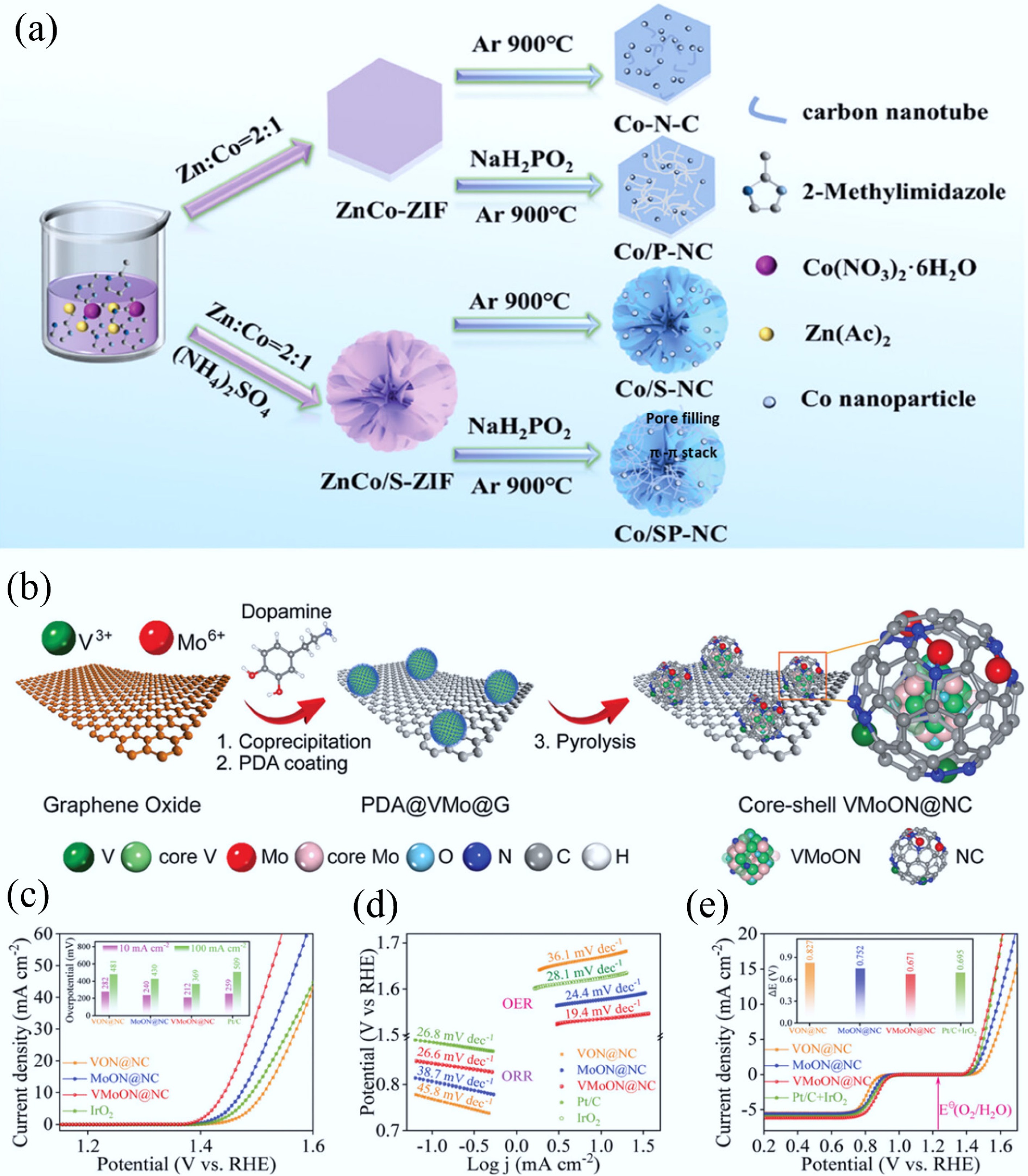
Figure 5.
(a) The preparation process of 3D N/P/S-tri-doped nanoflower with highly branched CNTs bifunctional catalyst[34]. (b) The preparation of the core–shell VMoON@NC 3D electrode architecture. (c) OER polarization curves of the as-obtained catalysts. (d) Tafel plots for ORR and OER catalysts. (e) Overall polarization curves within the ORR and OER potential window of the Mo SACs[33].
Surface modification
-
Surface modification strategies, including functional group grafting, surface metal, or quantum dot loading, have been reported as effective approaches to enhance the properties of CMs, thus propelling their application in energy storage systems and environmental remediation. Roy et al. fabricated carboxyl and amino groups modified GO adsorbent, and its adsorption performance was evaluated for MB and methyl orange (MO)[75]. The results showed that carboxyl-functionalized GO selectively adsorbed MB at pH 9, whereas amino-functionalized GO selectively adsorbed MO at pH 2. Meanwhile, amino/carboxyl functionalized GO extracted 80.9% MO at pH 2 and 92.6% MB at pH 9 from real textile effluent. The removal efficiency of amino- or carboxyl-functionalized GO still maintained 91% MB and 88% MO after five cycles, respectively. This study demonstrated that the reasonable introduction of functional groups into CMs can effectively improve the selectivity for target pollutants.
Beyond functional group modification, the introduction of single-atom catalysts (SACs) presents unprecedented opportunities to boost the catalytic activity of CMs, thus enabling their breakthroughs in energy conversion and environmental governance. Balamurugan et al. reported a bifunctional catalyst featuring single Mo electroactive sites[33]. The catalyst comprised nanoscale (~2.3 ± 0.6 nm) vanadium molybdenum oxynitride cores encapsulated by N-doped carbon shells (Fig. 5b). During pyrolysis, single Mo atoms were released from the core and anchored to electronegative N-dopant species in the graphitic carbon shell. The resulting Mo SACs excelled as active OER sites in pyrrolic-N and as active ORR sites in pyridinic-N environments. Owing to the coexistence of both pyrrolic-N and pyridinic-N dopants in the graphene shell, these Mo SACs exhibited the lowest overpotential, the lowest Tafel slope, and the smallest potential difference in comparison with other catalysts, demonstrating their superior bifunctional (OER and ORR) responses (Fig. 5c–e). Likewise, Poudel et al. designed a unique nanostructure via the coupling of Co single-atoms and Ni NPs on a pyri-N- enriched carbon network substrate[76]. According to DFT calculations, the atomic dispersion of Co single-atoms enables optimal exposure of active sites on the pyri-N dominated multidimensional carbon skeleton, while the synergistic effects with Ni NPs substantially suppress electron delocalization near metallic centers, which promotes the adsorption of oxygen intermediates, thus giving rise to a lower charge transfer barrier. Within this system, this hybrid catalyst presents superior OER/ORR activity.
Carbon-based composites
-
Carbon-based composite materials, mainly involving carbon-carbon, carbon-polymer, and carbon-metal oxide composites, have emerged as a more versatile and efficient alternative for sustainable energy generation and environmental remediation owing to the synergistic advantages of carbon and other functional components. For instance, Jain et al. prepared a hybrid GO/CNTs aerogel of GO and waste-derived CNTs for phenol adsorption[77]. The phenol adsorption efficiency of GO/CNTs aerogel was compared with that of GO, magnetic GO, and GO aerogel. The experimental data showed that the GO/CNTs aerogel exhibited the highest phenol adsorption efficiency of 204 mg/g. The mechanism analysis indicated that the higher adsorption capacity of GO/CNTs was related to the fact that the introduction of CNTs into the GO sheets can increase the interlayer distance of GO sheets, which leads to a higher BET surface area (539.2 m2/g) and richer pores (1.39 cm3/g), and more adsorption active sites as compared to the above GO-based adsorbents.
Due to their high stability and hydrophobic surfaces, CMs are frequently utilized to prepare carbon/metal oxide hybrid catalysts for applications in harsh environments. For instance, to avoid the dissolution or inactivation of first-row transition metal oxides at high proton concentrations, Yu et al. reported a carbon-decorated Co3O4@C electrode with excellent electrocatalytic OER activity and long-term stability in acidic conditions, which was supported by a hydrophobic carbon-based matrix[78]. This robust and scalable electrode exhibited excellent OER performance in 1 M sulfuric acid solution (pH < 0.1), maintaining a 10 mA/cm current density for > 40 h without appearance of performance fatigue. Besides, CMs, owing to their high conductivity and electric double-layer capacitance, are often combined with metal oxides that exhibit high pseudo-capacitance but weak electrical conductivity, thereby forming composites with complementary electrochemical properties. Lin's group fabricated a porous graphene nanosheets-CNTs@MnO2 film by combining porous graphene nanosheets, CNTs, and MnO2 nanosheets[79]. The pores on the graphene surface shorten the diffusion path of electrolyte ions, while the CNTs@MnO2 between graphene layers serve as 'spacers' to prevent graphene aggregation, ensuring interlayer ion transport. Meanwhile, the inclusion of CNTs improved the mechanical and conductive properties, while the MnO2 nanosheets grown on the CNTs underwent redox reactions, giving rise to pseudocapacitance and increasing the specific capacitance of the composite film. Consequently, this film displayed a gravimetric specific capacitance of 320 F/g and a volumetric capacitance of 275 F/cm3 at a current density of 1 A/g in 1 M Na2SO4 solution.
By virtue of their high electrical conductivity, rapid Faraday reactions, and high theoretical capacitance, pseudocapacitive-based polymers are frequently used in combination with double-layer capacitance-based CMs to further improve the overall capacitive performance. Noh et al. reported polyaniline-capped carbon nanosheet (CS) with high conductivity and porosity via vapor deposition polymerization[80]. During the preparation process, vaporized aniline monomers were slowly polymerized on the mesoporous carbon surface and partially filled the carbon pores, improving conductivity. The resultant composite exhibited efficient hybrid energy storage mechanisms integrating electric double-layer capacitance and pseudocapacitive behaviors. In a three-electrode system, the material delivered a high specific capacitance of 469.2 F/g at a current density of 0.5 A/g, which was ~3.7 times higher than that of mesoporous carbon alone.
-
The in vivo behavior and environmental fate of CMs are fundamental aspects that determine their potential risks to both human health and ecological systems. Once released into the environment or introduced into biological systems, CMs may undergo complex processes including absorption, distribution, metabolism, and excretion (ADME) within organisms, as well as transformation, migration, and accumulation in various environmental compartments[81].
The physicochemical properties of carbon nanomaterials, including surface functionalization, size, and structural defects, play a critical role in influencing these ADME profiles[82]. Surface functionalization can influence the in vivo distribution and accumulation of carbon nanomaterials by modulating their interactions with proteins. Specifically, appropriate surface modifications can reduce recognition by the reticuloendothelial system, thereby facilitating their clearance from the body[83]. Aggregation leads to a significant increase in particle size, which alters the ADME characteristics of carbon nanomaterials and results in their accumulation in key organs, exacerbating toxic effects[83]. Small-sized carbon nanomaterials typically exhibit improved dispersibility and stability, which may enhance their biocompatibility. However, their small size also facilitates cellular uptake, potentially increasing the risk of bioaccumulation. In addition, structural defects increase the susceptibility of carbon nanomaterials to attack by hydroxyl radicals, promoting oxidative degradation into smaller fragments[84]. These defect sites may also enhance their binding affinity with essential proteins, disrupting protein structure, and inducing potential toxic responses[32].
Moreover, the ability of carbon nanomaterials to enter food chains and undergo trophic transfer can lead to biomagnification and long-term ecological effects. A comprehensive understanding of these dynamic processes is essential for accurate risk assessment and the development of safer-by-design CNMs[82].
In vivo distribution and translocation
-
CNMs can be introduced into living organisms via multiple pathways, including intravenous injection for biomedical applications, as well as unintentional ingestion or inhalation resulting from environmental exposure. Once internalized, CNMs undergo a series of complex biological processes. However, due to their similarity in chemical composition to biological matrices, directly investigating their in vivo behavior remains a considerable challenge[82]. Among available techniques, labeling strategies remain the most reliable approach for tracking the biological fate of CNMs, including radiolabeling, conjugation with fluorescent probes, and doping with metal oxide NPs[82]. Radiolabeling can be achieved through skeletal incorporation of radioactive isotopes into the carbon framework, which minimizes interference with the physicochemical properties and biological behavior of the nanomaterials[85]. This approach enables long-term and trace-level tracking of their biodistribution and evaluation of their toxicological effects[85]. In addition, post-administration labeling strategies have been developed using DNA-conjugated gold NPs to selectively bind carbon nanomaterials after exposure[86]. This approach improves the accuracy of biodistribution analysis while minimizing the impact of surface modification. Notably, the exposure route of CNMs is a critical factor influencing their ADME characteristics, as different routes expose the CNMs to varying biological environments, such as protein composition and ionic strength, which may affect their initial morphology, surface charge, and colloidal stability, thereby influencing their biological behavior[81].
CNMs can enter biological systems through various routes, including intravenous injection, oral administration, and intratracheal instillation. These different exposure pathways can significantly influence the distribution and translocation of CNMs in vivo. For instance, CNMs administered via intravenous injection tend to primarily accumulate in the liver. As the largest solid organ in the human body, the liver receives approximately 13% of the total blood supply and features substantially reduced blood flow velocity, which allows sufficient time for the uptake and transformation of nanomaterials by hepatic cells[87]. The long-term fate of 14C-labeled few-layer graphene in mice following intravenous injection has been studied[85]. The results showed that smaller lateral graphene (SLG) primarily accumulated in the liver, whereas larger lateral graphene (LLG) enhanced the erythrophagocytic activity of Kupffer cells, leading to the elevated intracellular iron levels that initiated Fenton reactions, generating hydroxyl radicals responsible for the oxidative degradation of graphene into CO2 (Fig. 6a–c)[85]. Similarly, the biodistribution of 14C-labeled MWCNTs after intravenous administration indicated the liver as the primary organ of accumulation (Fig. 6d–e)[88]. In addition, orally administered CNMs can be absorbed through the gastrointestinal tract into systemic circulation and subsequently distributed to organs such as the kidneys, stomach, and liver. rGO delivered orally is reported to be distributed across multiple organs[89]. Furthermore, CNMs delivered through intratracheal instillation tend to accumulate in the lungs but may also be transported to the gastrointestinal tract via swallowing or mucociliary clearance, and potentially further redistributed to the blood, liver, and kidneys.
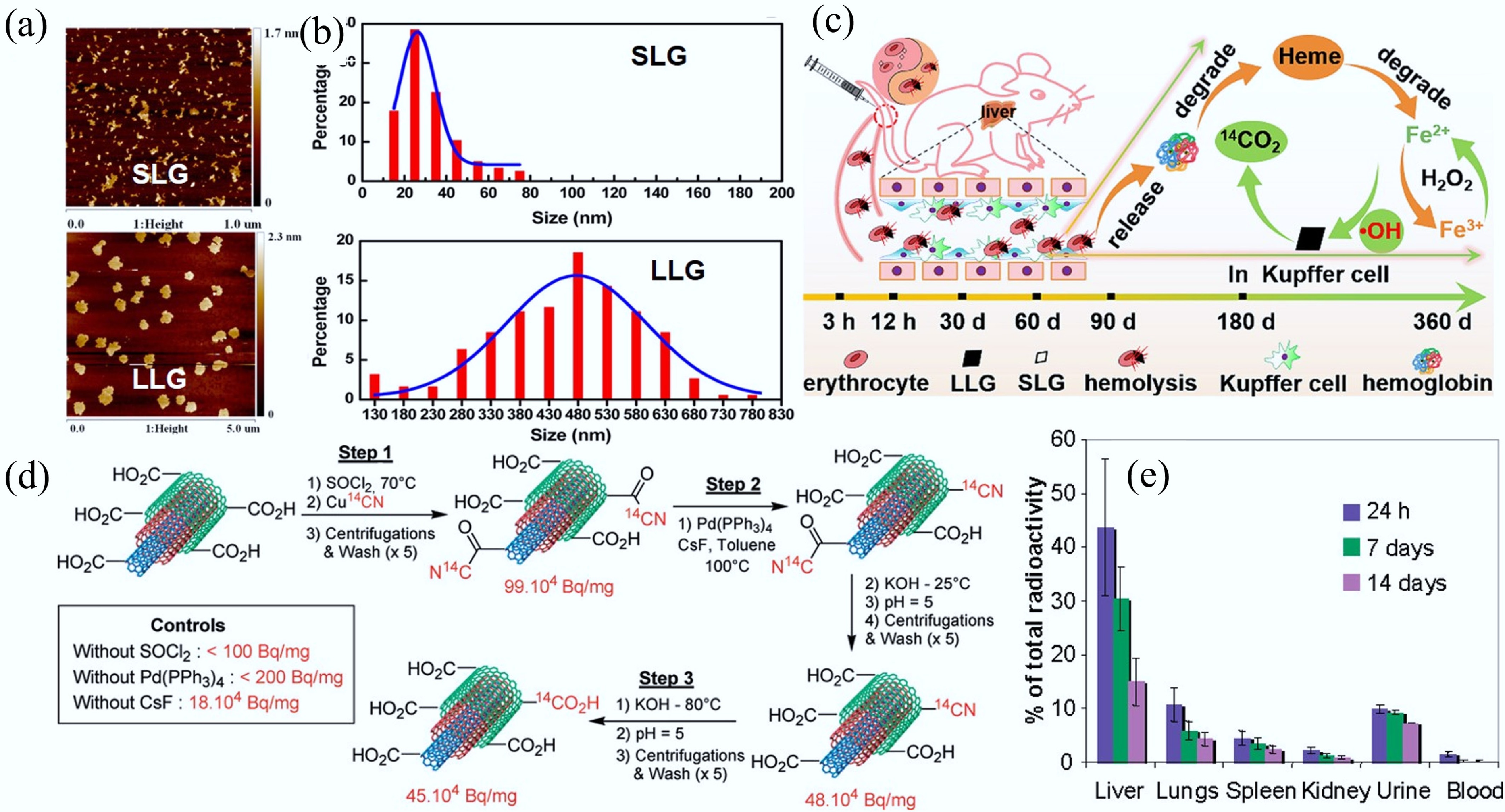
Figure 6.
(a) Representative atomic force microscope topography of SLG and LLG. (b) Histogram of SLG and LLG size distribution. (c) Schematic illustration of the process of LLG triggering the Fenton reaction in Kupffer cells[85]. (d) Preparation process of 14C-labelled multi-walled CNTs. (e) Biodistribution of intravenously administered 14C-labeled multi-walled CNTs at different time points after exposure. Each group contained six animals (n = 6)[88].
The biodistribution of CNMs is predominantly determined by their physicochemical properties, including lateral size and surface functionalization. Studies have shown that smaller graphene nanosheets demonstrate enhanced tissue penetration compared to larger nanosheets. Lu et al. investigated the distribution of graphene with different lateral sizes in zebrafish and found that large-sized graphene predominantly accumulated in the digestive tract (98.3% ± 1.3%), whereas small-sized graphene was detected in both the intestines and the liver, indicating that small-sized graphene could penetrate the intestinal wall to enter epithelial cells and blood[90]. Surface modification also significantly influences in vivo distribution. Polyethylene glycol (PEG)-coating on GO reduces retention in the liver, lungs, and spleen compared with uncoated GO, which is attributed to steric hindrance that facilitates clearance from these organs[91].
Environmental transport and transformation
-
Soil is a complex mixed system composed of various components and environmental substances, and serves as an important sink for CNMs. Soil properties can significantly influence the aggregation and transport of CNMs. When CNMs penetrate into the soil, they can interact with soil minerals to form nanomaterial-mineral aggregates, leading to their immobilization[92]. Under neutral conditions, GO readily binds to positively charged goethite through electrostatic attraction, promoting heteroaggregation with minerals. Dong et al. investigated the transport and transformation of 14C-labeled graphene in soil. The results showed that red soil with higher iron oxide content exhibited an adsorption capacity for graphene 10.2 times greater than that of black soil[92]. The presence of iron oxides and hydrogen peroxide triggered Fenton reactions that degraded graphene into CO2, facilitating its removal from the soil. Clay minerals can interact with CNMs, thereby affecting their mobility. Three typical clay minerals, namely kaolinite, montmorillonite, and illite, inhibit the transport of GO in quartz sand primarily through positively charged edge sites, with kaolinite showing the greatest effect due to its large edge surface area[93]. In addition, ionic composition in soil can influence the transport behavior of CNMs. Xia et al. used quartz sand to study the transport behavior of rGO in porous media. They found that the retention mechanisms of rGO were highly dependent on the type of cations present. Divalent cations (Ca2+) promoted retention through cation bridging, whereas monovalent cations led to retention via deposition in the secondary energy minimum[94].
In aquatic environments, CNMs are subject to various physical, chemical, and photochemical processes that influence their environmental behavior and fate. CNMs exhibit a certain degree of hydrophobicity, and their colloidal behavior is governed by the physicochemical properties of the surrounding medium (such as pH, ionic strength, and natural organic matter), which ultimately influence their aggregation and sedimentation[95]. At lower pH levels, carboxyl groups on the edges of GO become protonated, increasing its hydrophobicity and promoting aggregation. In contrast, under alkaline conditions, the deprotonation of carboxyl and phenolic hydroxyl groups enhances the colloidal stability of GO in solution[96]. In addition to pH, the ionic strength and valency of metal cations also affect the environmental behavior of CNMs. Wu et al. found that divalent cations such as Ca2+ and Mg2+ were more effective than monovalent Na+ in promoting the aggregation of GO sheets, which was attributed to cross-linking through bridging interactions between divalent cations and edge functional groups on GO sheets[97]. The adsorption of natural organic matter onto CNMs can modify their surface chemistry by introducing hydrophilic functional groups, thereby enhancing their colloidal stability and altering their behavior in aqueous environments[96, 98]. In natural waters, photochemical transformation can be a major environmental fate pathway for CNMs. For instance, C60 exhibits strong light absorption and undergoes photodegradation with a half-life of approximately 19 h, leading to the formation of water-soluble products and eventual mineralization[99]. Similarly, carboxylated CNTs exposed to UVA irradiation undergo decarboxylation and generate surface functional groups and vacancies, which reduce surface potential and colloidal stability[100]. The behavior of CNMs in aquatic environments also influences their toxicity. Hu et al. found that hydration and visible light exposure altered the morphology of graphene, increased its surface negative charge and aggregation, and ultimately reduced its toxic effects on algal cells[101].
In atmospheric environments, CNMs are prone to aging induced by ambient air pollutants, including physical adsorption or condensation of contaminants and heterogeneous reactions with trace gases such as SO2 and NOX[102]. This aging process involves a reduction in disordered carbon and C-H functional groups in CNMs[102]. In a separate study, Liu et al. simulated atmospheric aging by examining the oxidation of single-walled carbon nanotubes (SWCNTs) by ozone and hydroxyl radicals, and found that oxidation facilitated carboxyl functionalization without altering cytotoxic endpoints[103].
Trophic transfer and bioaccumulation
-
The bioaccumulation and trophic transfer of nanomaterials may pose serious risks to higher trophic level organisms and even human health. Once released into the environment, CNMs may enter biological systems. Unlike conventional chemicals, for which lipid-based accumulation is often used as a proxy for bioaccumulation potential, alternative frameworks for nanomaterials prioritize assessing whether these materials can be adsorbed through the gastrointestinal tract and translocated to other tissues[104]. Current evidence suggests that CNTs are generally not absorbed by the intestinal tract and thus exhibit low bioaccumulation potential[104]. In contrast, graphene has been shown to bioaccumulate in aquatic species such as Daphnia magna and zebrafish. Dong et al. assessed the bioaccumulation of graphene using the bioconcentration factors (BBF). The log-transformed BBF values were 3.66, 5.1, 3.9, and 1.62 for Escherichia coli, Tetrahymena thermophila, Daphnia magna, and Danio rerio, respectively (Fig. 7a), indicating that graphene accumulated in all tested organisms[31]. Notably, they further evaluated trophic transfer using the trophic transfer factor (TTF). In the food chain from E. coli to T. thermophila, the TTF value was 8.6, suggesting a high potential for trophic transfer and highlighting it as a significant pathway for graphene accumulation (Fig. 7a–c)[31]. Similarly, fullerenol NPs exhibit significant biomagnification at lower trophic levels, with a BMF of 3.2 from Scenedesmus obliquus to D. magna, while the BMF from D. magna to D. rerio was less than 1, indicating limited biomagnification at higher trophic levels[105]. In the aquatic food chain (Chlorella vulgaris-Artemia salina-Danio rerio), the toxic effects of GO on Artemia saline and D. rerio were evaluated through both direct exposure and trophic transfer pathways (Fig. 7d–f)[106]. Compared to direct exposure, trophic transfer of Nano-GO resulted in higher mortality in D. rerio, potentially due to the penetration of GO through the intestinal barrier and the subsequent disruption of reproductive function. This suggests that trophic transfer may enhance the bioavailability and toxicological impact of GO in aquatic organisms[106].
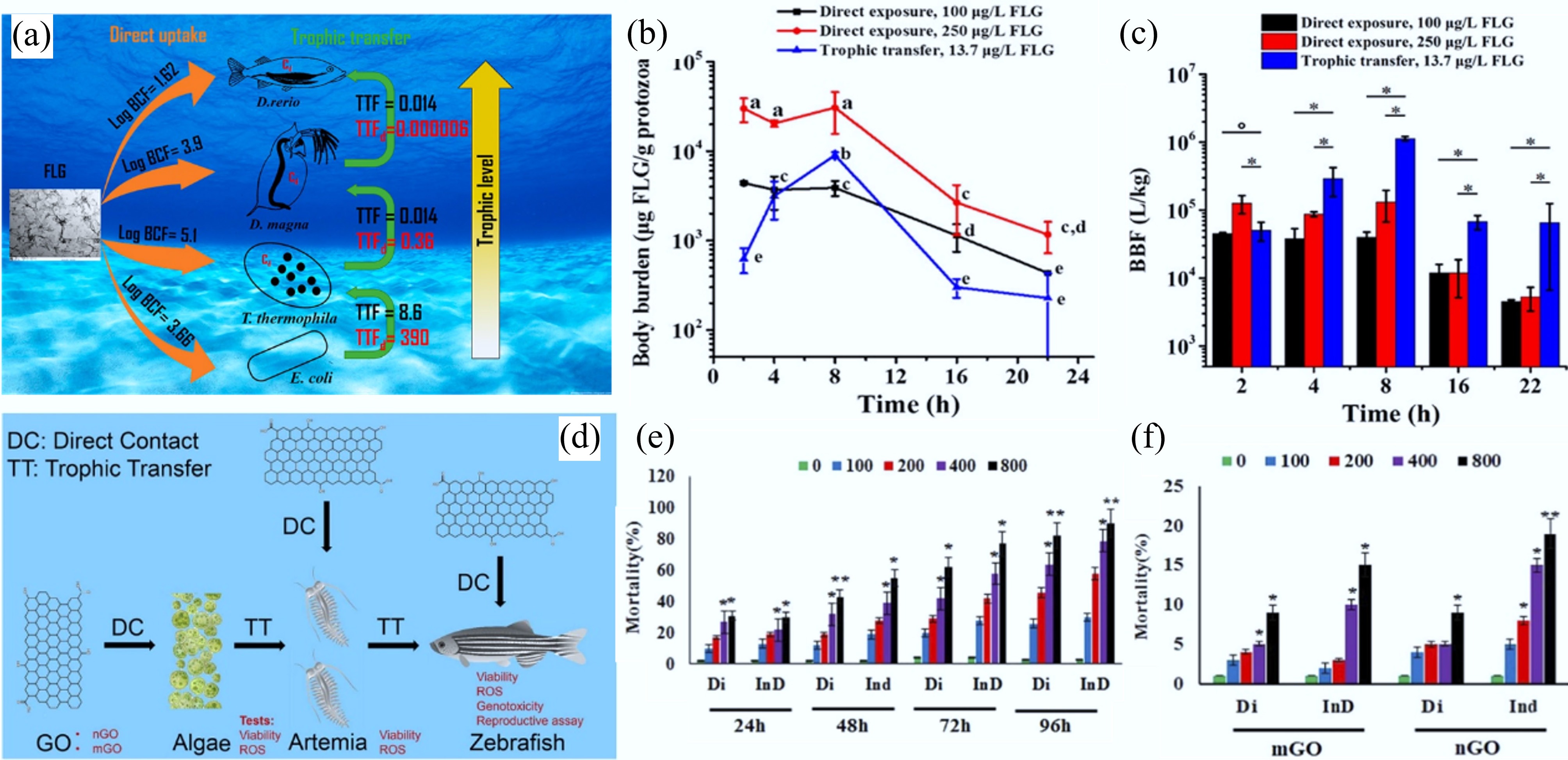
Figure 7.
(a) Schematic diagram of the bioaccumulation of 14C-labeled few-layer graphene (FLG) in an aquatic food chain through direct uptake or trophic transfer. (b) Body burden of T. thermophile during the direct exposure to FLG with concentration of 100 or 250 μg/L and trophic transfer from FLG associated E. coli. Each data point represents the mean of three independent replicates, with error bars showing the standard deviation (SD). Statistical significance is determined using Tukey's multiple comparison test. Groups labeled with the same letter do not differ significantly (p ≥ 0.05). (c) BBFs of FLG at different time points during T. thermophila growth in the presence of FLG, administered either directly in the medium (direct exposure) or with FLG-encrusted E. coli (trophic transfer). Bars represent BBFs derived from the mean values of FLG measured in triplicate. Asterisks (*) denote statistically significant differences[31]. (d) Schematic diagram of direct contact and trophic transfer of GO nanosheets in an aquatic food chain. (e) Mortality of Artemia salina caused by Nano-GO. (f) Mortality of D. rerio caused by Micro-GO and Nano-GO (in the Figures, direct ingestion and trophic transfer pathways are denoted as Di and InD, respectively). In (e) and (f), statistical significance is indicated by (*) and (**), corresponding to p ≤ 0.05 and p ≤ 0.01, respectively. All experiments were conducted in triplicate[106].
Factors influencing toxicological effects
-
During the synthesis of CNMs, considerable variation in particle size is often observed. After entering the environment, CNMs may undergo transformations such as enzymatic degradation, photodegradation, the Fenton reaction, and interactions with other substances[107]. Such processes can cause fragmentation, oxidation, and size reduction of CNMs, potentially resulting in distinct toxicological effects.
Physical characteristics
Size
-
Both the synthesis process and environmental transformations can alter the size of CNMs. Smaller nanomaterials generally exhibit better dispersibility and stability, making them more suitable for intracellular delivery. Reducing the lateral sizes of GO nanosheets through oxidative treatment enhances their biocompatibility and decreases cytotoxicity compared to untreated GO[108]. However, smaller CNMs may also increase the potential for bioaccumulation. Lu et al. investigated graphene nanosheets with different lateral sizes in zebrafish. The results showed that smaller graphene nanosheets more easily crossed the intestinal wall and entered epithelial cells and the blood, which reduced their excretion efficiency (Fig. 8a, b)[90]. The toxicological behavior of CNTs is also influenced by their length[109]. Shorter CNTs (less than 1 μm) are more readily able to penetrate cell membranes[110]. Due to their fibrous nature, CNTs may induce length-dependent effects similar to those of asbestos[111]. When CNTs of varying lengths were administered into the pleural cavity, longer CNTs induced acute inflammation, whereas shorter CNTs were effectively cleared[112]. In the case of fullerenes, smaller nano-C60 particles exhibit enhanced DNA polymerase inhibition and higher cytotoxicity, indicating a size-dependent toxic effect[113].
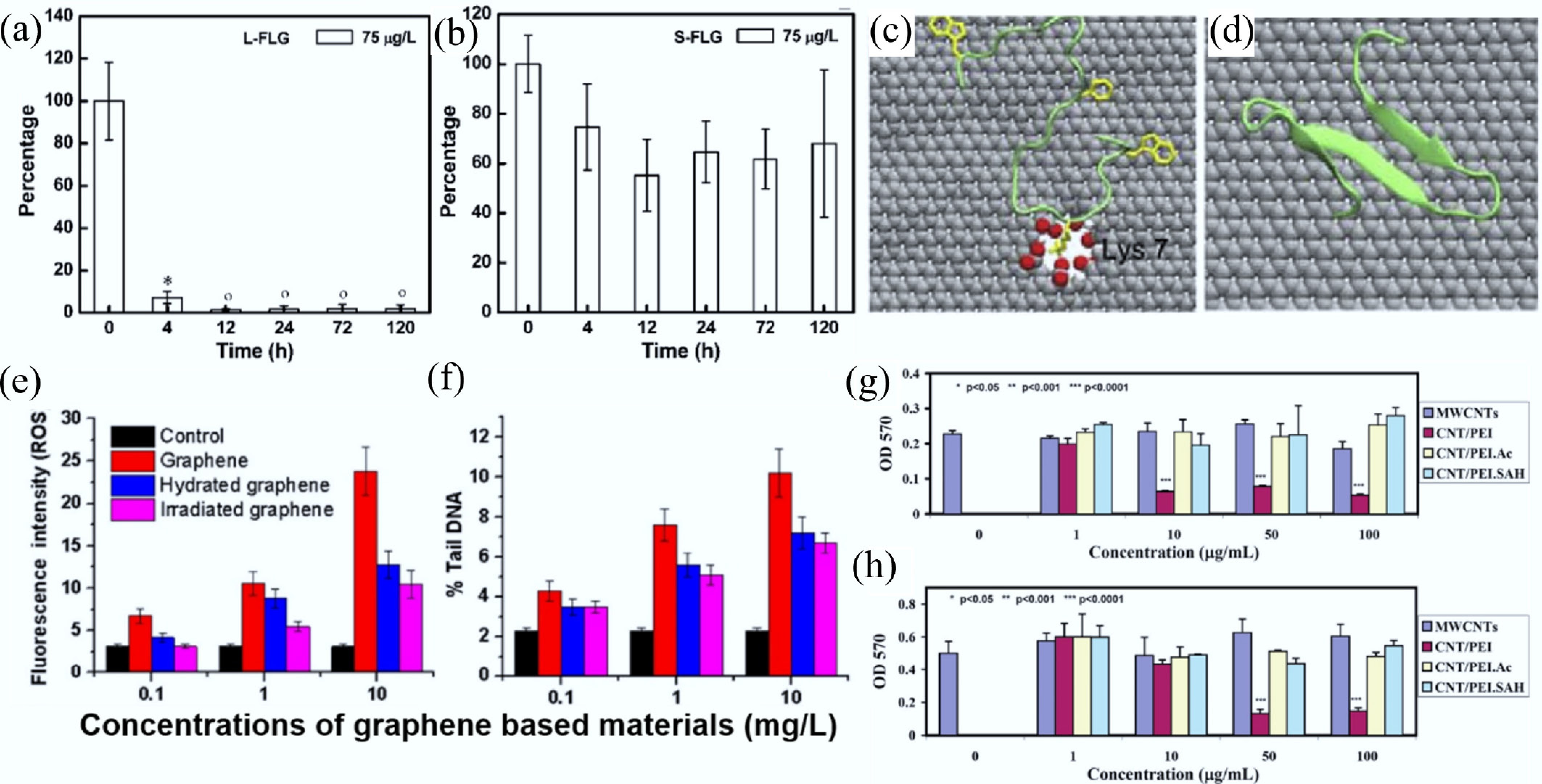
Figure 8.
Depuration of (a) Larger few-layer graphene and (b) Smaller few-layer graphene in zebrafish. Data are presented as mean ± SD (n = 5). Statistical significance was determined by Tukey's test (p < 0.05). Symbols (°) and (*) indicate values not significantly or significantly different from zero, respectively[90]. Representative contact configuration of YAP65WW protein adsorption onto (c) defective graphene and (d) ideal graphene surfaces (carbon, silver; oxygen, red; hydrogen, white). The critical residue Lys-7 involved in the binding process is labeled[114]. Effects of hydration and visible-light irradiation on the reduction of (e) oxidative stress and (f) DNA damage induced by pristine graphene. All experiments were conducted in triplicate, and error bars represent mean ± SD[101]. MTT assay results of cell viability for (g) FRO (a human thyroid cancer cell line) and (h) KB (a human epithelial carcinoma cell line) after 24 h treatment with differently functionalized MWCNTs. Each treatment was performed in triplicate, and error bars represent mean ± SD. Statistical significance was determined using the ANOVA test and is indicated by (*), (**), and (***) for p < 0.05, p < 0.001, and p < 0.0001, respectively[123].
Structural defects
-
Structural defects in nanomaterials may arise during synthesis or as a result of environmental transformation. These defects can alter the local electron density and mechanical properties of the nanomaterials. Graphene-based nanomaterials with appropriately sized defects have been applied in gas separation and seawater desalination[114]. Local defects in graphene can induce the unfolding of the YAP65WW-domain. Protein residues are tightly anchored to the defect sites through favorable electrostatic interactions, which constrain the protein conformation and ultimately lead to domain denaturation (Fig. 8c, d)[114]. In addition, Muller et al. found that the acute pulmonary and genotoxic effects of CNMs were closely associated with their structural defects. High-temperature annealing, which can repair such defects in CNMs, significantly reduces their toxicity. For MWCNTs, the extent of structural defects was positively correlated with their adhesion to cell membranes[115]. Dangling carbon bonds at defect sites served as reactive sites for cell membrane interactions, potentially disrupting membrane integrity and inducing cytotoxic effects[116].
Surface functionalization
Surface functional groups
-
When CNMs are released into the environment, they may undergo transformations through redox reactions, photochemical processes, and/or hydrolysis, resulting in the formation of oxygen-containing functional groups on their surfaces[107,117]. Such surface modifications can alter the hydrophilicity or hydrophobicity of CNMs and consequently affect their biological toxicity. Graphene oxidation under hydration and light irradiation introduces oxygen-containing functional groups, which increase the negative surface charge and promote aggregation, thereby significantly reducing the oxidative stress and nanotoxicity of graphene to algal cells (Fig. 8e, f)[101]. In contrast, studies on MWCNTs showed that oxidation by ROS increased surface oxygen functional groups, which enhanced cellular uptake and led to reduced cell proliferation, indicating increased cytotoxicity[117,118]. Pérez-Luna et al. further compared three types of surface-modified MWCNTs (pristine, oxidized, and alkylated) and examined their interactions with giant unilamellar vesicles. The results demonstrated that these interactions were predominantly driven by hydrophobic forces, indicating that the type of surface functional group may modulate hydrophobicity[119]. In addition to environmental transformations, intentional functionalization methods such as covalent modifications (including hydroxylation and carboxylation) and noncovalent modifications (including hydrogen bonding and π–π interactions) have been widely applied to improve the performance of CNMs. However, the potential toxicological consequences of such modifications should not be overlooked. Huang et al. investigated the toxic effects of carboxylated, aminated, and hydroxylated graphene on Daphnia magna. They found that aminated and hydroxylated graphene disrupted protein synthesis and function by interfering with transcription and translation pathways, leading to toxic effects[120]. In contrast, surface modification using biocompatible polymers (e.g., PEG and dextran) has been shown to reduce direct interactions with cell membranes and thus mitigate cytotoxicity[121].
Surface charge modification
-
The interaction between CNMs and cell membranes can induce cytotoxic effects, as nanomaterials may penetrate and be internalized through the negatively charged lipid bilayer. Therefore, the surface charge of CNMs influences their binding and internalization by the cell membranes, thereby affecting their toxicity. Studies have shown that CNMs with positive or neutral surface charges are more likely to associate with cell membranes[122]. For example, Shen et al. investigated the cytotoxicity of MWCNTs with different surface charges, and those with positive charges exhibited toxic effects even at low concentrations. This may be attributed to the strong interaction between positively charged MWCNTs and negatively charged cell membranes (Fig. 8g, h)[123]. Similarly, positively charged GO nanosheets have been reported to promote mitochondrial fission and induce cell death through apoptosis and autophagy[124].
Nanocomposite
Metal and metal oxides
-
CNMs possess a large SSA, making them excellent carriers for metals and metal oxides by providing abundant binding sites. This facilitates the formation of composite materials with enhanced catalytic, antibacterial, and electrochemical activities, thereby expanding the application scope of CNMs[125]. However, such modifications can also alter their toxicological profiles. Yin et al. investigated six rGO-based composites (rGO-Au, rGO-Ag, rGO-Pd, rGO-Fe3O4, rGO-Co3O4, and rGO-SnO2) and their toxic effects on algae. The results indicated that soluble metal ions released from the embedded metals or metal oxides could damage cell membranes and trigger oxidative stress, resulting in increased cytotoxicity[125]. Similarly, the Pb3O4@MWCNTs nanocomposite exhibited higher toxicity toward environmental bacteria compared to Pb3O4 or MWCNTs alone, likely due to the easier release of Pb2+ from the Pb3O4@MWCNTs[126]. In contrast to these findings, Valimukhametova et al. developed a series of lightly metal-doped (iron oxide, silver, thulium, neodymium, cerium oxide, cerium chloride, and molybdenum disulfide) NGQDs, which exhibited excellent fluorescence imaging capabilities under visible and near-infrared (NIR) light. Importantly, their low metal doping levels contributed to relatively low cytotoxicity[127]. Therefore, the dual impact of enhanced functionality and altered toxicity underscores the need for balanced design strategies when developing metal-modified CNMs.
Polymers
-
Polymer modification improves the dispersibility and stability of CNMs and has been widely studied in drug delivery and bioimaging. PEG, chitosan, and cellulose derivatives are commonly used for the functionalization of CNMs. It has been reported that PEGylated graphene nanosheets mainly accumulate in the liver and spleen after intravenous administration. They are subsequently excreted through the kidneys and feces. Long-term studies in mice showed no significant toxic effects[128]. Similarly, the incorporation of chitosan-functionalized CNTs into poly(acrylamide-co-acrylic acid) hydrogels result in a composite material. The results showed that the composite did not induce oxidative stress or cytotoxicity in intestinal cells, suggesting favorable biocompatibility[129]. Carboxymethyl cellulose is a cellulose derivative with tunable hydrophilicity, surface activity, and thickening properties. It has been used to modify hydrophobic carbon dots (CDs) to enhance their hydrophilicity. Due to the lipophilic nature of CDs, unmodified CDs can cross cell membranes and accumulate in lipid compartments, leading to oxidative stress and an inflammatory response. After modification with carboxymethyl cellulose, CDs were found to promote cell proliferation and increase cell viability[130]. Collectively, these findings suggest that polymer modification is beneficial for modulating the biological interactions of CNMs and enhancing their biosafety.
Mechanisms of carbon nanomaterials toxicity
Physical damage
-
Graphene-based CNMs, such as graphene, GO, and rGO, are 2D materials characterized by sharp edges. The physical interactions between graphene-based nanosheets and cell membranes are significant factors contributing to their cytotoxicity. The sharp edges of graphene-based nanosheets can insert into and cut bacterial cell membranes, causing membrane damage. Guo et al. investigated the neurotoxicity of graphene with different surface functionalizations and found that all functionalized graphene induced neurotoxic effects through physical disruption of membrane lipids[131]. In addition, the fibrous structure of CNTs enables them to directly pierce the membranes of Escherichia coli, leading to bacterial death[132]. Purified SWCNTs exhibited strong antibacterial activity, primarily attributed to membrane damage caused by direct contact between SWCNTs and bacterial cell membranes, which induced cytotoxic effects[133].
Oxidative stress
-
In addition to physical damage, a second major mechanism by which carbon nanomaterials induce toxicity is oxidative stress. Exposure to CNMs often leads to excessive ROS generation, which disrupts the antioxidant defense system and eventually induces oxidative stress responses. This oxidative imbalance results in damage to critical biomolecules such as proteins, DNA, and lipids, which can subsequently induce apoptosis, necrosis, or developmental toxicity[134,135]. The degree of oxidative stress is significantly influenced by the surface functionalization, physicochemical properties (e.g., size, metal impurities), and intracellular accumulation of CNMs[109,136]. Specifically, CNTs and graphene-based nanomaterials have been shown to induce excessive ROS generation upon interacting with cells, accompanied by reduced antioxidant enzyme activity. This can cause lipid membrane damage, DNA fragmentation, mitochondrial dysfunction, and disruption of cellular metabolism[137]. Additionally, photoactive nanomaterials such as nano-C60 aggregates can further induce ROS production through light-driven photochemical reactions, resulting in lipid peroxidation and increased cytotoxicity[138]. Together, these findings underscore the central role of ROS-mediated oxidative stress as a common toxicological pathway for CNMs.
Genotoxicity of carbon nanomaterials
-
Another critical toxicological mechanism associated with carbon nanomaterials is genotoxicity, which involves damage to chromosomes or DNA. The large surface area and surface charge of CNMs may contribute to their genotoxic potential, leading to DNA damage. GO nanosheets administered via intravenous injection have been shown to induce mutations in mice[139]. Although GO cannot penetrate the cell nucleus, it may still interact with DNA during mitosis, when the nuclear envelope temporarily disassembles, thereby increasing the risk of chromosomal aberrations[137,140]. The aromatic carbon rings of GO can bind to DNA base pairs through π–π stacking interactions, causing distortion at the DNA termini and contributing to genetic instability[141]. Additionally, carbon nanomaterials may induce genotoxicity indirectly through oxidative stress, inflammatory responses, and cell cycle disruption, all of which can lead to DNA damage[137] and promote the generation of ROS. For instance, CNTs can stimulate inflammatory cells to produce ROS[142], resulting in oxidative DNA damage such as oxidation of DNA bases and strand breaks, as well as lipid peroxidation-mediated DNA adduct formation, all of which are associated with genotoxic effects[109, 143].
Inflammatory response
-
In addition to the mechanisms described above, the inflammatory response has also emerged as an important pathway by which carbon nanomaterials exert toxic effects. CNMs may induce an inflammatory response through various exposure routes. In vivo studies have shown that subcutaneous injection of CNMs can trigger inflammation and tissue damage by releasing key cytokines and chemokines such as IL-6, IL-12, and TNF-α[137,144]. Similarly, tracheal exposure to SWCNTs in rats caused inflammation and cellular injury[135,145]. In vitro, CNMs have been found to upregulate the expression of immune- and inflammation-related genes in skin fibroblasts, potentially triggering inflammation[135].
In summary, to mitigate the potential toxicity of carbon nanomaterials and guide the development of safer and more sustainable materials, recent efforts have focused on green synthesis and rational surface engineering strategies. Green synthesis approaches, which utilize eco-friendly solvents and mild reaction conditions, aim to reduce the introduction of hazardous reagents and impurities during fabrication. For example, electrochemical methods can replace the harsh conditions of the Hummers method for synthesizing GO nanosheets, or plant extracts can be used as alternative green reducing and stabilizing agents for graphene. In addition, surface modification is a commonly employed strategy to improve the safety of carbon nanomaterials for biomedical applications. Functionalization with biocompatible polymers (e.g., PEG, poly[acrylic acid]), proteins, and DNA can reduce the toxicity and improve biocompatibility. Moreover, appropriate surface modification can enhance the dispersibility and colloidal stability of carbon nanomaterials, which is essential for controlling particle size and optimizing biodistribution, thereby reducing the risk of accumulation in critical organs. Furthermore, the biodegradation of carbon nanomaterials is significantly influenced by the molecules grafted onto their surfaces. Such grafting can facilitate their elimination from biological systems, reducing bioaccumulation and potential toxic effects. These design principles collectively offer a foundation for engineering carbon nanomaterials with reduced environmental and biological risks.
-
Carbon-based materials, including graphene, biochar, CDs, CNTs, CFs, and AC, etc., constitute a diverse class of functional materials characterized by tunable physicochemical properties, hierarchical architectures, and broad application versatility. Structurally, they span from 0D CDs, 1D CNTs and CFs, and 2D graphene to 3D AC and biochar frameworks, primarily composed of sp2 and/or sp3-hybridized carbon atoms. Their distinct morphologies and degrees of crystallinity engender diverse structural, optical, and electronic properties, which are highly relevant to applications in environmental governance, energy storage and conversion, catalysis, sensing technologies, and beyond (Fig. 9).
Figure 9.
Comprehensive analysis of structural characteristics and functional properties in sustainable CMs.
The photophysical and electronic behaviors of these materials are closely governed by their dimensionality, surface chemistry, and defect density. Photophysical properties, such as light absorption, photoluminescence, and photoinduced charge transfer, are particularly pronounced in CDs and graphene derivatives, while porous CMs like biochar and AC often act as optical supports or carriers for photoactive species. Meanwhile, electronic properties, including electrical conductivity, charge carrier mobility, and Fermi level modulation, are highly tunable through strategies such as heteroatom doping, structural ordering, or hybridization with functional nanomaterials. A summary of the key photophysical properties across these carbon-based materials is provided in Table 2. In the following sections, we focus on the photophysical and electronic properties of each carbon material class, emphasizing how structure and composition influence their performance in light-driven processes, charge transport, and related applications.
Table 2. Summary of photophysical properties of carbon-based materials
Carbon material Absorption range (nm) Emission range (nm) Quantum yield (QY) Bandgap (eV) Graphene UV - NIR (200–2,500) 400–800 1%–74% ~ 0–3.6 eV Biochar UV - NIR (250–800) N/A N/A 0.1–1.0 CDs UV-NIR (200–1,800) 450–800 1%–85% 1.7–3.3 CNTs UV - NIR (200–2,500) 900–1,600 N/A ~ 0.1–0.5 Carbon fibers UV - NIR (250–2,500) N/A N/A ~ 0–0.01 AC UV - NIR (250–2,500) N/A N/A N/A Photophysical properties
Graphene
-
Graphene's photophysical properties, such as high carrier concentration[146], low power loss, high optical transparency, broad spectral absorption range, and ultrafast optical response, make it an ideal material for a wide range of optoelectronic devices, involving broadband photodetectors, solar cells, nanosensors, and flexible electrodes[147–149].
By twisting and stacking two semiconductor films at a specific angle (the optical magic angle, 2.65°) to form an optical structure similar to magic angle graphene, the energy-momentum dispersion band of light becomes flat, resulting in a complete localization and stopping of the propagation of light[150,151]. Combined with a semiconductor gain medium, a nano-laser without a conventional optical cavity is realized. With a mode volume of only 0.01λ3 (λ is the wavelength), which is far below the diffraction limit, and a threshold power as low as 0.17 mW, it outperforms existing devices such as topological lasers[152].
Graphene quantum dots are optically tunable, with fluorescence emission ranging from the UV to the NIR[153,154], with a significant excitation wavelength dependence, and can be tuned by factors such as size, surface functional groups, and edge defects[155]. The luminescence emission mechanism involves models of quantum confinement effects, edge states (e.g., sawtooth-edged carbene structures), and surface states (e.g., oxygen-containing functional groups or defects)[156,157]. To enhance the penetration and specificity of in-vivo imaging, the fluorescence properties can be optimized by adjusting the size, edge structure, or number of layers[158,159]. In addition, the electronic structure can be altered to adjust the fluorescence wavelength and intensity by introducing heteroatoms such as nitrogen, boron, and sulfur[152,160,161].
Biochar
-
While pristine biochar is generally considered optically inert, it has increasingly been explored as a functional support in photophysical systems due to its tunable surface chemistry, high surface area, and structural compatibility with light-active species[162]. Rather than serving as an intrinsic luminophore, biochar primarily functions as a photonic interface or photochemical platform—facilitating light absorption, energy transfer, and stabilization of photoreactive components in hybrid architectures[163].
Biochar produced under controlled pyrolysis often exhibits a semi-conjugated carbon framework containing both sp2- and sp3-hybridized carbon domains[164]. The presence of π-conjugated aromatic structures, combined with abundant oxygen- or nitrogen-containing surface groups, introduces localized energy states that can participate in electron delocalization and act as mediators for light-induced processes[165].
Importantly, biochar's role as a support for photocatalytic species has attracted growing attention[163,166]. Its porous and chemically reactive surface provides anchoring sites for semiconductors (e.g., TiO2, ZnO), CDs, MOFs, and plasmonic NPs, enabling efficient dispersion and stabilization of these photoactive entities[167]. The synergistic interactions at the interface, such as π–π stacking, hydrogen bonding, or redox coupling, can facilitate charge separation, inhibit photogenerated carrier recombination, and enhance visible light harvesting[168–170].
CDs
-
CDs are a class of 0D carbon-based nanomaterials known for their strong PL activity[171]. They can absorb a broad range of photons from the UV to the visible region, typically exhibiting prominent absorption in the 230–300 nm range[172]. Their fluorescence is generally excitation-dependent, with tunable emission wavelengths that shift based on the excitation source[173]. The reported quantum yields of CDs vary widely, ranging from less than 1% to as high as 94.5%, underscoring their structural and synthetic diversity[174,175]. Despite extensive research, the photoluminescence mechanisms of CDs remain under active debate. The ambiguity primarily arises from the diversity of synthetic routes, complex structural characteristics, and methodological variations across different studies[176–179]. CDs can be synthesized from a wide array of carbonaceous precursors through either 'bottom-up' or 'top-down' strategies, with reaction parameters and post-synthesis treatments significantly influencing their physicochemical properties[180]. Moreover, research on CDs often targets specific applications, such as bioimaging[181], sensing[182], or electrocatalysis[183], each employing distinct characterization methods and performance criteria, further complicating cross-study comparisons. A prevailing view attributes the PL behavior of CDs to surface or near-surface electronic states, which are heavily influenced by the presence and nature of surface functional groups[184–186]. Oxygen- and nitrogen-containing groups (e.g., carboxyl, hydroxyl, amino) may alter the electronic environment and introduce localized emissive states[187,188]. Other hypotheses suggest that the PL originates from intrinsic π–π* transitions within sp2-conjugated domains or from fluorescent molecular by-products embedded within the carbon matrix during synthesis[189]. Quantum confinement effects have also been proposed to account for the size-dependent emission characteristics observed in some CDs[190]. According to this model, smaller-sized CDs possess wider band gaps and thus exhibit blue-shifted emission[191]. However, conflicting findings have reported red-shifted emission with decreasing particle size[192], indicating the possible involvement of additional structural or surface-related factors, such as trap state densities or surface oxidation levels[193].
Alternative interpretations emphasize extrinsic emissive mechanisms, positing that PL arises from defect states, surface traps, or molecular-like fluorophores[194–196]. This perspective is supported by evidence showing that surface passivation significantly enhances fluorescence intensity and that the emission is highly sensitive to environmental factors such as pH, solvent polarity, ionic strength, and redox conditions[197–199]. These properties make CDs excellent candidates for environmental and biological sensing applications. Despite the ongoing controversy surrounding their emission mechanisms, CDs typically exhibit bright, tunable fluorescence with broad emission bands (full width at half maximum, FWHM, of ~50 to 100 nm)[200,201]. Strong luminescence is generally observed when the particles are well dispersed, while quantum efficiency tends to decrease at longer wavelengths[202,203]. At the single-particle level, CDs often display narrower emission bands and multi-exponential decay kinetics, suggesting the presence of multiple emissive centers[204]. Some studies have reported the loss of emission tunability in single particles, supporting the idea that the broad emission observed in ensembles arises from the superposition of signals from heterogeneous populations[205]. Nonetheless, there are also reports of wavelength-tunable emission at the single-dot level, although distinguishing truly individual particles from small aggregates remains experimentally challenging.
CNTs
-
The photophysical properties of CNTs are a direct reflection of their unique 1D nanostructure. The absorption spectra of CNTs exhibit characteristic absorption peaks in the UV, Vis, and NIR regions, corresponding to their electronic energy level jumps. Due to the 1D nanostructure of SWCNTs, their electronic density of states is not a continuous function of energy, but rather a discontinuous spike, called a Van Hove singularity (a phenomenon where the density of electronic states becomes infinite at certain energy levels), symmetrically distributed on both sides of the Fermi energy level[206]. Each absorption peak position corresponds to a specific chiral index (n, m) of CNTs. Spectroscopy can therefore be used to rapidly determine the detailed composition of bulk SWCNT samples, providing distributions of tube diameters and chiral angles[207]. Resonance Raman, fluorescence spectroscopy, and UV-Vis-NIR spectroscopy are important tools for characterizing the optical properties of SWCNTs.
SWCNTs, in addition to their property of absorbing NIR light, are themselves capable of emitting NIR light, so that NIR fluorescence microscopy can be used to localize or image SWCNTs within cells. For practical applications of SWCNTs in environmental monitoring, medicine, energy, etc., they are usually functionalized. The optical properties of SWCNTs can be controlled by defect engineering, e.g., the covalent bonding of aminoaryl functional groups attached to SWCNT introduces sp3 defects into the sp2 carbon lattice, producing optically permissible defect states, which result in defective PL (E11−) with a significant redshift compared to pristine E11[208]. Similarly, oxygen doping alters the NIR band gap of fluorescent SWCNTs, causing them to have enhanced NIR fluorescence[209]. This makes them more suitable for bioimaging probes.
Carbon fibers
-
The optical absorption behavior of CFs is fundamentally governed by their unique electronic structure and hierarchical morphology. As a semi-metallic material with sp2-hybridized carbon networks, CFs exhibit exceptional broadband absorption spanning ultraviolet to infrared wavelengths. Spectroscopic analysis reveals nearly total absorption (α > 95%) across the visible spectrum (400–700 nm)[210]. This remarkable performance originates from inter-band transitions between π–π* states in the graphitic crystallites and free carrier absorption by delocalized electrons[193,211].
Microstructural parameters significantly influence absorption properties. Fibers with a higher graphitization degree show 10%–15% greater absorption of the NIR light compared to standard commercial grades[210]. This enhancement results from increased crystallite size and improved electron delocalization. Surface treatments also modify absorption characteristics: electrochemical oxidation introduces defect states that create a weak absorption tail extending to 900 nm, while plasma deposition of CNTs can boost visible absorption to 98% through light trapping in hierarchical structures[212,213].
CFs exhibit intrinsic anisotropic optical absorption characteristics due to their highly oriented graphitic microstructure. This structural anisotropy manifests as pronounced polarization-dependent absorption behavior, where light absorption varies significantly depending on the polarization direction relative to the fiber axis[214]. The dichroic absorption properties become especially enhanced in well-aligned fiber assemblies, highlighting their potential utility in polarization-sensitive optical applications.
Recent developments in microstructure engineering of CFs, including the fabrication of porous and hollow architectures, have enabled tunable spectral absorption characteristics[215]. These morphological modifications allow for tailored light-matter interactions, particularly in the NIR region, suggesting promising opportunities for spectrally selective photonic devices[216]. The ability to precisely control absorption properties through both intrinsic alignment and designed morphology positions CFs as versatile materials for advanced optical applications.
AC
-
The photophysical properties of AC originate from its unique combination of a disordered carbon skeleton structure, surface functional groups, and nanoscale pores, exhibiting optical behaviors significantly different from those of CMs such as graphene and CQDs[217]. Characterizing the photophysical properties of AC requires a comprehensive approach, integrating multiple techniques including Raman spectroscopy, UV–Vis–NIR absorption spectroscopy, and Fourier transform infrared spectroscopy (FT-IR) to unravel its complex optical response mechanisms.
Raman spectroscopy is a key technique for assessing the structural disorder in CMs. The intensity ratio of the D-band (~1,350 cm−1, associated with disordered or defective carbon) to the G-band (~1,580 cm−1, related to sp2-hybridized graphitic carbon) reflects the degree of graphitization and defect density[218]. For example, Erdogan et al. demonstrated that AC-based conductive coatings exhibit a higher G/D intensity ratio, indicating a higher graphitization level and good potential for thermal conductivity applications[219]. UV–Vis–NIR absorption spectra of AC typically show broad, featureless absorption, attributed to mixed sp2/sp3 carbon domains and multiple scattering effects[80]. The degree of graphitization affects the absorption: higher graphitization results in sharper peaks, while lower graphitization leads to broader absorption due to disorder and defects. In addition, porosity influences UV–Vis absorption by increasing light scattering and broadening the spectrum, with larger pores and surface functional groups further contributing to this effect. Meanwhile, FT-IR spectroscopy provides insight into AC surface chemistry. As shown by Dziejarski et al., modified AC materials exhibit characteristic O–H stretching bands around 3,450 cm−1 (free hydroxyl groups), along with C–H bending vibrations in the 3,000–2,850 and 1,380 cm−1 regions, reflecting the presence of aliphatic groups influenced by activation methods[220].
While carbon-based materials like graphene, biochar, and CDs exhibit exceptional photophysical properties that have propelled their applications in optoelectronic devices, sensors, and catalysis, there is still a lack of consensus on the exact mechanisms driving their photoluminescence. For example, in graphene quantum dots, although their tunable fluorescence is promising, the diverse synthetic routes and structural characteristics lead to inconsistent results regarding emission wavelength and efficiency. The role of surface defects and heteroatom doping remains contentious in CDs, complicating efforts to optimize them for specific applications. Moving forward, there is a need for more standardized synthetic methods and a deeper understanding of the photophysical processes at the molecular level, which will aid in designing more efficient and application-specific materials.
Electronic properties
Graphene
-
The electronic energy band structure of graphene exhibits a typical Dirac cone feature[221], with the conduction and valence bands intersecting linearly at the K and K' points (the boundary of the Brillouin zone) to form a semimetal property with a zero bandgap, and the special structure enables them to exhibit unique electrical properties[222,223].
By applying a gate voltage (Vg) to graphene, the type of carriers in graphene can be flexibly tuned to achieve a bipolar carrier transport mode[224,225]. In addition, graphene has an electron mobility of up to 200,000 cm2/(V·s) at room temperature[226,227]. Due to the quantum tunneling effect at Dirac points (the points where the conduction and valence bands of graphene meet, leading to a zero energy gap), graphene maintains a quantized conductivity of about 4e2/h ≈ 6 mS even when the carrier concentration tends to zero.
High transmittance (> 97.7%) and conductivity (square resistance < 100 Ω/sq) make graphene an ideal material to replace ITO, and a foldable electronic screen can be prepared through the development of a graphene touch screen, in addition to preparing a graphene transparent conductive film used in solar cells can significantly improve the photoelectric conversion efficiency[228–230].
Micro-supercapacitors suitable for electric vehicles and grid energy storage can be developed by utilizing graphene's high SSA (2,630 m2/g) and electrical conductivity[231,232]. Using graphene as an electrode material can increase the energy density and charge/discharge rate of batteries[233]. Graphene-based gas sensors are also a hotspot in current research, where the huge SSA of the nanomaterial allows for adsorption of gas molecules and improved detection sensitivity[234]. A room-temperature hydrogen gas sensor with a laser-induced graphene electrode-loaded platinum NPs responded ΔR/R0 = 4.4% to 1,000 ppm hydrogen, with a detection limit as low as 200 ppb, good linearity in the range of 100–10,000 ppm, and a response that remained stable after 50 cycle tests[235].
Biochar
-
Biochar is increasingly recognized as a redox-active and electronically tunable material, making it attractive for applications in energy storage, catalysis, and electrochemical sensing[236]. The electronic properties of biochar are governed by its carbon hybridization state, degree of conjugation, surface defect density, and heteroatom doping level[237].
In general, pristine biochar exhibits poor conductivity due to its predominantly amorphous carbon framework interspersed with disordered sp3 carbon and oxygen-containing functional groups[164]. Doping biochar with heteroatoms such as nitrogen, phosphorus, or sulfur introduces localized electronic states that modify the electronic band structure and facilitate charge carrier generation and migration[237]. Nitrogen, for instance, can be incorporated as pyridinic, pyrrolic, or graphitic N, each conferring different electronic characteristics—such as enhanced electron-donating capacity or improved conductivity through modulated Fermi level alignment (the energy level that separates occupied and unoccupied electron states at absolute zero temperature, which significantly influences the material's electronic properties)[238].
CDs
-
CDs exhibit tunable electronic properties arising from their π-conjugated carbon cores, surface functional groups, and heteroatom doping[239]. The sp2-hybridized domains enable delocalized electron transport, while surface groups introduce localized states that influence charge separation and recombination[240].
Doping with elements such as N, S, P, or B modifies the electronic band structure by introducing new energy levels or shifting the Fermi level, thus enhancing charge transfer and adjusting conductivity[241,242]. These effects are crucial for applications requiring tailored redox behavior or improved electron mobility. Under photoexcitation, CDs can act as either electron donors or acceptors, engaging in ultrafast charge transfer with surrounding metal ions or organic molecules[204, 243]. Such interactions often lead to fluorescence quenching due to suppressed radiative recombination. Two models have been proposed for CD emission: (1) core-to-surface charge migration, where the core absorbs photons and transfers excitons to surface traps for emission[244–246]; and (2) direct surface-state excitation, where emission arises from localized surface excitons[194, 247]. The true behavior likely involves a combination of both, depending on CD structure and synthesis.
CNTs
-
CNTs, as a carbon-based nanomaterial with a unique structure and excellent properties, have high SSA, good electrical conductivity, and chemical stability, which contribute to effective electron transfer[248,249]. The extremely small size and highly symmetrical structural features make the quantum effects in CNTs very obvious. Theoretical studies show that the electrical properties of CNTs depend on their diameter and helicity. Due to the quantum effect, the energy required for electrons to jump from the valence band to the CB in SWCNTs can be varied continuously from near zero (metal-like) to 1 eV (semiconductor) with different diameters and chiral angles, and the conductivity can be metallic, semi-metallic or semiconducting, and thus the conduction properties of CNTs can be altered by varying the diameter of the tubes and their chiral angles[250]. The electronic band gap of nanotubes increases as their length shortens in response to quantum confinement effects along the length axis. In nanoelectronics, 10 nm CNT transistors show the best low-voltage performance compared to other devices of similar size[251]. CNTs can be easily assembled on electrode surfaces to provide additional channels for electron transport and facilitate electron transfer, thereby improving charge transfer efficiency, enhancing the electrochemical sensing capability of the system, and acting as complementary active sites for electrochemical reactions to improve their electrochemical performance[252,253]. In addition, another reason why CNTs have received significant attention in the development of improved sensing platforms is their electrocatalytic nature, which is largely attributed to defects in the edge planes of the CNTs' structure. Edge-like planar defects at the open end of MWCNTs and pore defects in the tube walls can serve to enhance the current. As the large surface area and edge plane defects in MWCNTs create excellent electrical conductivity and superior electron transfer rates, they contribute positively to the enhancement of current and catalysis[254].
Carbon fibers
-
CFs exhibit excellent electronic properties due to their high degree of graphitization and extended sp2-hybridized carbon networks[255]. Their electrical conductivity is primarily anisotropic, with high axial conductivity enabled by π-electron delocalization along the fiber direction[256]. These properties make CFs suitable for applications in lightweight conductive components, electromagnetic shielding, and structural energy devices[257]. Surface functionalization and heteroatom doping further modulate the electronic behavior of CFs[258]. Oxidative treatments introduce surface groups (e.g., –OH, –COOH), which can enhance interfacial charge transfer, especially in composite systems or electrochemical electrodes. Meanwhile, doping with elements such as nitrogen or boron alters the electronic band structure, enhances carrier density, and improves electrocatalytic activity[259]. These features position CFs as versatile electronic materials for advanced applications in flexible electronics, batteries, and electrocatalysis.
AC
-
AC exhibits tunable electronic properties derived from its disordered sp2/sp3 carbon network and surface functional groups[260]. Its electrical conductivity is primarily governed by the degree of graphitization, while oxygen- and heteroatom-containing functionalities (e.g., N, S, and P) modulate the electronic density of states and introduce redox-active sites[22]. Through thermal treatment and heteroatom doping, AC can achieve enhanced charge transport and electron transfer capabilities, making it an effective conductive support in electrochemical energy storage, catalysis, and sensing applications.
The electronic properties of carbon-based materials such as graphene, biochar, and CNTs are highly promising, especially with their tunable conductivity, high surface areas, and potential for integration into a wide range of devices. However, despite their successes in various applications, challenges remain in fully harnessing their electronic capabilities. For instance, while graphene's high mobility and conductivity are advantageous for use in flexible electronics and sensors, its zero bandgap limits its use in switching devices without external modification. Furthermore, the effects of heteroatom doping and structural defects in biochar and CNTs need to be more thoroughly explored to maximize their electronic behavior. Future work should focus on optimizing doping strategies and minimizing defects to enhance material performance, while also considering scalability and sustainability in commercial applications.
-
The intensification of industrial processes and social development has resulted in significant environmental pollution, which poses a considerable threat to human and biological health and causes substantial damage to ecosystems. Of particular concern are persistent organic pollutants (POPs), which exhibit both strong transport and degradation characteristics[261,262]. POPs are frequently characterized by a broad array of intricate chemical structures and a considerable array of sources[263]. These include, but are not limited to, human-produced substances, such as pesticides, organic solvents, organic dyes, pharmaceuticals, food additives, and plastic additives[264]. These organic materials are mainly composed of benzenoids, heterocyclics, acids and derivatives, lipid-like molecules, and organic oxygen compounds, which are mainly present in water[265]. POPs exhibit high migratory tendencies and water solubility, facilitating their entry into the water column and subsequent dispersion in soil. Additionally, some POPs volatilize into the atmosphere[266]. The elimination of POPs from water is imperative because of their persistent accumulation in plants and animals and their subsequent uptake by humans. Many technologies have been utilized to address organic pollution, including adsorption, mechanical-vapor recompression, precipitation, biodegradation, solvent extraction, membrane separation, ion exchange, chemical oxidation, and coagulation[267]. The aim of these technologies is to remove organic pollutants from water bodies; however, these technologies cannot be implemented without the support of materials. A significant body of research has been dedicated to investigating the potential of various materials to remove organic pollutants from water bodies. These materials include layered double hydroxides, perovskites, graphene, MOFs, and covalent organic frameworks (COFs), among others. The use of these materials in organic-removal processes has been documented in numerous studies. The presence of carbon is a distinguishing feature of many materials, and carbon-based materials have been extensively employed for the removal of organic pollutants. This is due to the environmental friendliness of the materials, broad species coverage, and favorable physicochemical properties. This section focuses on the preparation and application of carbon-based nanomaterials, polymer carbon-based materials, and metal-organic synergistic carbon-based materials in different fields, including adsorption, photocatalysis, advanced oxidation reactions, and Fenton reactions, for the removal of organic matter, with the aim of providing ideas for future research.
Carbon-based nanomaterials
One-dimensional carbon nanomaterials
-
As environmentally friendly 1D nanofunctional materials, CNTs have great application prospects in the environmental field. CNTs typically consist of one or multiple layers of SP2 hybridization graphene (Fig. 10a), featuring a hexagonal network of carbon atoms[268]. These nanotubes have nanoscale diameters and typically measure a few micrometers in length[269]. This property is conducive to their high SAA, which provides ample active sites for pollutant adsorption. The pi-conjugative structure enables the strong affinity with organic pollutants that are able to induce π–π coupling interaction[268].
Figure 10.
(a) CNTs with different forms, monolithic (right) and multilayered CNTs (left)[270]. (b) Synthesis of CNTs[271]. (c) ROS generation mechanism based on the electrochemical nZVC-CNT/ peroxymonosulfate (PMS) filter[272]. (d) Schematic illustration of reaction mechanism for Rhodamine B (RhB) degradation by Fe@NCNT-BC-800/PS[273]. (e) TEM image of Fe@NCNT-BC-800[273]. (f) Electron density distribution of Tetraethylamine[274].
CNTs were first synthesized by Iijima in 1991 using an arc evaporation method[275], and numerous efficient methods have been proposed as research has advanced. Similar to the arc evaporation method, the laser-ablation method[276] also involves the evaporation of solid carbon material at high temperatures, ranging from 3,000 to 4,000 °C, to obtain CNTs[271]. CVD has also been employed in the synthesis of CNTs, using hydrocarbon gases (e.g., CH4, C3H2) as the carbon source in a reductive atmosphere (e.g., NH3, H2) on catalyst substrates (e.g., Ni, Fe)[165]. In comparison with the arc evaporation and laser-ablation methods, CVD has the advantage of requiring a lower reaction temperature (600–1,100 °C) and is capable of producing purer products[271] (Fig. 10b). Ghorbani et al. proposed an efficient metal dusting process for the preparation of MWCNTs, achieving a high yield (700%–1,000%) and low cost[277]. CNTs possess a high SAA, distinctive molecular structure, and excellent electrical conductivity. These properties render them highly versatile, and they are extensively utilized in a number of fields such as the removal of organic pollutants through adsorption, photocatalysis, and AOPs. Zheng et al. established an efficient electrocatalytic continuous-flow filtration system based on CNT-supported nanoscale zerovalent copper (nZVC-CNT) for the ultrafast activation of PMS and oxidation of organic Congo red (CR)[272]. In this system, nZVC-CNTs were fabricated through the in-situ reduction of Cu2+ to Cu0 and further acted as the cathode in the electrocatalytic continuous-flow filtration system. The large SSA and excellent electrical conductivity of CNTs supplied a platform for Cu0 and numerous active sites. Compared with conventional carbon cloth, the CNTs exhibited a 24.2% increase in CR degradation. The conductive sp2-hybridised carbon accelerated the charge transfer from the pollutants to PMS, •OH and 1O2 were produced through the activation of PMS by CNT. Concurrently, the redox cycle of Cu2+/Cu+ reacted with PMS to generate more 1O2, thereby facilitating dye degradation, as shown in Fig. 10c. Furthermore, both radical and nonradical pathways made a contribution to the degradation process. The system demonstrated consistently high efficacy over a broad pH range in complex aqueous matrices. In another study, soybean dregs were utilized as raw materials to construct N-doped and Fe NPs loaded onto bamboo-like CNTs (Fe@NCNT-BC) by Zhu et al.[273], which exhibited the ability to degrade RhB (Fig. 10e). The degradation rate of RhB reached ~100% within 10 min, and upon persulfate-activation-based AOPs, CNTs were demonstrated to display a substantial and extensive array of activator (PMS, PS, etc.) activation, thereby rendering them a subject of interest in the AOPs. Both radical (·OH, SO4−) and nonradical (1O2) paths were observed to achieve the degradation of RhB (Fig. 10d).
In addition to their application in AOPs, heteroatom N-doped multiwall CNTs were fabricated by Yi et al.[274] to adsorb three emerging polar/ionizable organic contaminants. The strong electronic polarizability, presence of numerous active functional groups, and unique π conjugation provide high affinity toward a number of pollutants through various reaction mechanisms. The introduction of N atoms increased the polarizability of the MNCTs, thereby accelerating the depletion of electron clouds, which enhanced the π–π coupling interaction. The DFT calculation demonstrated bisphenol A (BPA) and Tylosin as π-electron donor and acceptor respectively, while tetracycline exhibited both characterizations (Fig. 10f). Two main mechanisms were involved in the adsorption of these three pollutants: the π–π EDA mechanism and cation-π interaction. The electronegativity of the N atom was higher than that of the C atom. This results in an enhancement of the polarization of the N-MCNTs, leading to a clear separation of the positive and negative centers. As the π-electron donor and acceptor, BPA and tylosin were adsorbed by N-MCNTs through reactions with the π-electron-depleted and π-electron-rich regions, respectively. Moreover, the electron donor and acceptor parts in tetracycline have the ability to engage in interaction with their counterparts in N-MCNTs, thereby achieving adsorption. Besides, the N-doping-induced enhancement of the π electron density enables the interaction of the N-MCNTs with the protonated amino group in tylosin or tetracycline through cation-π interaction. Benefiting from the large specific area and enhanced polarized π-electrons, the KF values of N-MCNTs are 2.6–3.5 times those of MCNTs, and the qmax (maximum adsorption capacity) values of N-MCNTs are 3.7–4 times those of MCNTs.
In addition to common organic pollutants, CNT also demonstrates considerable potential for the removal of specific pollutants such as perfluoroalkyl substances (PFAS). As the most common PFASs substrate, PFOA and PFHxS are characterized by their chemical stability and resistance to natural biodegradation. Their prolonged existence and mobility within the environment are of particular concern, as they present a significant hazard to human and biological health[278]. CNTs with different outer diameters and surface modifications (–OH, –COOH functionalized) were used by Deng et al.[279] for adsorption removal studies of PFASs in water. To illustrate this, the experimental results demonstrated that, in the case of comparing the sizes, the smaller the outer diameter of the CNT, the greater the adsorption of PFOA in the same adsorption time. Among the CNTs, SWCNT exhibited the optimal adsorption performance. The present study posits that hydrophobic interaction is the primary driving force for PFASs adsorption onto CNTs, in contradistinction to π–π bonds, hydrogen bonds, and electrostatic interactions. It is noteworthy that the adsorption selectivity of six PFASs for SWCNT follows a sequence that is characterized by the increasing hydrophobicity of the compounds, namely PFOS > PFHxS > PFOA > PFBS > PFHxA > PFBA.
Two-dimensional carbon nanomaterials
-
As a typical 2D carbon-based material, graphene was first fabricated in 2004[56,280], and graphene-based materials (graphene, GO, and rGO) have been used in a wide range of applications in catalysis, environment, energy, sensing, and other fields. Thus, graphene materials have been industrially mass-produced, and Zhu et al. summarized the mass production of graphene materials for some industrial applications[56]. Graphene has a planar hexagonal lattice structure with aromatic (sp2) domains; thus, it can easily adsorb specific organic pollutants through robust π–π interaction[281]. Common graphene-synthesis methods include the top-down and bottom-up techniques[282]. The top-down method mainly refers to the exfoliation of bulk graphitic materials to obtain 2D graphene, including scotch-tape exfoliation and liquid-phase exfoliation. The bottom-up methods utilize chemical reactions to directly synthesize graphene, including CVD and molecular-beam epitaxy[282–284].
Notably, the low water solubility of graphene significantly impacts its practical applications, particularly in the context of pollutant removal from water bodies[289]. The treatment of graphene with strong oxidizing agents to form GO introduces abundant oxygen-containing groups[290], thereby enhancing its hydrophilicity and broadening its scope of application in the treatment of water pollutants (Fig. 11a). A straightforward method for producing GO facilitates its large-scale preparation[291]. The structural differences between GO and graphene can be attributed to the presence of additional oxygen atoms bonded to certain carbons[285]. The rich active oxygen-containing functional groups (hydroxyl, carboxyl, epoxy, and quinone), as well as the ultrathin 2D morphology provide sufficient surface area and active sites for reactant adsorption and activation[292]. Consequently, this renders the material highly effective for the removal of organic pollutants. GO is synthesized by the oxidation of graphene, followed by exfoliation[293]. The oxidation method involves the oxidation of graphene using different component oxidizing agents and the stripping process of van der Waals bonds breaking in H2O or other solutions[294,295]. Hummers' method, which utilizes a combination of strong acids as oxidizing agents, has been extensively employed. This method does not involve the use of metal salts and is straightforward. However, it releases substantial amounts of gaseous pollutants such as NO2 and N2O4. David et al. proposed a method for oxidizing graphene with a short reaction time of only 2 h; however, the introduction of heavy metals may pollute the environment[296]. As research progresses, additional synthetic methods with unique advantages and drawbacks are being proposed.
Figure 11.
(a) Production method of GO[285]. (b) Transformation pathway of graphite to GO and rGO[282]. (c) Schematic illustration of the formation of CNTs and carbon-doped SrTiO3 and its TEM image[286]. (d) Schematic illustration of sulfamethoxazole (SMX) degradation mechanism upon 3D-printed-WO3-UiO-66@rGO photocatalyst system[287]. (e) Mechanism of the 4-CP degradation upon N-rGO-CoSA[288]. (f) Graphene synthesis via the CVD method[282].
Furthermore, a divergence in the thermal stability of graphene (approximately 400–500 °C) and GO (approximately 200 °C) is evident[290,297]. The ease with which GO decomposes makes it a candidate for carbon doping in other materials. Chang & Hu[286] proposed carbon-doped SrTiO3 using GO as the carbon source to decompose into carbon species anchored on SrTiO3 (Fig. 11c), while some of the GO transformed into curly graphene sheets. The k value of the best samples was 1.5–200 times higher than that of the other samples. Utilizing GO as a carbon source and operating at the appropriate temperature resulted in the decomposition of GO into the lattice of SrTiO3 rather than its adhesion to the surface of SrTiO3 in the inorganic carbon form.
However, excessive oxygen-containing groups occupying the interlayer or edge carbon atoms may also lead to a decrease in the electrical conductivity of graphene, which, in turn, affects its charge-migration efficiency during catalytic reactions[282]. By reducing GO, the number of oxygen-containing groups on its surface can be precisely regulated to control its hydrophilicity and electrical conductivity. The rGO obtained by the reduction of GO (Fig. 11b) retains some of the advantages and properties of both graphene and GO, such as better hydrophilicity, electrical conductivity, and active surface functional groups. Generally, rGO is prepared by reducing GO using NaBH4, ascorbic acid, and HI, or by a thermochemical reduction method[282]. Long et al.[288] fabricated a single-atom catalyst with a Co–N configuration on the ample N sites of N-doped rGO (N-rGO-CoSA) to efficiently remove organic pollutants via periodate (IO4−)-based AOPs (Fig. 11e). N-doped rGO exhibits an ultrathin 2D nanosheet morphology, and its single-atom configuration was confirmed using HAADF-STEM and XAFS characterizations. The distribution of Co atoms on the rGO surface exhibited an approximate size of 0.2 nm. The EXAFS fitted results indicated that a single Co atom was bonded to two N atoms and two O atoms in rGO. In this case, the active site of the poise was provided by rGO, which anchored the Co atoms and adsorbed 4-CP via hydrophobic interactions and π–π interactions. Both N-rGO and Co-N sites were active, and the formed N-rGO-CoSA/periodate* complexes could catch electrons from 4-CP, achieving oxidation through a nonradical pathway. About 95% of 4-CP was removed within 12 min using the N-rGO-CoSA/periodate* complexes, demonstrating their potential for the removal of a wide range of other organics in complex real-world water environments.
The superior performance of rGO can be attributed to its ultrathin structure, high light-utilization rate, and excellent electrical conductivity. These properties facilitate the rapid separation and migration of photogenerated charges between its layers, making it a highly effective material for constructing heterojunctions with other semiconductors. This integration enhances the overall photocatalytic efficiency for various applications. Huong et al.[287] fabricated WO3-UiO-66@rGO photocatalysts via 3D printing for the first time for the photocatalytic degradation of SMX (Fig. 11d). The introduction of rGO significantly enhanced the utilization of visible light and shortened the band gap, which excited the photoinduced electrons from the valence band to the conduction band. Owing to its large SSA and superior mechanical strength, rGO served as support material for WO3 and UiO-66. Furthermore, the electrical conductivity of rGO enhanced the charge-transfer efficiency and could receive the electrons from WO3-UiO-66 to generate active radicals through the delocalized π–π conjugated forms[298]. The 3D-printing technology enhanced the mechanical strength and recycling ability; 3D-WO3-UiO-66@rGO exhibited a high kinetic constant of 0.02955 min−1 and maintained 100% of its catalytic activity even after 10 cycles of SMX degradation.
Furthermore, the interaction of rGO with GO has been demonstrated to exhibit superior performance, as evidenced by the removal of specific pollutants from water bodies. Khaliha et al. demonstrated that a mixture of GO and rGO can be used for the removal of PFASs from a water body and constructed a two-step water treatment method using MF on polyethersulfone hollow fiber to separate GO/rGO from water[299]. The GO/rGO combination was used to remove the PFAS mixture from real wastewater at a concentration of 10 μg/L. The adsorption capacity reached 138 μg/g within 0.5 h, at a solid-liquid ratio of 1 g/L.
Polymer carbon-based materials
COFs
-
COFs were first reported in 2005 by Dierks & Yaghi[300], and are a class of polymer materials composed of light elements (C, N, O, and H) connected by covalent bonds that possess a 2D or 3D mesh structure[301]. As illustrated in Fig. 12a, the research process of COFs has been demonstrated. The material exhibits the characteristics of a high surface area, tunable pore size, and ample functional groups, which suggests significant potential for development in the field of pollutant removal. COFs are formed by the polymerization of organic monomers; therefore, they have a number of π-conjugations, resulting in an excellent affinity to some organic pollutants through π–π interactions. Furthermore, the covalent bonds and long-range order form its rigid skeleton, which ensures its structural stability and resistance to strong acids and bases during long-term practical applications[302]. COFs are composed of two main parts, linkers (building blocks) and bonds (formed between these units during the process of network formation)[303], and can be synthesized via numerous methods. The solvothermal method, which is operated in stainless-steel autoclaves under high temperature and pressure, is widely utilized owing to its easy-to-operate characteristics[304]. For example, COF-NUC-1 was synthesized by heating the freeze-pump-thaw cycle-treated mixture of ETMP-Br, TFPB, DMAP, and deionized water at 160 °C for 3 d[305]. In a separate study, Pan et al.[306] synthesized COF-LZU1 at ambient temperature. While this method is less onerous, it requires greater temporal investment. Many other methods may be used to synthesize COFs, such as electrophoretic deposition, the ionothermal method, the mechanochemical method, and the microwave method. However, conventional solvothermal synthesis is a time-consuming process that requires elevated reaction temperatures, which significantly restricts its practical application. Consequently, it will take a significant period of time for COFs to achieve mass production and industrialized applications.
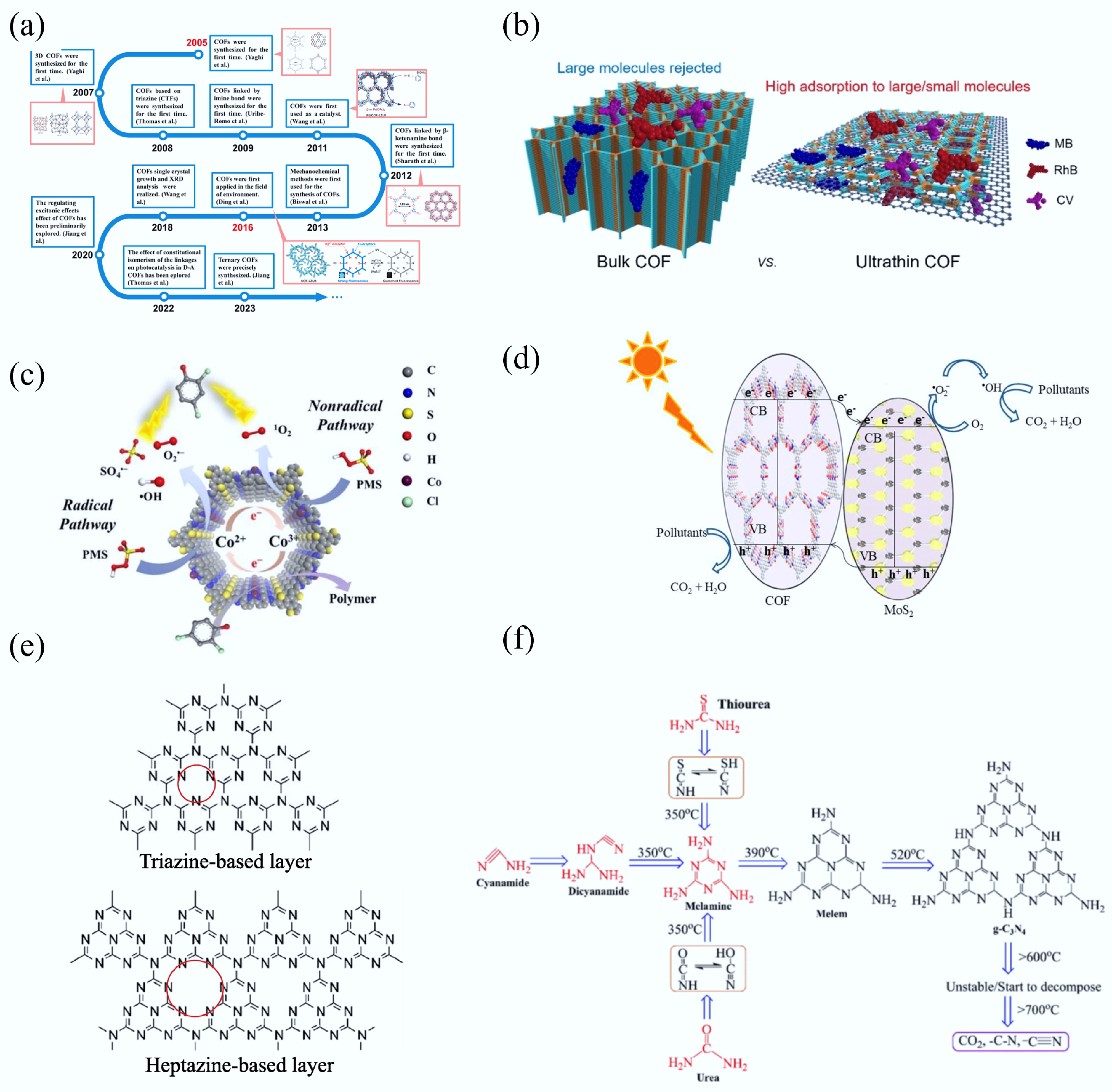
Figure 12.
(a) Development procedure of COFs[307]. (b) Schematic illustration of dye adsorption mechanism over bulk COF and ultrathin COF[308]. (c) Schematic illustration of 2,4-DCP mineralization mechanism over JLNU-317-Co/PMS[36]. (d) Schematic diagram of photocatalytic reaction mechanism over MoS2/COF[309]. (e) Layers description based on triazine and heptazine units[310]. (f) Schematic diagram of structure related to different precursors and pyrolysis temperature[311].
In summary, the focus on COFs in the domain of adsorption and the elimination of organic pollutants can be attributed to several key factors. First, these materials possess substantial SSA, which is advantageous for interactions with target molecules. Second, the presence of diverse surface functional groups offers enhanced reactivity, thereby facilitating the effective removal of pollutants. Third, the materials demonstrate notable stability, which exhibits the potential for practical applications in environmental applications. Finally, the porosity of COFs can be tuned by adjusting the reaction conditions, which enhances their versatility for different applications.
COFs for organic pollutants removal
-
Li et al.[308] fabricated a COF with an ultrathin thickness of approximately 2 nm, which was combined with graphene to create a COF/graphene aerogel (CGA) via a facile hydrothermal method for the adsorption of RhB (Fig. 12b). Ultrathin 2D COFs have increased SSA and, thus, increased adsorption active sites, while avoiding the interception of pollutants by micropores or small mesopores. The microporosity and charged surfaces of COFs provide a fundamental driving force for pollutant adsorption. CGA exhibits superior removal capacity, with > 99% of the dye captured within a span of 3 min. The substantial presence of carbon and nitrogen endows the COF with the capacity to regulate the local charge distribution through defective engineering. Furthermore, these elements anchor the loaded metal components through the defect sites. The substantial SSA of the material facilitates the homogeneous dispersion of the elements, thereby enabling atomic-level catalysis. Ma et al.[36] fabricated JLNU-317 through a straightforward solvothermal method, and Co2+ was anchored to JLNU-317 by stirring (Fig. 12c). This process results in the formation of the JLNU-317-Co catalyst, which is highly effective in the degradation of 2,4-dichlorophenol through PS-AOPs. The π-conjugation of JLNU-317 accelerates the charge transfer to active PS. DFT theory calculations illustrate the redistribution of electrons induced by the incorporation of Co. The asymmetrically distributed charge of JLNU-317-Co favors efficient conversion between Co2+ and Co3+ in the reaction process, thereby triggering a Fenton-like reaction. The degradation of 2,4-dichlorophenol was achieved through both free and non-free radicals (SO4•−, •OH, •O2−, and 1O2). However, the application of this material to photocatalytic pollutant removal is restricted by its low electron-hole mobility and rapid carrier recombination[312]. This problem can be resolved by combining the material with other semiconductors. The 2D-2D integration was designed by combining TpPa-1 COF with inorganic MoS2 (Fig. 12d) to achieve the photocatalytic degradation of TC and RhB, as reported by Khaing et al.[309]. The material demonstrates excellent structural stability when subjected to high-temperature hydrothermal reactions (200 °C for 12 h). Charge transfer is promoted due to the interlayer π–π conjugation and ultra-thin 2D morphology of the COF. The heterojunction mode conforms to the type-II transfer mode, whereby electrons in the conduction band (CB) of the COF are transferred to the CB of MoS2, leaving holes in the VB of the COF. Thus, MoS2/COF achieves pollutant degradation via direct oxidation by holes and the radical path (•OH), with a degradation rate of 85.9% after 60 min of irradiation.
Graphite phase carbon nitride (g-C3N4)
-
g-C3N4 is a C- and N-containing polymer composed of arranged 2D arrays of triazine or tri-s-triazine (s-heptazine) moieties[313]. As shown in Fig. 12e. Notably, van der Waals forces exist nonspecifically between the layers of g-C3N4. In addition, a strong sp2 hybridization covalent bond is exhibited between the C and N atoms. The graphitic π-conjugated bond, in conjunction with the absence of a metal, endows the material with favorable thermal stability, optical properties, charge-mobility efficiency, and a substantial SSA[310]. The synthesis of g-C3N4 can be accomplished through a straightforward thermal process that utilizes thiourea, melamine, cyanamide, dicyanamide, and urea, among other reagents, to yield the bulk phase, which gives it the potential to be produced large quantities at a low cost (Fig. 12f). The 2D g-C3N4 nanosheets were obtained through the exfoliation of bulk g-C3N4 via three different methods: ultrasound sonication, thermal exfoliation, and chemical exfoliation. In the raw-material synthesis process, varying reaction conditions must be considered, as this will result in the construction of different isoforms.
Its narrow Eg (~2.7 eV) and symmetrically distributed ECB (~ −1.3 eV), and EVB (~1.4 eV) enable it to drive most photocatalytic oxidation/reduction reactions, making it popular in photocatalysis[319,320]. Xu et al.[314] fabricated an S, K-doped g-C3N4 for efficient BPA degradation under alkaline conditions (Fig. 13a). The EVB value of g-C3N4 exhibits insufficient oxidation potential for the degradation of some pollutants, and the generation of active •OH is unfavorable for some natural alkaline environments. Moreover, 1O2 is more suitable for the preferential oxidation of high-priority pollutants in common wastewater due to a longer carrier lifetime compared to that of •OH and its electrophilic properties[321]. The K and S dopants provide g-C3N4 with a more negative surface charge to adsorb O2. A previous study demonstrated that the enhanced ECB of g-C3N4 accelerates oxygen reduction to generate 1O2 (Fig. 13c). The degradation rate for BPA of 0.61 h−1 at pH 8 was achieved over S, K-doped g-C3N4 under visible-light irradiation.
In addition to photocatalysis, g-C3N4 has been applied to organic pollutant degradation via Fenton and AOPs. Jing et al.[315] prepared coral-like B-doped g-C3N4 to degrade 4-CP through a photocatalytic self-Fenton reaction. In this system, two degradation paths were achieved, with e− in the CB and h+ in the VB of g-C3N4 as the responsible agents (Fig. 13b). Hydrogen peroxide was generated in situ to promote the Fenton reaction via the ORR on g-C3N4 without the need for additional reagents. Furthermore, the VB holes were capable of directly degrading 4-CP. A mineralization rate of 70.3% was observed for this synergistic system, which was 2.6 and 4.9 times higher than those of the Fenton process and photocatalysis, respectively. The π-conjugation of g-C3N4 gives it a strong affinity to some active substances and promotes AOPs. Cao et al.[317] integrated g-C3N4 with CuS to activate PMS and induce AOPs for TC degradation. The transfer of photoinduced electrons between g-C3N4 and CuS accelerated the Cu(II)/Cu(I) redox cycle to produce the reactive oxygen species 1O2, •O2−, •OH, and h+. CuS/g-C3N4 exhibited the highest adsorption energy, which results in accelerating the spontaneous progress of the reaction (Fig. 13e). As a result, 97.46% of TC was removed within 30 min with 35% CuS/g-C3N4 and PMS under visible-light irradiation.
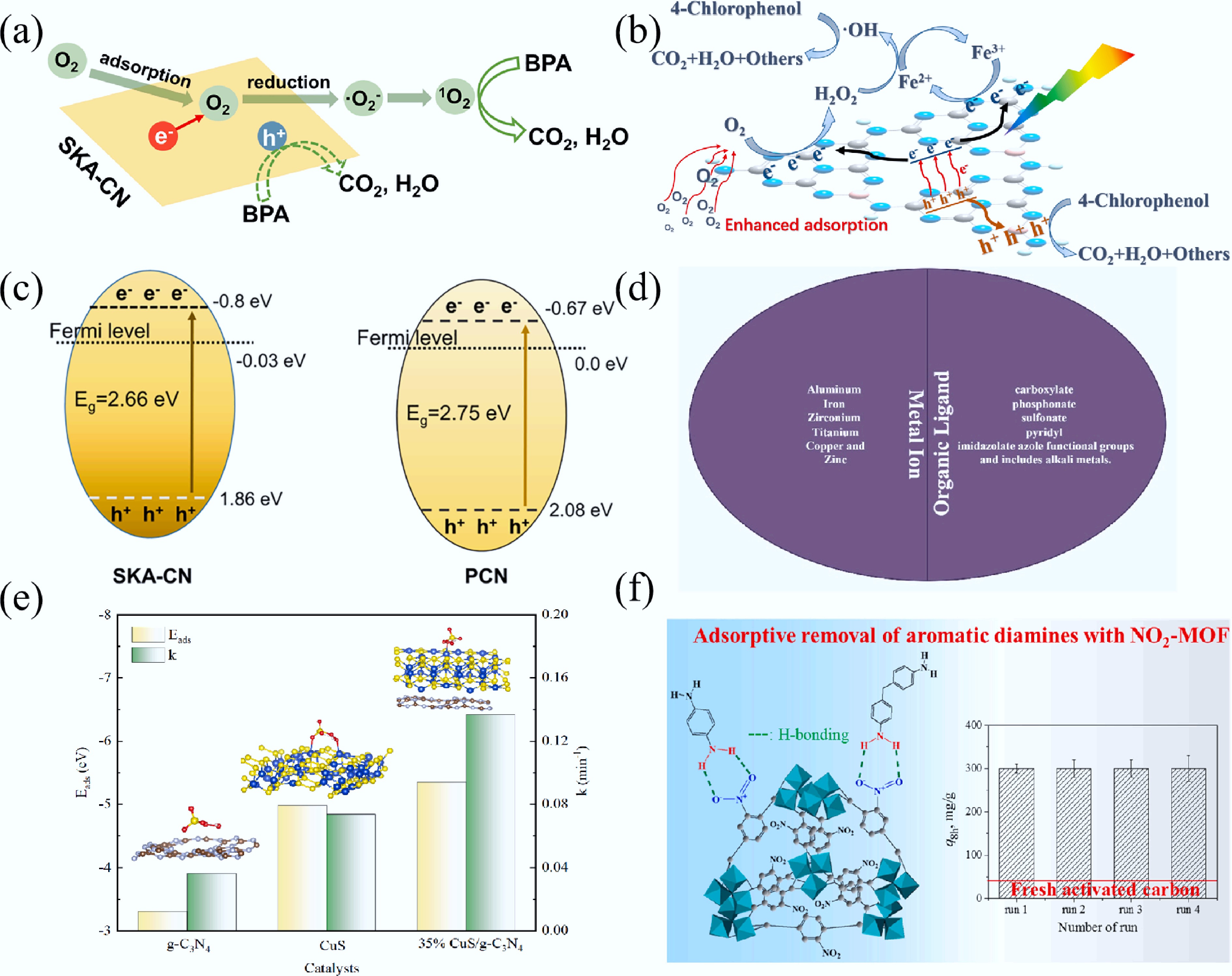
Figure 13.
(a) Schematic illustration of BPA degradation through 1O2 generated by SKA-CN[314]. (b) Schematic illustration of photocatalysis-self-Fenton degradation of 4-CP over Coral-B-CN[315]; (c) Band diagrams of SKA-CN and PCN[314]. (d) Composition of MOFs[316]. (e) Relationship between Eads and k (inset: geometric configurations for PMS adsorption on catalysts)[317]. (f) Illustration of the H-bonding between diamines and NO2-MOF and the recyclability[318].
Metal-organic synergistic carbon-based materials
MOFs
-
MOFs, composed of metals (or metal clusters) bonded with organic linkers are characterized by tunable pore sizes, large surface areas, and excellent porosities[322] (Fig. 13d). In a manner analogous to that of COFs, the porosity and pore size can be regulated through the utilization of diverse synthesis methods and the employment of a variety of organic ligands. This attribute confers the potential for application across multiple domains. Following the initial report on MOFs in 1995 by Yaghi et al.[323], numerous synthesis methods have been proposed. The most prevalent preparation methods are hydrothermal or solvothermal synthesis[324], which involve subjecting metal salts and organic ligands to high-temperature and high-pressure reactions in various solvents. The microwave method involves a solvothermal reaction in which polar molecules collide to produce a thermal reaction within an alternating electromagnetic field[325]. This reaction typically requires a shorter preparation time for nanosized crystals. The room-temperature solution reaction method is characterized by its ease of execution, safety, and reliability. In many cases, this is accomplished by stirring the mixture at room temperature. However, this method can be time-consuming, and the products have undesirable stability, as evidenced by the preparation of ZIF-67[326]. A plethora of MOFs exists, often exhibiting significant disparities in synthesis processes and production costs. However, ZIFs synthesis, accomplished through stirring at ambient temperature, harbors the potential for large-scale production at minimal expense.
MOFs for organic pollutant removal
-
In contrast to COFs, the incorporation of metals is advantageous in terms of the asymmetric distribution of charge within the metallic and nonmetallic centers. This phenomenon leads to the augmentation of the adsorption and reaction-active sites. Lee et al.[318] constructed nitro-functionalized MOFs (MIL-101(Cr)-NO2) to remove methylenedianiline (MDA) and PPD through adsorption. Experiments confirmed that the enhanced adsorption capacity is attributed to the formed hydrogen bond between -NO2 of the MOF and -NH2 of the PPD and MDA. The π–π interaction between the aromatic rings and pollutants contributed to the adsorption of MDA, and the activity remained even after four cycles (Fig. 13f). Hydrogen bonding-based absorbents exhibit unique directionality towards -NH2-containing pollutants. This study provides a foundation for the fabrication of different functionalized MOFs to achieve the efficient adsorption of specific pollutants. The π-clean aromatics of MOFs enable convenient tuning, which is a significant advantage. The metal center of the framework introduces supplementary active sites and endows MOFs with a superior photoresponse and charge mobility. The manipulation of the metal center facilitates the employment of a range of scenarios. In addition to its use as an adsorbent, its charge-transfer ability is involved in catalytic applications. Tong et al.[327] fabricated an FeCu bimetallic MOF (FeCuBDC) to achieve the efficient photo-Fenton synergistic degradation of phenol. The fabrication of FeCuBDC was accomplished by introducing Cu into FeBDC using solvothermal method. The incorporation of Cu into its organic links has been demonstrated to enhance the light-utilization rate, particularly in the region > 420 nm. This process has also been shown to reduce the band gap, thereby facilitating excitation by light, as evidenced by the ultraviolet-visible diffuse reflectance spectroscopy curves. The valence cycling of Fe and Cu in bimetallic sites greatly facilitates carrier migration between the catalyst and target reactants, and the phenol is degraded by the photoinduced holes and •OH generated by the Fenton reaction (Fig. 14a). The potential applications of MOFs in the removal of pollutants are significant, and their distinctive characteristics and structure, make them valuable for indirect utilization. As a constituent of the extensive category of MOFs, ZIFs are composed of metals (typically Zn or Co) and imidazolates. The unique micromorphology and suitable metal content of Co-ZIFs have led to their widespread use as a source of metal loading. Lei et al.[328] synthesized hollow octahedral ZIF-67 particles with diameters of approximately 200 nm. These particles were used as the metal source to load Co NPs onto cow-manure biochar (Fig. 14c). This enabled the efficient degradation of carbamazepine through the recycling of Co2+- and Co3+-based PMS advanced-oxidation reactions. Using MOFs as templates prevents the agglomeration of metal NPs and increases the number of reactive sites.
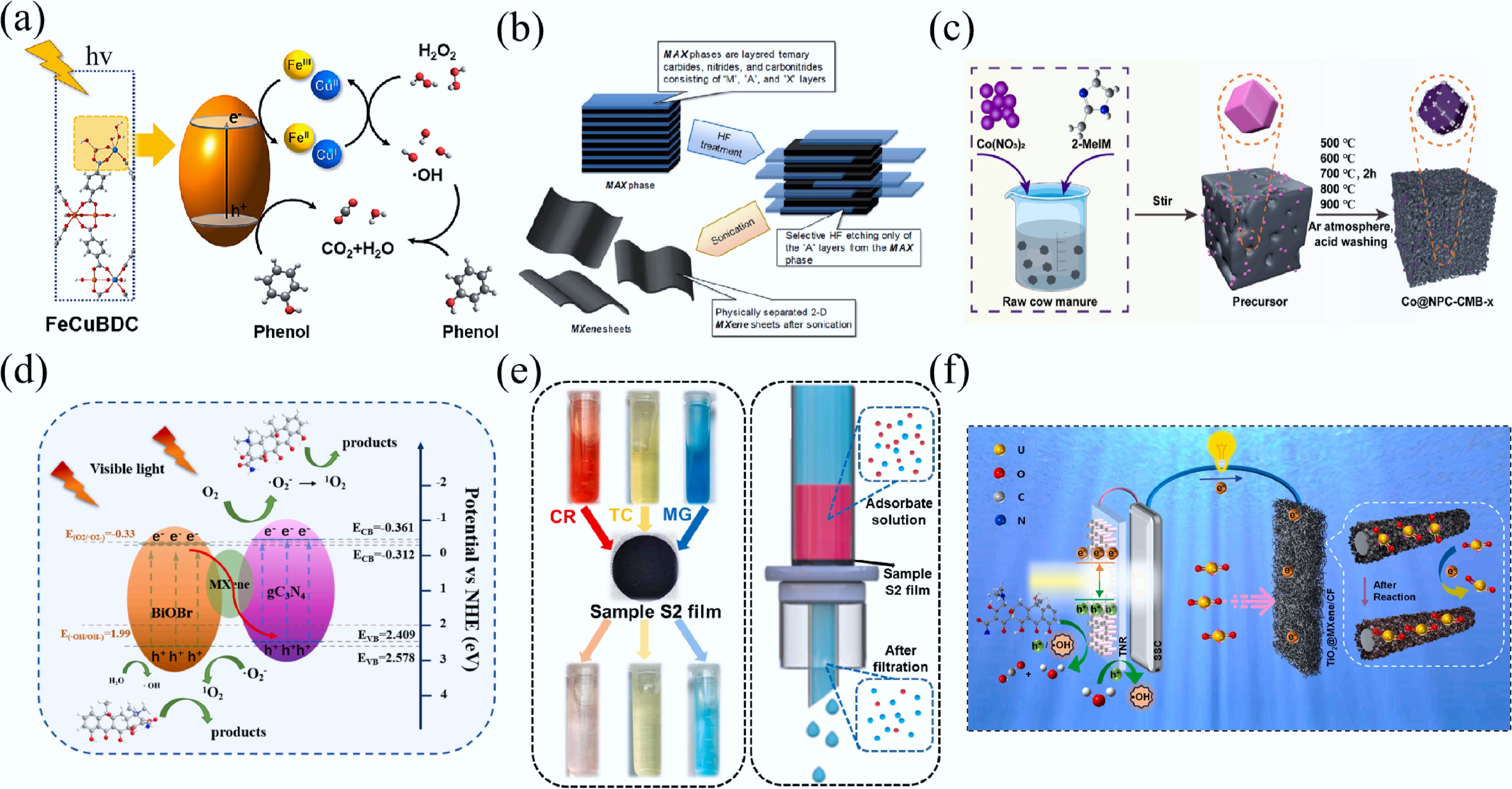
Figure 14.
(a) Schematic illustration of phenol degradation over FeCuBDC photo-Fenton system[327]. (b) The exfoliation of MAX phases and formation of MXenes[332]. (c) The schematic of the Co@NPC-CMB-x synthesis[328]. (d) Schematic illustration of charge transfer process on CBM for TCH degradation[333]. (e) Photographic image of sample S2 films for filtration of adsorbates and the schematic illustration of the filtering mechanism[334]. (f) Proposed mechanism of the self-driven solar coupling system (SSCS) for simultaneous uranyl ions (UO22+) reduction, organic pollutant oxidation and electricity production under sunlight[335].
MXenes
-
MXenes, which are novel 2D materials, were first fabricated in 2011[329], leading to a surge of interest in their applications in energy storage, environmental restoration, and optical devices. These materials are famous for the substantial surface area, remarkable chemical stability, and notable thermal and electrical conductivities[330]. MXenes are composed of ternary carbides and nitrides, and their generation is achieved by etching element A in the MAX phase (Fig. 14b). The common formula for MXenes is Mn+1AXn, where M represents transition metals, A represents group A elements (mostly IIIA and IVA, such as Al and Si), X represents nitrogen or carbon, and n is an integer ranging from one to four[331]. After etching, multilayered Mn+1XnTx (n = 1–3) is produced, where T is identified as exterior termination groups, such as fluorine, oxygen, chlorine, and hydroxyl (–F, –O, –Cl, and –OH), and x indicates various surface functionalities[330].
The predominant etching method involves the use of HF to remove weakly bonded layers[332]. However, HF contributes to environmental pollution and poses a significant threat to human health. In contrast, a less aggressive method employing fluoride-based salts (NaF, LiF, and KF)[331] in conjunction with HCl has been proposed as an alternative. The use of disparate etching methodologies can give rise to divergent terminal groups, thereby influencing their chemical and physical properties. It is evident that alternative etching methods have been developed that are both more efficient and environmentally friendly. Xie et al.[336] proposed a method that utilizes alternative reagents such as NaOH and H2SO4. Tu et al.[337] introduced an etching method based on a eutectic molten salt. Following the etching of the A layers, the resultant material is an organ-shaped MXene, which then undergoes an exfoliation process, typically, sonication, to obtain 2D MXene nanosheets. At the present research stage, HF etching has been identified as the most efficient means of processing, with the capacity to achieve 50 g of MXenes etching in a single pass. However, further investigation is required to fully explore the large-scale intercalation and delamination processes[338].
The introduction of metal sites and terminal functional groups enables MXenes with higher adsorption and more active sites to interact with organic pollutants via electrostatic and hydrophobic interactions, ion exchange, and hydrogen bonds[339]. Moreover, the combination of high electron density, metallic conductivity, and ultrathin nanostructure produces the material with a high charge-transfer ability. These characteristics make MXenes a leading candidate in the field of organic matter removal. Gao et al.[333] designed an MXene (Ti3C2Tx) quantum-dot synergistic g-C3N4 and BiOBr ternary catalyst CBM (g-C3N4/BiOBr/MXene), to achieve efficient tetracycline hydrochloride (TCH) degradation. The high electronic conductivity of the MXene quantum dots led to the formation of Schottky junctions between g-C3N4 and BiOBr effectively preventing the recombination of photoinduced carriers. The overall charge transfer was in accordance with the Z-scheme transfer mode, with the MXene functioning as a charge-transfer channel. The highly separated e- and h+ were concentrated in the CB of g-C3N4 and VB of BiOBr, respectively, resulting in higher oxidation and reduction potentials. The efficiently separated carriers enabled the simultaneous generation of reactive oxygen radicals via the ORR and WOR pathways to degrade TCH with a 99% removal rate by CBM within 30 min (Fig. 14d). The excellent electronic conductivity and light-absorption ability of MXenes makes them widely utilized in electrocatalysis. A bifunctional electrode material, TiO2@MXene/CF cathode-based SSCS, was fabricated by Guo et al.[335] to simultaneously degrade TCH and reduce UO22+. TiO2 was grown in situ on MXene, and TiO2@MXene was attached to carbon felt to form the cathode. An additional applied bias voltage was demonstrated to contribute to the separation of the photogenerated carriers. This process caused electrons to migrate from the TiO2 nanorod array (TNR) anode to the TiO2@MXene/CF cathode, thereby reducing UO22+. In contrast, holes were left to directly oxidize TCH through •OH (Fig. 14f). The incorporation of MXenes has been demonstrated to enhance electronic reception, thereby increasing the number of active sites and facilitating the degradation of organic pollutants. The removal efficiencies of UO22+ and TCH have been shown to reach 99.7% and 97.5%, respectively.
In addition to applications in the optical and electrical fields, the large surface area of MXenes, coupled with their excessive surface functional groups, makes them advantageous for the adsorption of pollutants. To increase their surface area and protect MXenes from oxidation, Wang et al.[334] grew in-situ ZIF (ZIF-67/ZIF-8) particles on the interlayers and surface of alkalized MXene to achieve the efficient degradation of CR, TC, and malachite green (MG). The efficient adsorption performance derived from the synergistic effects of electrostatic interaction, hydrogen bonding, and π-π stacking resulted in an excellent adsorption capacity. One of the samples (S2) exhibited optimal adsorption performances with qm of 539.7, 1,053.3, and 7,111.3 mg/g for CR, TC, and MG, respectively. The stability and practical applications were validated through a filtration system using S2, and the removal rate of these three pollutants remained at 90% even after five cycles (Fig. 14e).
Metal ions
Introduction to metal ion pollution
-
Metal ion pollutions in wastewater mainly consist of heavy metals, which rank among the most critical carcinogens threatening global water safety due to their high toxicity, and non-degradability[340–342]. Given that heavy metals can bioaccumulate and biomagnify through biological chains, thereby constituting a substantial threat to human health and ecological integrity[343,344]. Thereby, their effective removal is of critical importance[345,346]. The sources contributing to heavy metal accumulation are geological, soil, surface water, anthropogenic, and industrial activities[347]. Natural geological sources, such as weathering and hydrothermal alteration of all types of rocks, allow for increased mobilization and concentration of heavy metals, including Ni, Co, Mn, Zn, Cu, V, Sc, Y, and rare earth elements[348]. Soils act as one of the major reservoirs for heavy metals, with their concentrations greatly influenced by rock type. Typically, heavy metals are accumulated in the B-horizon (the third layer soil) due to downward migration of dissolved metals from the A-horizon (topsoil), where they are retained by iron oxyhydroxides and clay through ion interactions within the B-horizon[349]. Surface waters, including springs, streams, and rivers, transport heavy metals across long distances, leading to a large area of migration. The identity and concentration of heavy metals are influenced by geological, biological, and physicochemical factors, such as pH, redox potential, and temperature, which can enhance solubility and lead to bioaccumulation and toxicity through adsorption onto sediments, algae, and their entry into the food web[350]. In the case of human activities (agriculture, urban runoff, and waste disposal), pollutants can be generated in large quantities, aiding in the redistribution of heavy metals through environmental pathways, leading to toxicity in plants, animals, and humans. Heavy metals from fertilizers (As, Cd, and Zn), sewage (B, Cd, Cu, Pb, Ni, and Zn), and historical pesticides (As, Pb, and Hg) are key contributing factors that significantly add to this pollution. Moreover, one of the major heavy metal pollution sources is industrial activities, especially mining, electroplating, chemical industries, textile industry, and paper manufacturing. Particularly in regions lacking strict environmental regulations, where these processes are allowed to release higher levels of toxic metals into soil, water, and the atmosphere[351]. Considering the environmental hazards, the most commonly recognized and intensively investigated "heavy metals" include Pb, Hg, Cd, As, Cr, Ni, Cu, Zn, and Mn. Table 3 summarizes the major environmental sources and toxic effects on human health of the aforementioned heavy metals[352].
Table 3. Heavy metals and their sources, toxic effects
Heavy metal Major environmental sources Toxic effects on human health Ref. Pb Battery manufacturing, paint, contaminated soil, old plumbing Neurotoxicity, cognitive impairment in children, kidney damage, hypertension [738] Hg Coal combustion, gold mining, seafood (methylmercury) Neurotoxicity, tremors, vision/hearing loss, developmental defects in fetus [739,740] Cd Industrial emissions, phosphate fertilizers, smoking, mining Renal dysfunction, bone demineralization (Itai-itai disease), cancer [741] As Groundwater (natural/geogenic), mining, pesticides The most poisonous heavy metals, skin lesions, cancer (lung, bladder, skin), cardiovascular diseases [742] Cr Electroplating, leather tanning, pigments, steel manufacturing Carcinogenic, respiratory tract irritation, skin ulcers [743] Ni Stainless steel production, electroplating, combustion Dermatitis, lung fibrosis, carcinogenic potential (inhalation) [744] Cu Mining, plumbing, industrial waste, pesticides Gastrointestinal distress, liver/kidney damage at high doses [745] Zn Galvanized metal, industrial discharges, fertilizers Nausea, vomiting, immune system suppression (at excessive levels) [746] Mn Welding fumes, industrial emissions, contaminated water Neurological disorders resembling Parkinson's disease [747] Since heavy metals accumulate from many different sources and have a great impact on human health and the environment, the remediation of these metals can be challenging. A variety of methods that have been developed as potential solutions to these challenges are as follows, ion exchange[353], membrane separation[354,355], precipitation[356], electrodeposition and hybrid approaches[357]. However, since most of the above removal strategies suffer from drawbacks such as effectiveness, high cost, low design/operation flexibility, and secondary pollution generation[358], therefore adsorption technologies have been extensively utilized as alternative remediation methods, such as CMs[359,360]. CMs are widely regarded as one of the most effective adsorbents for heavy metal removal due to large surface, high porosity and versatile adsorption mechanisms. Moreover, the rich oxygen-containing functional groups on the surface of CMs enable electrostatic interactions, complex formation, and ion exchange with the metal ions[359,361]. In this context, various carbon adsorbents such as AC, CNTs, graphene, GO, BC, and other emerging CMs like CDs have been developed and applied for the removal of heavy metals. Key considerations for selecting adsorbents for industrial applications include cost-effectiveness and compatibility with the specific technology[362].
Mechanisms of metal ion removal by CMs
-
In general, the capture of heavy metal ions is due to the interactions between metal ions and functional groups on the surface of CMs[363]. These inter-molecular interactions are complicated and often include single or multiple mechanism as physical adsorption, ion exchange, surface complexation, electrostatic interaction, and precipitation (Fig. 15), which are not only influenced by operation conditions (such as solution temperature, solid-to-liquid ratio, pH value, reaction time, and initial concentration of metal ions), but also depend on the target metal ions, ionic environment of the solution, and the properties of adsorbents[364].
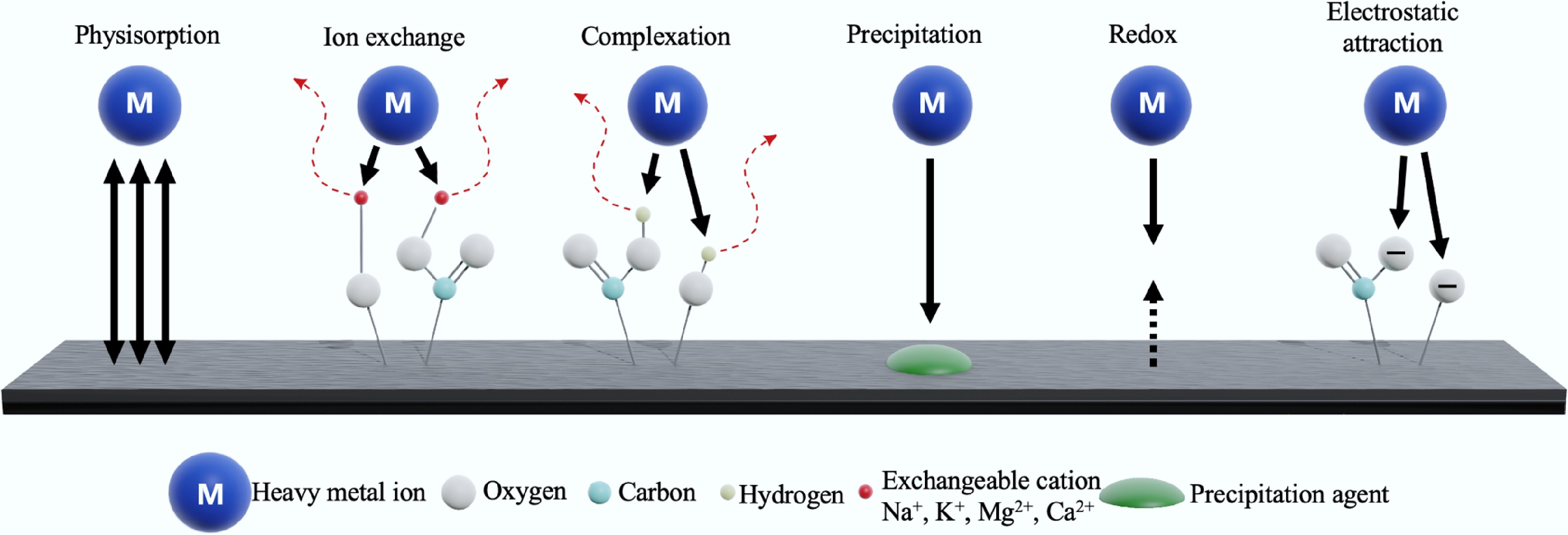
Figure 15.
Schematic diagram illustrating major metal ion removal mechanisms on carbon materials surfaces.
Physical adsorption
-
During the process of metal ions entering micropores and mesopores of porous CMs and being deposited onto the surface, the surface properties and the pore structure of the CMs contribute significantly to uptake capacity, and adsorbents can normally be regenerated[365].
Ion exchange
-
The interaction between heavy metal cations and exchange sites, where the heavy metal cations interact with protons from oxygen-containing functional groups such as carboxyl (−COOH) and hydroxyl (−OH) on carbon adsorbents, is normally attributed to ion exchange[366]. Cation exchange capacity (CEC) is the main indicator for removal efficiency, which is significantly influenced by pH. Moreover, the ionic radius, temperature, contact time, and kinetics are also important factors for the ion exchange process in heavy metal removal by CMs[367].
Surface complexation
-
In adsorption processes, surface complex formation is commonly regarded as the interaction between cations and molecules or anions that possess lone electron pairs. This mechanism provides insight into the physicochemical interactions occurring between heavy metal ions and the surface of CMs. Surface complexation is typically stronger than physical adsorption or ion exchange and plays a dominant role at the following conditions: i) functional groups like –COOH, –OH, and –C=O are present; ii) metals form coordination bonds with surface oxygen or nitrogen atoms; iii) specific adsorption sites chemically interact with metal ions[368].
Electrostatic interaction
-
This process refers to the interaction between charged metal ions (typically cations like Pb2+, Cd2+, and Zn2+) and oppositely charged surface sites of CMs, especially when functional groups are present[369]. Unlike physical adsorption, surface complexation, and ion exchange, the electrostatic adsorption only plays a secondary role in the adsorption of heavy metals onto carbon adsorbents and is purely Coulombic, and pH-dependent[363].
Precipitation
-
Precipitation involves the formation of insoluble metal compounds (commonly hydroxides, carbonates, or sulfides), usually either on or near the surface of CMs, or within surface pores[370]. CMs often indirectly promote precipitation by increasing local pH, providing nucleation sites for metal hydroxide crystals, or hosting inorganic functional groups (e.g., Si–OH, CaCO3 in biochar) that react with metal ions. Precipitation is one of the primary mechanisms for heavy metal removal and often operates in conjunction with other processes, including ion exchange, electrostatic attraction, and surface complexation[371].
Types of CMs for metal ion capture
AC
-
Although AC has a large surface area and high porosity, making it a promising adsorbent, many of the pores are too small for heavy metal ions. Therefore, it is necessary to enlarge the pores to enhance adsorption efficiency. One strategy for pore enlargement is chemical activation (such as NaOH activation), which is one of the most effective methods for enlarging the micropores of AC[359]. There are various mesoporous-AC materials, such as: chitosan flakes AC fiber (CS/ACF) generated via CS loading and activation in sodium hydroxide activation for the adsorption of Pb2+-ethylenediaminetetraacetic acid (EDTA) complex[372], zeolite-AC (Z-AC) composites through CO2 activation and hydrothermal treatment method using coal gangue as raw materials for Cu2+ and Rh-B adsorption[373], plum (Vojvodina, Serbia) stone (kernels and shells) AC generated by H3PO4 treatment and annealing for Cd2+, Pb2+ and Ni2+ removal[374]. Additionally, Vasiraja et al.[375] developed a green and cost-effective AC, which was ZnCl2-activated prosopis juliflora stem carbon (ZAPJSC) with a high surface area of 252.7 m2/g. The ZAPJSC adsorption performance of textile industry effluent was tested, where heavy metals, including Pb, Cu, Fe, Co, and Mn, were removed with removal rates of 9.42%, 11.52%, 100%, 66.66%, and 71.42%, respectively. Moreover, it demonstrated its reusability by maintaining a removal efficiency of 73.34% after four cycles in MB removal. As mentioned above, the adsorption capacity of AC to Pb2+-EDTA was enhanced, leading to the development of SC/ACF for treating wastewater via electroplating[372]. SC/ACF can reach adsorption equilibrium within 30 min with a removal rate of 99.40% as shown in Fig. 16a. In the presence of competing ions, Cl−, HCO3−, SO42−, PO43−, F−, and EDTA2−, the removal rate of CS/ACF still remains above 97%. The key adsorption mechanism was characterized and analyzed as both ligand exchange and inner-sphere complexation. Additionally, CS/ACF was shown to have good reusability for Pb2+-EDTA adsorption over 10 regeneration cycles[372].
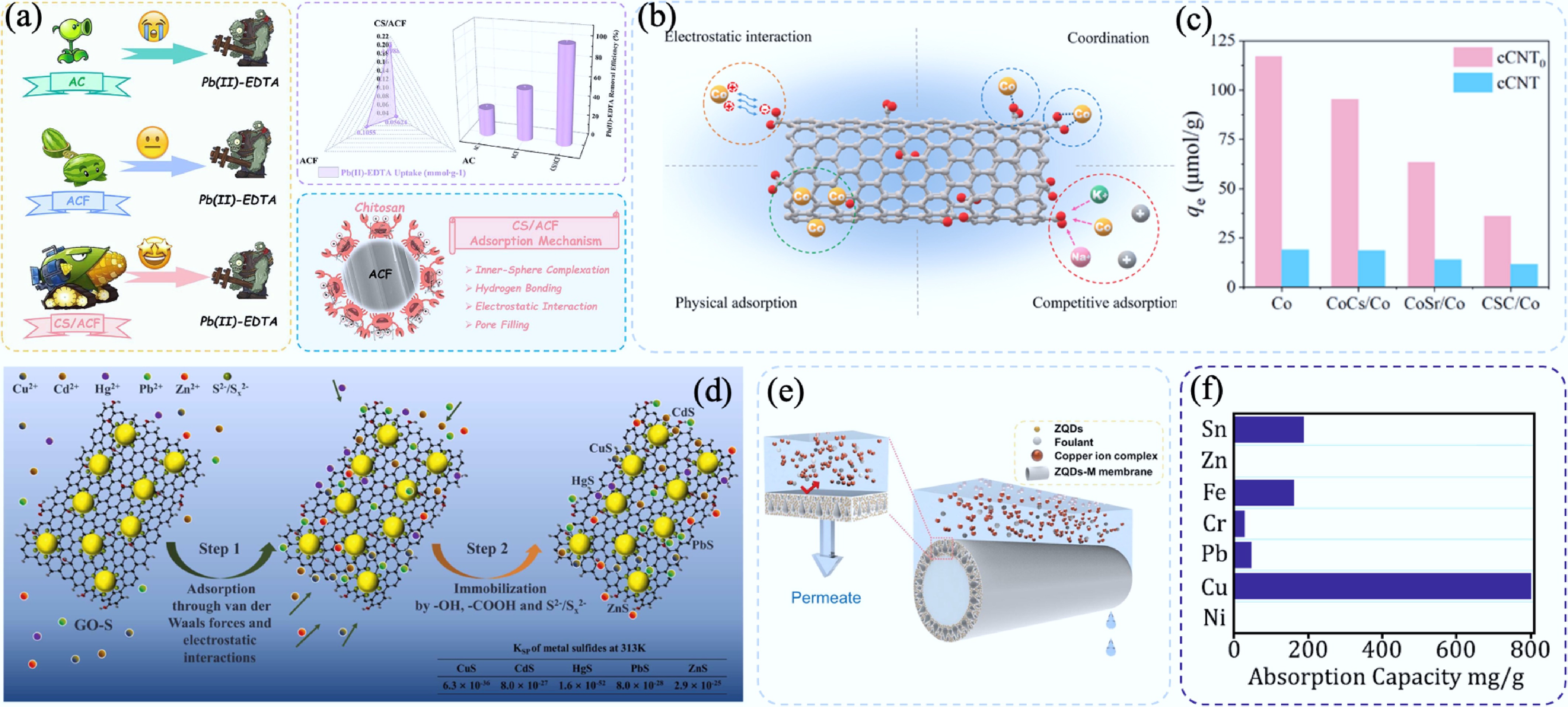
Figure 16.
(a) Schematic diagram of functionalized CS/ACF and its adsorption performance for Pb2+-EDTA[372]. (b) Schematic diagram showing the adsorption mechanism of cCNT and (c) adsorption capacity of Co2+ in simulated seawater[387]. (d) Schematic diagram of the mechanism of heavy metal ions adsorbed by GO-S[395]. (e) Schematic diagram of ZQDs-M removal Cu2+[401] and (f) adsorption capacities of CA for a variety of heavy ions[402].
Another strategy to improve heavy metal removal is to enhance the interaction between heavy metal ions and adsorbent, specifically by increasing the affinity of the active sites on the adsorbent. One effective approach involves forming surface complexes through carbon-sulfur bonding. According to Pearson's rule, certain heavy metals, such as Cd, exhibit a strong affinity toward sulfur-containing groups. Based on this principle, Tajar et al.[376] used nut shells as a precursor to synthesize AC and introduced sulfur using SO2 as the sulfurizing agent. The resulting sulfur-modified AC showed significantly enhanced affinity for Cd2+ in aqueous solutions. In the case of Pb2+, the presence of carboxyl functional groups on the surface of AC plays an important role in its removal[377]. A thiol functionalized activated CF was developed by Zhu et al., demonstrating qmax of Pb2+, which was fitted to be 700.77 mg/g by the Langmuir isotherm model[378].
AC is typically used in neutral or weakly acidic solutions, where it exhibits the strongest adsorption of heavy metals. Moreover, the adsorption capacity of reused AC generally remains stable over multiple cycles. Due to its high surface area (up to 3,000 m2/g) and excellent adsorption performance, AC is the most widely used commercial carbon material for heavy metal treatment.
CNTs
-
It has been proven that CNTs are a very efficient technology for treating heavy metals such as Pb2+[379], Cd2+[380], As(V)[381], and Hg2+[382] from water. There are two classifications of CNTs: SWCNTs, which consist of a single graphene sheet, and MWCNTs, which are composed of multiple concentric graphene layers, both providing a large surface area[383]. Lu & Chiu utilized commercial SWCNTs and MWCNTs purified in sodium hypochlorite solutions to achieve Zn2+ adsorption, reaching maximum adsorption in the pH range of 8–11 within 60 min[384].
CNTs are highly effective adsorbents due to their physio-chemical properties. However, CNTs require functionalization with specific groups to enhance adsorption capacity and selectivity toward heavy metals[385]. The most reactive functional groups commonly adopted for the surface functionalization of CNTs are carboxyl and hydroxyl groups[386]. Commercial carboxyl-functionalized CNTs (cCNTs) were utilized as adsorbent by Wang's group[387], showing promising adsorption capacity for Co2+ removal from wastewater, where the qmax was 25.1 mg/g at 30 °C. Good selectivity for Co2+ was observed when Sr2+ and Cs+ were present (Fig. 16b and c). Using DFT analysis, the adsorption mechanism was realized, consisting of a combination of electrostatic interactions and coordination[387].
Compared with AC, CNTs offer more functional adsorption sites and higher adsorption capacity for heavy metal ions, particularly in neutral or weakly acidic solutions. However, CNTs typically exhibit lower reusability and face challenges in regeneration. Overall, although AC remains the most common commercialization carbon material for heavy metals removal, the utilization of CNTs has recently emerged as a highly effective alternative.
GOs
-
The use of GOs is regarded as one of the most efficient approaches for heavy metal removal due to their large SSA and ability for enrichment with oxygenated functional groups[388]. In wastewater pollution treatment, GOs have been applied for the adsorption of heavy metals such as Pb2+[389], Cd2+[390], Cu2+[389], and Cr(VI)[391,392]. Zhang et al. developed a GO-based macroscopic adsorbent, DGSP-1, incorporating SA and poly(vinyl alcohol) (PVA), using 3D printing technology. The adsorption capacity of DGSP-1 for Cu2+ is 56.2 mg/g at pH 5 (30 °C), primarily due to the oxygen-containing functional groups in the cross-linker, leading to the potential utilized for mining wastewater[393]. The interaction between GOs and heavy metals is mainly limited to electrostatic interactions, which highly depend on the number of sorption sites available[394]. Enhancing the affinity of metals onto GOs can also be achieved through NP loading, as GOs serve as nucleation sites and anchoring platforms that restrict the growth of NPs more effectively than biochar, alumina, g-C3N4, or graphene[86,395]. Fan et al. fabricated sulfur NPs loaded GO (GO-S) using a light-driven in-situ synthesis method. The presence of abundant S2−/Sx2− species on the surface of GO-S enabled the adsorption and immobilization of heavy metal ions, including Cu2+, Cd2+, Hg2+, Pb2+, and Zn2+, through complex formation (Fig. 16d). Moreover, the adsorption capacity of GO-S was 1.7–12.4 times higher than that of raw GOs[395].
GOs have shown excellent removal performance for heavy metals due to their large surface area, diverse functional groups, and high charge mobility. They have demonstrated removal efficiencies of ~99% for Cr(VI), As(V), and Pb(II) down to the μg/L level, while also exhibiting antibacterial activity against E. coli and low toxicity toward zebrafish. Similar to CNTs, GOs have emerged as promising carbon-based materials for heavy metal ion removal.
Emerging CMs
-
Recently, CDs, CAs, and other low-dimensional CMs have emerged as a new class of heavy metal treatment adsorbents, offering high photo/thermal stability, outstanding biocompatibility, and low environmental toxicity[396,397]. CDs, specifically, are extensively utilized as fluorescent probes owing to their exceptional optical properties, biocompatibility, and low toxicity, and their strong fluorescence makes them highly promising for the trace detection of metal ions such as Cr(VI), Fe3+, and Hg2+[398]. Thus, CDs are often applied in the form of composites for the removal of heavy metal ions. Ryabchenko et al. developed a CD-loaded cellulose-condensed silica composite (CD/HPMC/SiO2) for the detection and removal of Pb2+ and Co2+ from wastewater. The composite achieved adsorption capacities of 258 and 268 mg/g for Pb2+ and Co2+, respectively. Additionally, CD/HPMC/SiO2 maintained over 90% removal efficiency after three reuse cycles[399]. Luo et al. integrated amino-modified CDs into a 3D porous hydrogel, which showed both high adsorption capacity (534.4 mg/g) and sensing ability for Cr(VI)[400]. Yang et al. synthesized a zwitterionic-modified CDs added cellulose acetate tubular membrane (ZQDs-M) and investigated its application for purifying Cu2+ contained wastewater (Fig. 16e). Initially, 99.8% of Cu2+ was chelated using sodium dimethyldithiocarbamate (SDDC), and the resulting Cu complexes were removed by ZQDs-M with a rejection rate of 95.4% and a pure water flux of 6,277.4 LMHB[401].
In the case of CAs, oxygen-containing functional groups on the surface of CAs also contribute to the adsorption of heavy metals from aqueous solutions[403]. Li et al. developed a novel hydroxyl-rich CA using cellulose colloid, which achieved an adsorption capacity of 55.25 mg/g for Cu2+ within 10 min, at pH 7 and 25 °C. Furthermore, after five adsorption-desorption cycles, the Cu2+ removal rate still remained at 96%[404]. Wang et al. synthesized a cellulose-based CA and applied it for sewage treatment to remove oils, organic solvents, dyes, and heavy metal ions. Specifically for Cu2+, the adsorption capacity reached 801 mg/g (Fig. 16f), highlighting the strong potential of CAs for industrial-scale sewage treatment[402].
Table 4 shows the removal of heavy metals by ACs, CNTs, GOs, CDs, and CAs, with a comparison of adsorption capacity and regeneration performance of different CMs. The presence of heteroatoms (such as N, O, or S) in CDs enhances their affinity for heavy metal ions by introducing additional active sites for adsorption via coordination or electrostatic interactions. However, pristine CDs are typically not employed directly for heavy metal removal, as their fluorescence properties are susceptible to quenching upon metal ion binding, which may limit their functional stability. To overcome this limitation, CD-based composites with high surface and porous structure are widely developed. These hybrid materials not only retain the advantageous surface chemistry of CDs but also offer improved adsorption performance, structural stability, and multifunctionality for efficient removal of heavy metal ions from aqueous environments. Similarly, CAs, particularly those doped with heteroatoms or functionalized with specific surface functional groups, provide a rapid, selective, and recyclable platform for heavy metal removal. Their highly porous and often hydrophobic nature significantly enhances contaminant separation efficiency and selectivity in aqueous systems.
Table 4. Removal of heavy metals onto carbon adsorbents
Carbon materials Dosage
(g/L)Heavy metals Initial concentration
(mg/L)pH Temperature* (°C) Time
(min)Adsorption capacity
(mg/g)Removal efficiency (%) Reusability Ref. ACs BAC 0.1525 Pb2+ 0.2 − 25 120 1.21 95% Reuse of spent AC [366] 0.1032 Cd2+ 0.50 86% RAC − Hg2+ − 5.5 25 360 109.05 ~100% ~100% by MWH after five cycles [382] CS/ACF 1 Pb2+−EDTA 0.2 mM 7 25 30 − 99.40% Stable after 10 cycles of regeneration [372] APS oxidized ACF 2 Pb2+ 4.5−5.0 25 120 559 − − [377] Thiol functionalized activated CF Pb2+ 100 5.5 25 120 700.77 − Adsorption capacity remain 150 mg/g after five cycles. [378] Sulfur-modified AC 5 Cd2+ 1,000 6.0 25 240 139 27.8 − [376] CNTs e-MWCNTs 100 As(V) 0.047-10.2 3 35 45 12.18 − − [381] MWCNTs/MnO2 0.2 As(V) 20 5.5 25 60 − 95% Can be regenerated [748] cCNTs 0.2 Co2+ 10 5.7 30 < 60 25.1 − − [387] CNTs 0.5 Pb2+ 2-14 7 25 6 h 17.5 87.8 − [379] CNT-S 1 Hg2+ 10 6 25 90 151.5 Around 13% and 17% dropped in second and third cycle, separately [740] GOs GO/UiO-66-NDC 2.5 Cr(VI) 10 mM 3 25 150 157.23 96.4% Dropped to 71.23% after six cycles [391] GO 0.1 Cu2+ − 5 25 120 294 − − [394] Zn2+ 345 Cd2+ 530 Pb2+ 1,119 GO-AG 0.03 Cd2+ − 3-4 25 12 h 1,792.6 − − [390] DGSP 1 Cu2+ 20 5 30 240 208 Remained at 71.54% of the initial removal efficiency after five cycles [393] GO-S 0.05 Cu2+ 150 5 25 60 678 − Remain at 83% of the initial removal efficiency after five cycles [395] Cd2+ 646 Hg2+ 565 Pb2+ 683 Zn2+ 490 CDs FH-5 − Cr(VI) − − − < 120 534.4 − − [400] Ba2+ 271.9 Pb2+ 789.6 Cu2+ 98.2 ZQDs-M − Cu2+−SDDC 4 ppm Cu2+
10 ppm SDDC10 25 − − 95.4%
(pure water flux of 6,227.4 L/[m2·h1·bar1])Remain 73.4% flux recovery ratio after three cycles [401] CD/HPMC/SiO2 1 Pb2+ 1,000 6 15 190 s 258.3 − Remain 90% for Pb2+ after three cycles, and for Co2+ after four cycles [399] Co2+ 5 20 300 s 168.1 CAs MCA 0.4 Cd2+ 60 mg/L 7 20 30 143.88 − Remain 93% of the initial removal efficiency after five cycles [403] Carbon aerogels 0.4 Cu2+ 400 25 12 h 801 − No obvious change after five cycles [402] CCA − Cu2+ − 7 25 10 55.24 − Remain 96% of the initial removal efficiency after five cycles [404] CA 1.6 Pb2+ − 4.5 − − 34.72 − − [749] Summary
-
CMs, especially ACs, CNTs, and GOs, are extensively utilized for heavy metal removal due to their high surface area and versatile surface chemistry. Among these, AC is the most widely commercialized adsorbent, valued for its porosity and large surface area, though its selectivity and regenerability can be limited. CNTs and GOs offer enhanced adsorption capacities and abundant functional sites, especially under neutral or mildly acidic conditions. Notably, GO also exhibits antibacterial activity and low toxicity, further broadening its applicability. In addition, CDs, while limited in direct application due to fluorescence quenching upon metal ion binding, have demonstrated significant potential when incorporated into porous composite materials. These composites improve both adsorption and structural stability. CAs, with their highly porous and often hydrophobic frameworks, enable rapid, selective, and recyclable heavy metal removal. Their performance can be further tuned via heteroatom doping and surface modifications. Table 3 summarizes the removal of heavy metals by ACs, CNTs, GOs, CDs, and CAs, with a comparison of the adsorption capacity and regeneration performance of different CMs.
The effectiveness of these materials is closely linked to their surface chemistry. Functional groups such as carboxyl, phenyl, and lactone moieties enhance surface charge and binding affinity toward metal ions. To further optimize performance, a variety of modification strategies, such as nitrogenation, oxidation, and sulfuration, are commonly applied. These approaches improve SSA, pore structure, adsorption capacity, thermal stability, and mechanical strength. As a result, carbon-based adsorbents are highly adaptable and hold great promise for future applications in water purification and decentralized treatment systems.
Radionuclides
Separation of uranium
-
As a pivotal strategic resource, uranium plays an indispensable role in nuclear energy production and national defense systems[405]. However, during nuclear reactor operations, residual uranium-containing materials may inadvertently enter ecosystems through aqueous pathways, posing significant ecotoxicological risks[406,407]. In response to these risks, the World Health Organization (WHO) and the US Environmental Protection Agency (USEPA) have established stringent regulatory thresholds, with the WHO guideline strictly limiting uranium concentrations in drinking water to 30 µg/L. Consequently, the development of efficient U(VI) removal technologies has emerged as both a scientific priority and a public health imperative[408].
Carbon-based materials have emerged as superior adsorbents for U(VI) remediation, owing to their economic viability, structural diversity, and tunable surface properties that facilitate enhanced contaminant sequestration[409]. Current research focuses on engineered carbon allotropes, including PC, CDs, CNTs, and GO, each demonstrating unique advantages in U(VI) adsorption. The foundational study by Kütahyalı & Eral[410] pioneered the use of ZnCl2-activated olive stone-derived AC (precursor/activating agent ratio = 1:2, SSA = 464.68 m2/g) for aqueous U(VI) removal. This work was expanded through systematic investigations of KOH-activated rice straw carbon (RSK carbon), which achieved optimal adsorption at pH = 5.5, with a 40 min equilibrium time, yielding a maximum capacity of 100 mg/g[411–413]. The findings revealed a concentration-dependent inhibitory effect of Fe3+ coexisting ions, reducing U(VI) removal efficiency to 77% and 20% at Fe3+ concentrations of 20 and 100 mg/L, respectively. This contrasts with recent strategies where controlled iron incorporation enhances U(VI) capture through surface charge modulation, demonstrating that ion-specific interfacial interactions govern adsorption dynamics.
The covalent interactions between carbon-based substrates and target ions exhibit substantially enhanced binding affinity relative to physical adsorption mechanisms, a critical advantage in selective contaminant sequestration. GO, synthesized through oxidative exfoliation of graphite (Fig. 17a, b), represents a quintessential 2D carbon derivative with an exceptional theoretical surface area of 2,630 m2/g[414]. Subsequent reduction strategies, encompassing chemical, thermal, microwave-assisted, flash photolytic, and microbial methodologies, transform GO into reduced GO (rGO, Fig. 17c), engineered with controlled oxygen residuals and strategically introduced carbon vacancies[419]. The spatially distributed oxygen functionalities, comprising epoxide and hydroxyl groups on basal planes alongside carboxyl and carbonyl moieties at sheet peripheries, establish molecular recognition architectures[420]. This remarkable performance originates from the abundant oxygen-containing functional groups on GO, including edge-localized carboxyl (–COOH) and surface-distributed hydroxyl (–OH) groups, which form stable surface complexes with U(VI) ions. Additionally, ion exchange between dissociated H+ ions and radionuclide species further contributes to U(VI) adsorption, as corroborated by the interfacial interactions[421]. Building on these advancements, researchers have strategically incorporated oxygen-containing functional groups (–OH, –COOH, –O−) into diverse carbon matrices, including AC[422], mesoporous carbon[423], CNTs[424], CNFs[425], and hydrothermal carbon (HTC)[426], to enhance U(VI) adsorption. Hierarchically PC adsorbents have become a focal point due to their structural advantages, with significant efforts dedicated to optimizing their design. Dong et al.[415] pioneered the synthesis of functionalized millimeter-sized hierarchically PC spheres (MMCs) via evaporation-induced self-assembly and suspension polymerization, which was shown in Fig. 17d. Through HNO3 oxidation treatment, abundant oxygenated groups were successfully grafted onto mesoporous carbon microspheres (OMMCs-S), achieving a remarkable U(VI) adsorption capacity of 232.45 mg/g at pH 4.5 and 298.15 K, nearly four times higher than that of unmodified MMCs. Meanwhile, after five cycles, the adsorption capacity still maintained 85% of the initial value, verifying the excellent structural stability and good reusability of OMMCs-S.
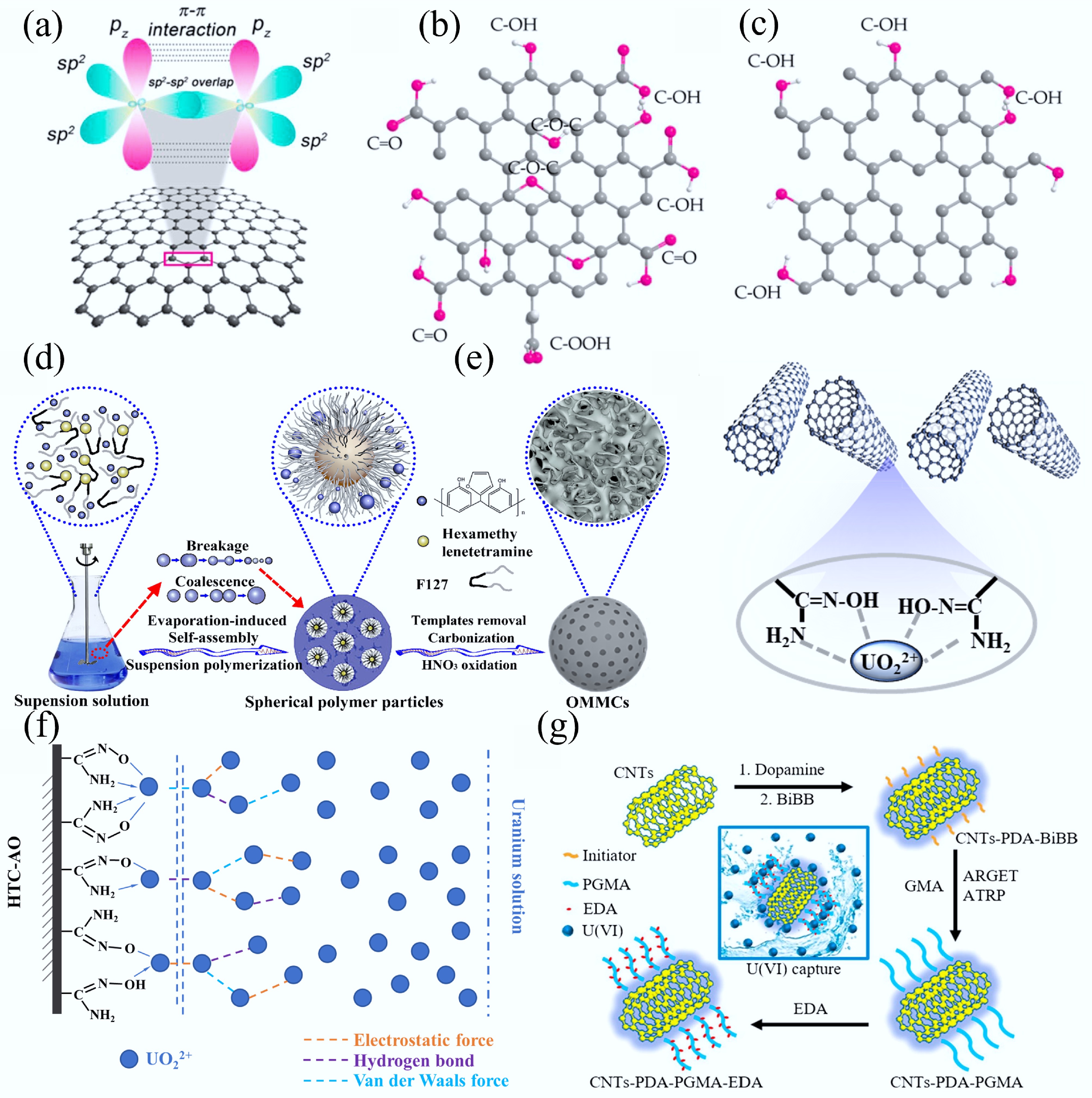
Figure 17.
Structural representations: (a) Pristine graphene. (b) GO. (c) Reduced GO[414]. (d) Schematic illustration of the synthesis of OMMCs[415]. (e) Probable sorption mechanism of U(VI) on AO-g-MWCNTs[416]. (f) The proposed monolayer and multilayer sorption modes for uranium sorption onto HTC-AO[417]. (g) Schematic diagram of the synthetic route of CNTs- polydopamine (PDA)-PGMA-EDA[418].
In environmental remediation and nuclear waste management, optimizing uranylophilic functional groups on carbon matrices proves essential for developing advanced radionuclide sequestration strategies[427]. Conventional surface engineering approaches, including thermal processing, oxidation, sulfidative/nitridative treatments, impregnation, and ligand grafting techniques, strategically enhance adsorption performance by modulating functional group density (e.g., carboxyl, hydroxyl, oxime, benzoyl, thiourea, pyridine, and amidoxime), or creating composite architectures with synergistic adsorbent phases[422,428]. These modifications amplify chemical affinity through targeted coordination site engineering while improving structural accessibility for uranyl ion binding. Tian et al.[429] demonstrated the covalent immobilization of 4-acetophenone oxime onto ordered mesoporous carbon CMK-5 through a thermally induced diazotization reaction, achieving effective U(VI) removal from simulated nuclear effluents. The functionalized oxime-CMK-5 exhibited a maximum Langmuir-model-derived adsorption capacity of 65.4 mg/g under acidic conditions (pH 4.0, 20 °C), directly correlated to its high ligand grafting density (2.1 mmol/g), enabling selective U(VI) coordination. In parallel, plasma surface modification techniques offer a versatile approach for introducing diverse functional groups onto material surfaces while preserving substrate bulk properties, providing an alternative pathway for tailored adsorbent engineering[430]. Shao et al.[424] pioneered the application of plasma-assisted grafting to functionalize MWCNTs with carboxymethyl cellulose (CMC), yielding MWCNT-g-CMC composites. The Langmuir model revealed a maximum U(VI) adsorption capacity of ~111 mg/g for MWCNT-g-CMC, significantly outperforming raw MWCNT (~14 mg/g) and N2 plasma-treated MWCNT (~26 mg/g) under identical conditions. This enhancement was attributed to the formation of strong surface complexes between UO22+ and -NH2 groups introduced during plasma treatment.
Based on this methodology, Wang et al.[416] subsequently synthesized amidoxime-grafted MWCNTs (AO-g-MWCNTs) via plasma techniques (Fig. 17e), further expanding the functionalization strategies for uranium adsorption. The bidentate coordination capability of amidoxime ligands on functionalized carbon nanotube surfaces, utilizing oxygen and nitrogen lone pairs, enables robust complexation with UO22+. AO-g-MWCNTs exhibited a maximum uranium adsorption capacity of 145 mg/g at pH 4.5. The distribution coefficients (Kd) for U(VI) reached 850 mL/g, significantly exceeding values (< 320 mL/g) for competing ions. This demonstrates superior selectivity of AO-g-MWCNTs toward U(VI) over coexisting metal species, including Mn2+, Co2+, Ni2+, Zn2+, Sr2+, Ba2+, and Cs+. Further refinement of functionalization strategies revealed that plasma-induced grafting (p-AO/CNFs) outperformed chemical grafting (c-AO/CNFs)[431], which could achieve capacities of 588.24 mg/g and 263.18 mg/g, respectively, significantly surpassing those of unmodified CNFs. In addition, p-AO/CNFs exhibited maximum adsorption capacities of 167.34 (Th[IV]), 135.87 (Eu[III]), 69.97 (Ni[II]), 67.64 (Co[II]), 57.57 (Sr[II]), 42.61 (Cs[I]), and 40.79 mg/g (Am[III]), demonstrating significantly higher selective adsorption of p-AO/CNFs toward U(VI) than toward other metal ions. Parallel advancements in hydrothermal carbon (HTC) functionalization introduced salicylideneimine (HTC-Sal), 5-azacytosine (HTC-Acy), amidoxime (HTC-AO), and phenolic (HTC-btg) ligands, with HTC-AO demonstrating superior performance (1,021.6 mg/g at pH 4.5, 298 K)[417]. HTC-AO achieved a significantly enhanced U(VI) adsorption capacity of 1.56 mmol/g, approximately 3-fold higher than both HTC-gly and HTC-CN. In the solution containing 12 competing ions (Sr2+, Ni2+, Zn2+, Ba2+, Co2+, Mn2+, Gd3+, etc.), the uranium-selectivity (SU) for HTC-AO was calculated up to 64.0%, about 2.5 times higher than that for HTC-gly and HTC-CN (Fig. 17f). Meanwhile, amidoxime-functionalized GO nanoribbons (AOGONRs) demonstrated significantly enhanced U(VI) adsorption performance compared to pristine GONRs, achieving a maximum capacity of 502.6 mg/g[432]. In simulated nuclear effluents containing 11 competing radionuclides, AOGONRs exhibited exceptional selectivity for U(VI) with a distribution coefficient (Kd) of 6 × 104 mL/g, highlighting their potential for practical applications. The adsorption mechanism involves synergistic electrostatic coordination between anionic functionalities (–SO3–, –COO–, –PO43–), and cationic UO22+ species, complemented by ligand-specific chelation effects[433].
Subsequently, Cai et al.[434] synthesized a phosphorylated GO-chitosan composite (GO-CS-P), demonstrating enhanced U(VI) adsorption performance. The maximum U(VI) adsorption capacities followed the order: GO-CS-P (779.44 mg/g) > pristine GO (573.91 mg/g) > GO-CS (346.16 mg/g). The reduced adsorption capacity of GO-CS was attributed to crosslinking-induced amidation, which diminished accessible carboxyl groups on GO. In contrast, spectroscopic analyses (FT-IR, XAS, XPS) confirmed that GO-CS-P's superior performance arose from inner-sphere complexation with phosphonate ligands, coupled with partial surface reduction of GO, which collectively enhanced U(VI) selectivity in multi-ion systems. Further studies highlight that functionalizing GO with inorganic acids (e.g., phosphoric or sulfuric acid) optimizes adsorption efficacy by introducing targeted active sites[435,436].
Technically, the ligand-functionalized moieties were immobilized onto AC substrates via either physical impregnation or covalent grafting methodologies. Notable agents such as trioctylamine[437], 2-hydroxy-4-aminotriazine[438], polyethylenimine[439], and benzoylthiourea[440] have demonstrated enhanced selectivity and affinity for UO22+ through tailored surface interactions. For instance, PANI-CMK-3 and CMK-3-PDA-4.4-10 composites, synthesized by in-situ polymerization of aniline and polydopamine on CMK-3 mesoporous carbon[441,442], achieved maximum U(VI) adsorption capacities of 118.3 and 93.6 mg/g, respectively. However, CMK-3-PDA-4.4-10 exhibited reduced reusability due to incomplete U(VI) desorption using 0.1 M HCl. Zhao et al.[440] engineered a benzoylthiourea-modified AC with O/S/N donor ligands, achieving an 82 mg/g U(VI) uptake capacity, threefold higher than that of pristine AC. Similarly, GO composites functionalized with polymers like polyaniline[443], polydopamine[444], PPy[445], polyacrylamide[446], and chitosan[434, 447] have shown significantly improved U(VI) adsorption performance through synergistic coordination mechanisms.
Electron transfer atom transfer radical polymerization serves as a green and controlled approach for nanomaterial surface functionalization[448]. Song et al.[418] pioneered a novel integration of mussel-inspired PDA chemistry with surface-initiated activators regenerated by electron transfer atom transfer radical polymerization, enabling controlled grafting of poly(glycidyl methacrylate) (PGMA) brushes onto CNTs, as depicted in Fig. 17g. The U(VI) removal efficiencies of pristine CNTs, CNTs-PDA, CNTs-PDA-PGMA, and CNTs-PDA-PGMA-EDA reached 6.4%, 31.3%, 37.4%, and 93.9%, respectively, under conditions of pH 5.0, 301 K, 0.25 g/L phase ratio, and 24 h contact time. Notably, the CNTs-PDA-PGMA-EDA composite achieved a maximum adsorption capacity of 192.9 mg/g, 15 times higher than that of pristine CNTs and surpassing all other modified counterparts. This exceptional performance originated from the synergistic coordination effects of abundant amine/hydroxyl groups introduced via EDA grafting and PGMA ring-opening reactions, which strongly complexed with UO22+.
The integration of magnetic metals/metal oxides into carbon matrices facilitates selective U(VI) recovery through redox deposition mechanisms while enabling efficient solid-liquid separation under external magnetic fields. For instance, Tan et al.[449] synthesized cobalt ferrite/MWCNT hybrids (CoFe2O4/MWCNTs) via hydrothermal methods, achieving a maximum U(VI) adsorption capacity of 212.7 mg/g at pH 6.0. Similarly, Liu et al.[450] developed PPy-coated dual-shell CoFe2O4/MWCNTs (PPy/CoFe2O4/MWCNTs) with a capacity of 148.8 mg/g at pH 7.0. Both systems followed Langmuir isotherm and pseudo-second-order (PSO) kinetic models, indicating monolayer chemisorption-dominated processes. Other carbon-based composites, including nano-flake-like Fe-loaded sludge carbon (Fe-SC)[451], magnetic ligand material (EDTA-mGO)[452], magnetic cucurbituril/GO (CB/GO/Fe3O4)[453], magnetic nanocomposite (CoFe2O4-rGO)[454], and manganese dioxide–iron oxide–reduced graphite oxide magnetic nanocomposite (MnO2-Fe3O4-RGO)[455] have also demonstrated exceptional U(VI) selectivity and adsorption performance in aqueous systems.
The integration of thermocatalytic and photoelectrochemical mechanisms in hybrid systems has significantly advanced U(VI) separation. GO-based catalysts play a pivotal role by simultaneously generating oxidative species (H2O2, •O2–) and reductive photogenerated electrons (e–), synergistically driving uranium valence transitions to precipitate as metastudtite (UO2O2•2H2O) with > 99% extraction efficiency via phase-transition isolation[456] (Fig. 18a). Qin et al.[457] engineered a GO/g-C3N4/PAO composite film by encapsulating GO/g-C3N4 heterojunctions with polyamide oxime fibers, achieving dual adsorption-photoreduction functionality (Fig. 18b). Under natural seawater conditions over 30 d, the material exhibited a U(VI) adsorption capacity of 10.39 mg/g, a 38.6% enhancement under light irradiation compared to dark conditions. In the presence of various competing ions, the removal rate of U(VI) reached 96%. The elution rate of GO/g-C3N4/PAO was 82% after five cycles. Further advancements include Fe2O3-GO composite (Fe2O3-GO)[458], K2Ti6O13 nanobelts hybridized GO nanosheets (GO/KTO)[459], 3D C3N5/reduced GO aerogels[460], TiO2-x/1T-MoS2/RGO heterojunctions[71] and 3D MXene-derived TiO2(M)@reduced graphene oxide (rGO) aerogel[461]. These architectures optimize charge carrier dynamics by suppressing recombination and directing electron transfer to U(VI). Through selective electron capture, U(VI) undergoes metastudtite (UO2)O2•2H2O precipitation via the reduction pathway:
$ \rm{UO}_2^{2+}+e^-\to\mathrm{UO_2}^+\, (0.05\; \mathrm{V}) $ $ \rm {UO}_{2}^{2+} + 4H^{+} + 2e^{-} \to {U}^{4+} + 2H_{2}O \,{(0.31\;V)} $ $\rm {UO}_{2}^{2+} + e^{-} \to {UO}_{2} {(s)\,(+0.41\;V)} $ $ \rm {O}_{2} + e^{-} \to {\cdot O}_{2}^{-}\, {(-0.33\;V)} $ $ \rm {\cdot O}_{2}^{-} + e^{-} + 2H+ \to H_{2} {O}_{2}\, {(0.68\;V)} $ $\rm {UO}_{2} (s) + 2H_{2} {O}_{2} \to (UO_{2} )O_{2} {\cdot 2H}_{2} {O}(s) $ Heteroatom-doping enhances ion-binding site density, charge transfer kinetics, and hydrophilicity of PC electrodes, thereby improving both catalytic activity toward U(VI) and electrochemical extraction performance. Yang et al.[35] recently developed an iron-nitrogen co-doped PC electrocatalyst (Fe–Nx–C–R) demonstrating a maximum U(VI) adsorption capacity of 282.2 mg/g at pH 4.0 under an applied potential of 0.9 V (Fig. 18c). The distribution coefficient (Kd) value was calculated to equal 3.6 × 104 mL/g, indicating an excellent affinity toward UO22+. The extraction mechanism involves dual pathways: surface amidoxime groups selectively coordinate UO22+ ions, while atomically dispersed Fe–Nx catalytic sites sequentially reduce UO22+ to UO2+ and subsequently oxidize it to Na2O(UO3•H2O)x deposits in sodium-containing media (Fig. 18d). Advanced carbon architectures, such as Mn/N co-doped carbon nanospheres[462], cotton-derived carbon-polyaniline composites[463], and self-standing porous aromatic frameworks[34], exhibit exceptional U(VI) extraction capacities from both artificial and natural seawater matrices.
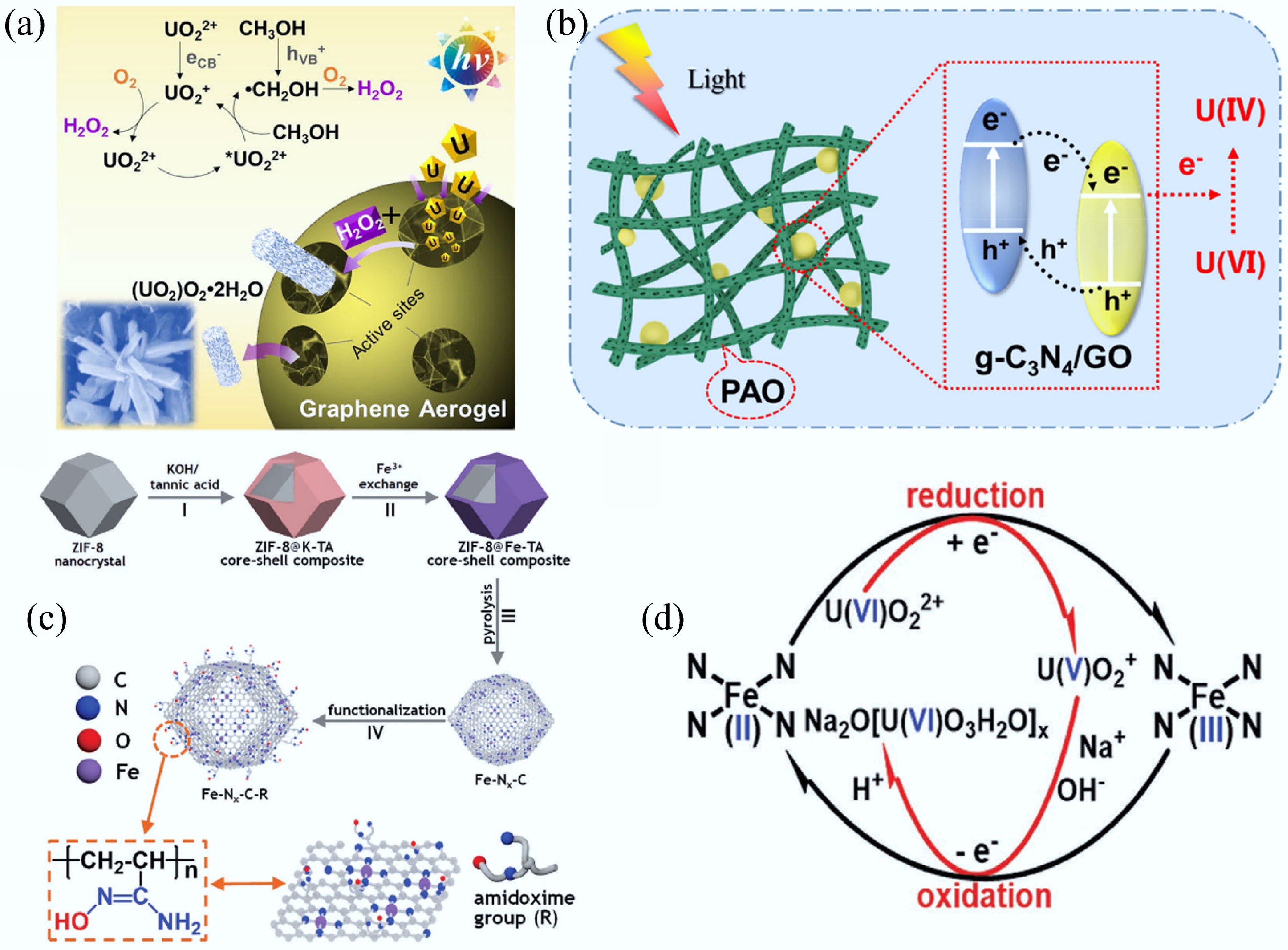
Figure 18.
(a) Illustration of graphene aerogel for the photocatalytic extraction of uranium under visible light irradiation and air atmosphere[456]. (b) Mechanism diagram of the photocatalytic extraction of U(VI)[457]. (c) Schematic illustration of the synthesis of Fe–Nx–C–R. (d) Schematic showing a plausible reaction mechanism for the Fe–Nx–C–R catalyzed extraction of uranium from seawater[35].
Separation of cesium and strontium
-
Cesium and strontium radionuclides, generated as byproducts of uranium fission processes, pose significant environmental risks due to their high water solubility and intense radioactivity, particularly when released through nuclear accidents or containment failures, as documented in the Chernobyl, Three Mile Island, and Fukushima incidents[464]. These heat-generating radioactive elements exhibit substantial biological toxicity and environmental persistence, necessitating urgent development of efficient and economically viable removal technologies for real water systems[465]. Adsorption-based treatment has emerged as a particularly promising solution for Cs+ and Sr2+ elimination from contaminated wastewater, offering advantages in operational cost-effectiveness and reduced secondary waste generation compared to conventional remediation methods[466].
Carbon-based adsorbents demonstrate practical potential in radionuclide remediation owing to their exceptional adsorption capacity, radiation resistance, and chemical stability across acidic to mildly alkaline conditions[467]. Notably, chemically activated carbonaceous materials synthesized via H3PO4 impregnation offer cost-effective solutions for contaminant sequestration[468], while advanced composites integrating transition metal hexacyanoferrates with microporous AC matrices exhibit superior performance in retaining Cs(I) and Sr(II) ions from nitric acid media[469]. Mechanistic studies reveal that Cs(I) adsorption efficiency depends primarily on the carbon surface's chemical functionality, facilitated by dual synergistic adsorption mechanisms, rather than on its porous structural characteristics.
The coordination architecture of adsorbents fundamentally determines their radionuclide sequestration performance, with crown ethers and calixarene derivatives demonstrating exceptional cesium selectivity through ion-dipole interactions and cation-π bonding mechanisms[470]. Adsorption efficiency in these supramolecular systems depends critically on host-guest structural complementarity, operating through either physical entrapment or chemical grafting pathways. A breakthrough millimeter-sized carbon-based supramolecular sorbent (C4BisC6/MMCs-P) was engineered through PS-divinylbenzene coating on hierarchical PC matrices coupled with calix[4]biscrown-6 immobilization, achieving a remarkable Cs(I) distribution coefficient of 225.79 mL/g, a ninefold enhancement over MMCs-P[470]. This structural innovation combines macroscopic dimensions for operational practicality with molecular-level recognition capabilities, enabling recyclable, high-selectivity cesium capture (Fig. 19a). Parallel advancements by Nisola et al.[471] developed magnetically responsive Fe3O4-rGO nanocomposites functionalized with azacrown ethers (15C5, 18C6) and dibenzo-24-crown-8 ligands. The aza-crown derivatives exhibited preferential strontium adsorption, while the expanded DB24C8@Fe3O4-rGO system demonstrated exceptional cesium selectivity, governed by precise cavity size matching with target ion dimensions.
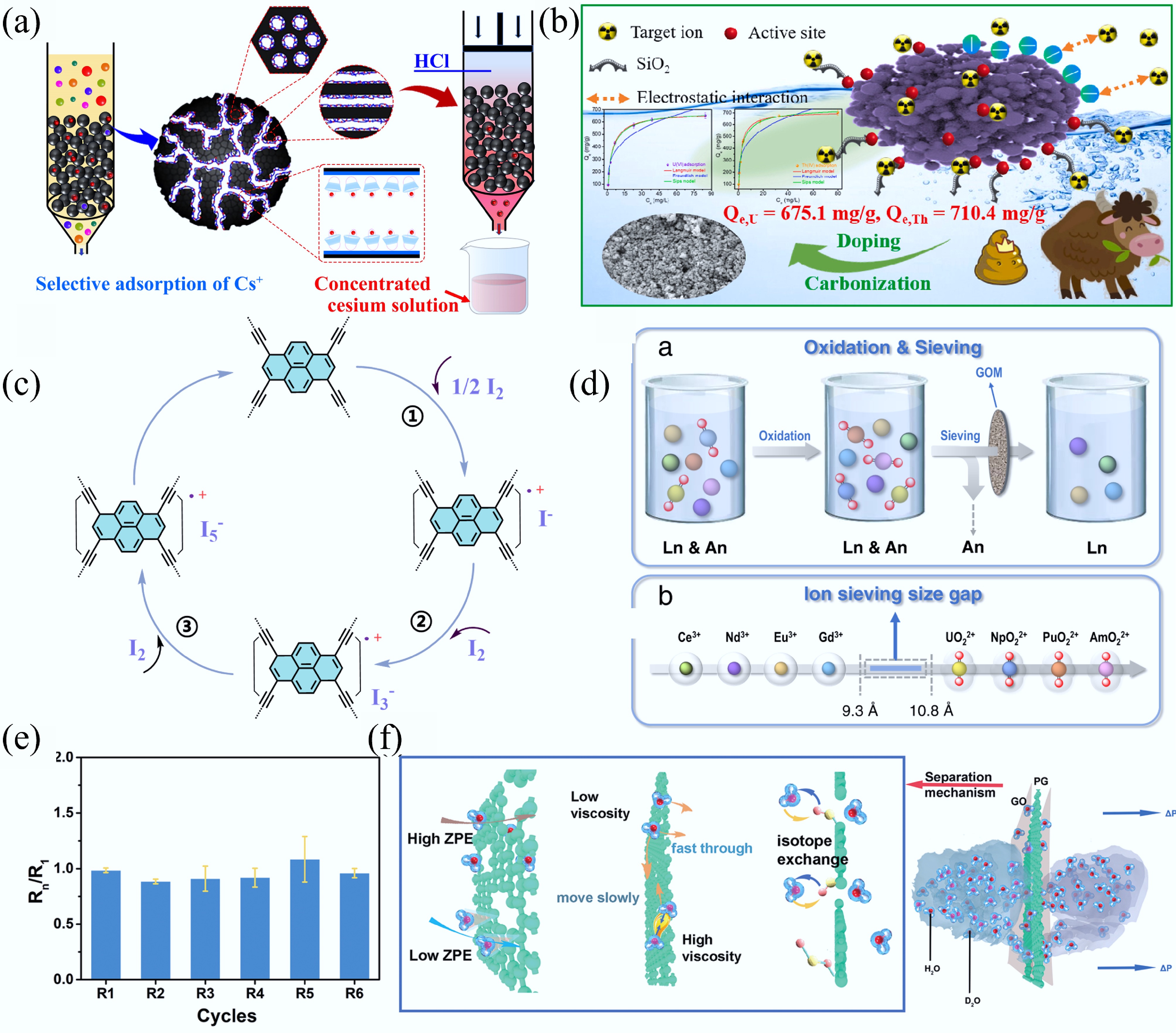
Figure 19.
(a) Mechanism for adsorption Cs(I) on C4BisC6/MMCs-P-5[470]. (b) Adsorption mechanism of U(VI) on TiO2@SiO2-CMBC[472]. (c) Mechanism of I2 uptake by PTEM[37]. (d) Scheme of actinides/lanthanides group separation and representative[473]. (e) Rejection rates with GO heterostructure membranes for the H2O/D2O. (f) Mechanism of GO/PG/GO separation of H2O/D2OM[474].
Moreover, the 3D GO aerogel architecture substantially enhances radionuclide adsorption performance by optimizing active site accessibility through its hierarchical macro-mesoporous network. This structural advantage is exemplified in MnO2-modified GO/PVA aerogels, where the integration of MnO2 creates additional Sr(II)-selective binding sites, achieving a maximum adsorption capacity (qm) of 26.85 mg/g with equilibrium attained within 6 h. Prussian blue (PB) emerges as a critical functional motif for cesium sequestration, combining high selectivity with robust adsorption kinetics through its unique cage-like structure. Notable implementations include Prussian blue/polyvinylpyrrolidone/reduced GO aerogel (PB@PVP/rGO)[475], and polyvinylidene fluoride-Prussian blue-GO modified membrane[476], where their engineered porous architectures demonstrate exceptional Cs(I) uptake capacities. The mechanistic studies on Cs(I) sequestration using GO hybrid composites have established fundamental guidelines for engineering advanced filtration matrices through structure-performance relationship optimization.
Separation of thorium
-
Thorium, a naturally occurring radioactive element ubiquitously distributed in trace quantities across diverse mineral matrices, exhibits unique chemical behavior as a tetravalent actinide analogue predominantly stabilized in the +4 oxidation state within aqueous systems[477]. Its nuclear potential emerges through neutron capture processes where 232Th transmutes into fissile 233U, positioning it as a strategic alternative nuclear fuel when coupled with conventional fissile isotopes. Beyond energy applications, thorium derivatives surpass uranium-based materials in industrial utility, with ThO2 serving critical roles in high-temperature ceramics, catalytic systems, and precision optical components. The environmental persistence of Th(IV) species poses significant ecological and health risks, with chronic exposure linked to pancreatic, hepatic, and pulmonary malignancies, prompting regulatory agencies, including WHO and USEPA, to establish stringent aqueous concentration thresholds (e.g., 22.5 µg/L maximum contaminant level for drinking water). Consequently, the extraction of Th(IV) ions from aqueous systems is critical for mitigating environmental contamination and facilitating thorium resource recovery[477].
Strategic surface functionalization of carbon-based matrices with oxygenated moieties significantly enhances Th(IV) ion sequestration through coordinative adsorption mechanisms. Notably, the synergistic interplay between Th(IV) species and oxygen functionalities (–OH, –COOH) induces concentration-dependent agglomerative transitions in GO nanoflakes[478], triggering macroscopic sedimentation phenomena that enable efficient solid-liquid phase separation—a critical advantage for practical remediation applications. The maximum Th(IV) uptake capacity reaches 430.94 mg/g under acidic conditions (pH 3.0), achieving adsorption equilibrium within 480 min. DFT simulations elucidate that this exceptional performance originates from dual mechanisms: (1) direct coordination bonding between Th(IV) and hydroxyl groups; and (2) electronic activation of adjacent sp2-hybridized carbon domains, which function as secondary adsorption sites through π-electron mediated interactions.
A ternary magnetic TiO2/Fe3O4/GO (TFGO) nanocomposite was engineered via a colloidal self-assembly protocol for high-efficiency Th(IV) sequestration[479], demonstrating exceptional reusability over 10 adsorption-desorption cycles without capacity degradation. Comprehensive multiscale characterization confirmed homogeneous Fe3O4 nanoparticle dispersion across TiO2-anchored GO matrices, with superparamagnetic responsiveness enabling rapid magnetic separation. The system achieved maximal Th(IV) uptake (29.97 mg/g) and removal efficiency (92.31%), under acidic conditions (pH 2.5), obeying a PSO kinetic model and Langmuir-Freundlich hybrid isotherm, indicative of monolayer adsorption on heterogeneous active sites. Adsorption strategies employing GO have attracted significant research attention, particularly due to the enhanced performance of GO-integrated metal oxide nanocomposites. The strategic integration of Fe3O4 with GO and TiO2 synergistically improves catalytic activity, regeneration capacity, contaminant removal efficiency, and operational cost-effectiveness. This design paradigm addresses nuclear wastewater challenges by integrating high-efficiency Th(IV) capture with practical magnetic separation and photocatalytic regeneration, leveraging the cost-effectiveness and environmental stability of TiO2 for sustainable implementation in radioactive contaminant management.
The engineered surface-oxidized wrinkled mesoporous carbon (WMC-O), fabricated through controlled acid treatment, demonstrates exceptional selectivity for Th(IV) extraction from rare earth element (REE) matrices, leveraging thorium's distinctive tetravalent oxidation state for preferential adsorption[480]. This optimized adsorbent exhibited a thorium distribution coefficient surpassing that of conventional surface-oxidized AC by two orders of magnitude (13 × 104 vs 35 × 102 at pH 2.15), while maintaining superior REE adsorption capacity (Kd > 3 × 105). The exceptional performance arises from synergistic structural advantages: a high SSA facilitating abundant active sites, a hierarchical mesoporous architecture enabling efficient ion transport, and oxygen-rich surface functionalities providing strong Th(IV) coordination through chelation and electrostatic interactions. The combination of attributes position WMC-O as a dual-functional material for simultaneous Th(IV) recovery and REE reclamation in nuclear fuel cycle applications, addressing critical needs in radioactive waste management and strategic metal resource utilization.
The engineered TiO2@SiO2-doped cow manure-derived biocarbon composite (TiO2@SiO2-CMBC)[472], fabricated via an impregnation-carbonization methodology demonstrates exceptional Th(IV) sequestration capabilities in aqueous systems, achieving a maximum adsorption capacity of 710.4 mg/g with 99.8% removal efficiency at pH 4.5. After five adsorption-desorption cycles, TiO2@SiO2-CMBC maintained 87.6% and 92.5% of the initial U(VI) and Th(IV) removal efficiencies, respectively. This exceptional recyclability demonstrates its significant potential for practical implementation in actual wastewater treatment systems. This hierarchical porous architecture exhibits rapid initial adsorption kinetics and robust performance in saline conditions (93.1% Th[IV] retention in 1.0 g/L NaCl). Mechanistic studies reveal that SiO2 components serve as coordination bridges between Th(IV) ions and active sites, enhancing interfacial binding through synergistic Lewis acid-base interactions and surface complexation. The TiO2 phase further stabilizes the composite matrix while contributing to electrostatic attraction mechanisms (Fig. 19b). This sustainable adsorbent design leverages agricultural waste valorization and dual metal oxide functionalization, offering a high-efficiency solution for Th(IV) recovery in complex ionic environments with industrial scalability potential.
Wang et al. engineered magnetic zinc ferrite/porous biochar composite (c-PBC/ZF), fabricated via hydrothermal methodology, which demonstrates exceptional Th(IV) sequestration capabilities with a maximum adsorption capacity of 41.47 mg/g at pH 4 (25 °C, 30 min contact time), achieving 97.95% removal efficiency at optimal 1:1 component ratio[481]. After six adsorption-desorption cycles, c-PBC/ZF maintained a high adsorption efficiency of > 90%, demonstrating excellent regeneration capability and reusability. This performance supports its long-term applicability in radioactive wastewater treatment systems, enabling effective recovery of Th(IV) radionuclides. Mechanistic investigations revealed that the adsorption process conforms to the Langmuir isotherm model and PSO kinetics, indicating monolayer chemisorption dominated by Th(IV) coordination with surface functional groups. The cubic spinel-structured ZnFe2O4 NPs embedded within the hierarchical porous biochar matrix impart superparamagnetic properties, enabling facile magnetic separation while enhancing adsorption performance beyond that of individual PBC and ZF components. FT-IR spectral characterization confirmed that Th(IV) sequestration by c-PBC/ZF is predominantly governed by Th–O covalent bond formation and coordination complexation with surface-bound hydroxyl and carboxyl functional groups. Finally, the results of cyclic experiments confirm the good stability of c-PBC/ZF, providing a new solution for the treatment of nuclear waste.
Separation of iodine
-
Radioactive iodine isotopes (129I, 131I), predominant volatile fission products in nuclear fuel reprocessing, exist as iodate (IO3–) and triiodide (I3–) ions in aqueous phases and as molecular iodine (I2) in off-gas emissions[482]. These species pose significant environmental risks due to their potential release into ecosystems and subsequent bioaccumulation through trophic transfer pathways, ultimately threatening human health via thyroid-specific radiotoxicity[483,484]. Effective containment strategies are therefore imperative. Current remediation approaches leverage physisorption, chemisorption, electrodialysis, ion exchange, precipitation, and dry dedusting techniques, tailored to address distinct iodine speciation challenges across liquid and gaseous waste streams.
Carbon-based materials exhibit exceptional radioactive iodine capture capabilities through synergistic physisorption (van der Waals forces) and chemisorption mechanisms, with performance modulated by structural and chemical modifications[485,486]. Two resorcinol-formaldehyde-derived CAs, RFac (790 m2/g, cylindrical pore geometry with acidic surface groups) and RMF-GO (375 m2/g, slit-cylindrical pores dominated by basic functionalities), demonstrated distinct iodide adsorption capacities under acidic conditions (pH 2.5). These materials achieved iodide uptake capacities of 82 mg/g (RFac), and 97 mg/g (RMF-GO) from solutions containing 500 mg/L initial iodide concentration[487]. Enhanced performance emerges in heteroatom-doped systems—sulfur/nitrogen-co-doped graphene aerogel (SN-GA) attains 0.999 g I2/g adsorption at 0.200 g/L initial concentration through π–π interactions between iodine's electrophilic orbitals and graphene's conjugated sp2 networks[488]. Surface functionalization with nitrogen (amine) or sulfur (thiol) groups amplifies binding via hydrogen bonding and covalent conjugation, as evidenced in B/N-doped nanocomposites, three-dimensional hydrolyzed collagen aerogel immobilized with catechin (Catechin@3DCF aerogel), and nitrogen-rich silk fibroin aerogels[489–491]. The pyrene-alkaline framework, a graphene-analogous 2D material with hydrophobic surfaces, demonstrates dual-phase adsorption capacities of 3.380 g/g (75 °C, gas) and 2.570 g/g (25 °C, aqueous), leveraging its electron-rich carbon matrix to form charge-transfer complexes with iodine molecules acting as Lewis acids[37]. Mechanistic studies reveal that iodine sequestration predominantly occurs through π-electron delocalization in graphitic systems, complemented by Lewis acid-base interactions at functionalized surfaces, establishing a multi-modal adsorption paradigm for nuclear waste remediation (Fig. 19c).
In addition, silver-functionalized adsorbents demonstrate superior iodine capture capabilities through chemisorption mechanisms, where Ag NPs exhibit strong affinities for both I– and I2 via Ag-I coordination bonding. GO/silver nanocomposites (GO/Ag), synthesized through carboxyl-directed Ag+ reduction, achieve rapid I– adsorption equilibrium (15 min) with a maximum capacity of 150.2 mg/g, leveraging silver's electrophilic interaction with iodide anions[492]. Similarly, silver-impregnated activated CFs attain 0.372 g /g I– uptake at pH 2 (400 mg/L initial concentration), attributed to enhanced ionic exchange at protonated surface sites[493]. In the presence of high concentrations of competing anions (SO42−, Cl−, HCO3−, CO32−), Ag@ACF retained 76% of its original adsorption efficiency, demonstrating exceptional anti-interference capacity against anionic species. For volatile I2 mitigation, silver-modified graphene aerogels (GA/Ag) exhibit dual-phase capture capability, underscoring silver's versatility in radioiodine remediation across diverse speciation and environmental matrices[494].
Separation of other radionuclides
-
Except for U(VI), Cs(I), Th(IV), Sr(II), and iodine species, carbon-based adsorbents demonstrate versatile remediation capabilities for other radionuclides, including La(III), 99TcO4–, Pu(IV), and deuterium (D). However, achieving selective extraction of these radionuclides from complex nuclear effluents poses significant technical challenges[495,496]. Biosorbents derived from microwave-assisted rice husk CDs and barium hydroxide composites[497] achieve rapid La(III) capture (1,500 μmol/g in 15 s) with 94.6%–96.2% removal efficiency across varied aqueous matrices through enhanced surface chelation. For 99TcO4– sequestration, a cationic polymeric network (R) containing imidazolium-N+ units modified hollow porous N-doped carbon capsules loaded with ruthenium clusters[498] materials exhibit exceptional selectivity and acid stability (3 M HNO3), leveraging synergistic effects between nitrogen-rich frameworks and ultrafine ruthenium clusters for targeted anion exchange. Wang et al.[473] prepared a GO membrane that enables precise actinide/lanthanide separation via tunable interlayer spacing, achieving separation factors > 400 for Pu(IV)/Ln(III) systems through size-exclusion of larger actinyl ions while permitting spherical lanthanide permeation (Fig. 19d). Moreover, advanced GO/PG/GO sandwich membranes[474] further demonstrate 97.02% D2O rejection with H2O/D2O selectivity of 35.2 (Fig. 19e), exploiting isotopic viscosity differences and hydroxyl-mediated isotope exchange within confined nanochannels to retard deuterium permeation while maintaining structural integrity under operational stresses (Fig. 19f). These multifunctional systems exemplify carbon-based materials' capacity to address isotopic separation challenges through engineered pore architectures, surface chemistry modulation, and quantum tunneling effects, establishing a comprehensive platform for advanced nuclear waste partitioning and isotope purification.
Summary and perspectives
-
Carbon-based materials have emerged as promising candidates for radionuclide separation due to their structural versatility, superior reactivity, and exceptional separation efficiency. This section systematically elucidates their separation mechanisms and performance parameters for typical radionuclides, including U(VI), Th(IV), Sr(II), and Cs(I), among others. Notably, our critical assessment of extant literature reveals four consistently dominant adsorption mechanisms for U(VI) across material categories: (i) functional group-driven electrostatic attraction, (ii) ion exchange, (iii) donor ligand-enabled coordination/chelation, and (iv) catalytic reduction, all of which have been rigorously validated through experiment and multimodal characterization. Although significant progress has been achieved, critical challenges remain for practical implementation: (1) current research predominantly focuses on functional group effects while neglecting the interplay between chemical complexation and topological configuration. A comprehensive investigation of pore architecture-morphology synergism appears crucial for performance optimization. (2) integration of photo-, electro-, and magneto-responsive components could enable smart separation systems through external field manipulation, warranting systematic exploration. (3) assessment under simulated harsh conditions (extreme pH, elevated temperature, intense radiation) remains imperative given the operational environments of nuclear waste management. In addition, both long-term chemical stability and suitability of adsorbents for industrial-scale columns are also important factors to apply treatment of real industrial effluent. (4) significant discrepancies persist between laboratory-scale experiments and real radioactive wastewater treatment scenarios, necessitating pilot-scale investigations and industrial prototype development.
-
As a prototypical greenhouse gas, CO2 has exhibited an ever-growing concentration in the atmosphere and brought severe concerns for researchers[217,499,500]. The CO2RR driven by sustainable electricity enables the conversion of CO2 into value-added chemicals[501–503]. This strategy not only contributes to carbon neutrality but also facilitates the storage of renewable energy in chemical bonds, thereby delivering dual environmental and resource benefits[504,505]. Characterized by a linear centrosymmetric structure, CO2 exhibits exceptional thermodynamic stability with a standard formation enthalpy of −394 kJ/mol, and the C=O dissociation energy of 1,072 kJ/mol[506]. Theoretical calculations indicate that activating CO2 to the CO2− anion requires a substantial overpotential of −1.90 V vs the Normal Hydrogen Electrode. Furthermore, the electrocatalytic reduction of CO2 to carbon-based fuels necessitates multi-step proton-coupled electron transfer (PCET) processes (Fig. 20)[507]. Taking CO production as an example, CO2 initially undergoes synchronous proton-electron transfer to form the adsorbed COOH* intermediate, followed by a second PCET step to yield CO and H2O. Specifically, when proton-electron transfer occurs without the participation of carbonaceous intermediates, adsorbed hydrogen atoms (H*) may form on the catalyst surface, subsequently recombining into H2 gas. Consequently, the cathodic CO2RR commonly competes with the HER. To effectively promote CO2 activation and inhibit the HER, many researchers have developed various catalysts for CO2RR for enhancing both activity and selectivity toward carbon-based fuels.
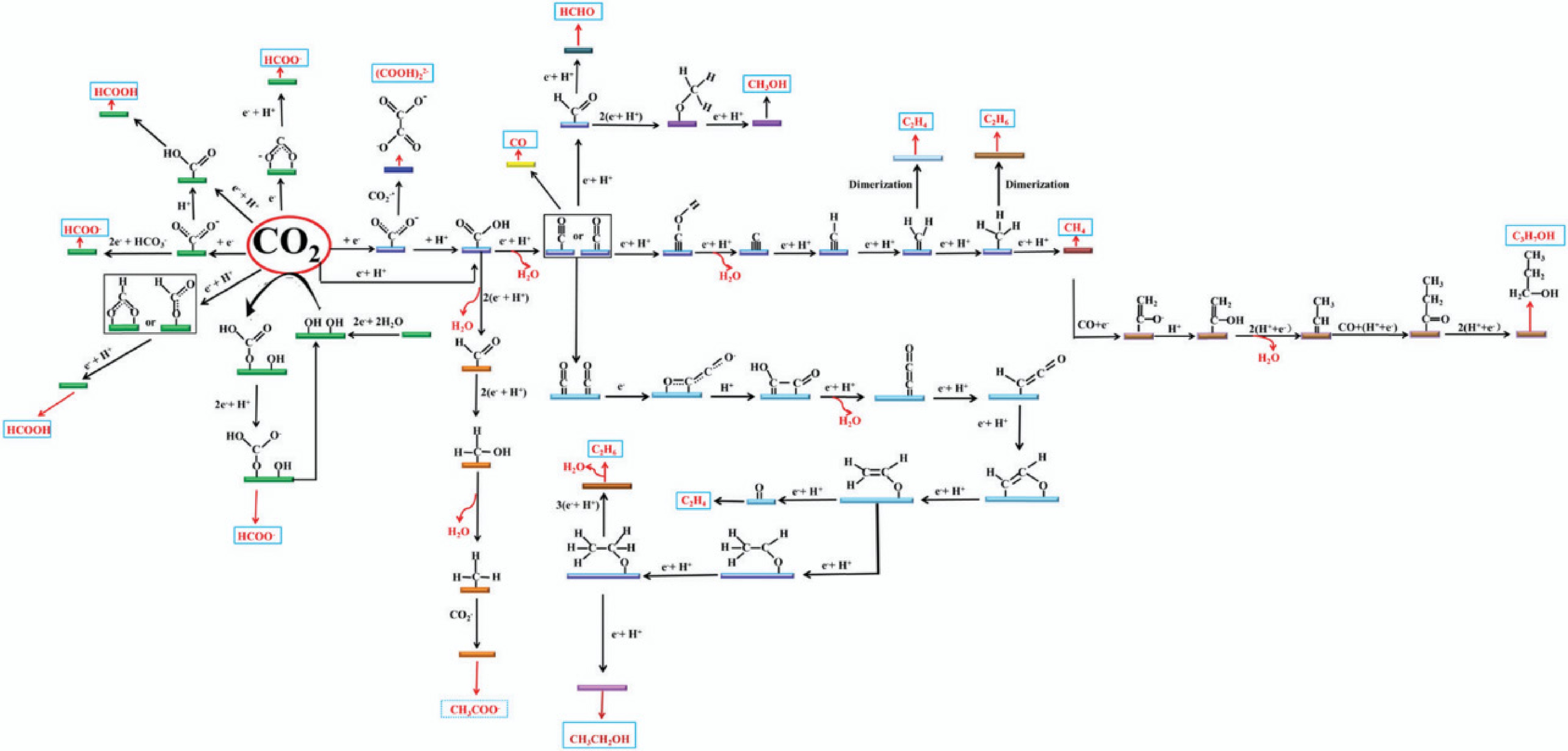
Figure 20.
The PCET pathway for CO2RR to carbon-based fuels[507].
The catalyst system for CO2RR encompasses metals and alloys, metal compounds, and carbon-based materials, etc. In recent years, carbon-based catalytic materials have exhibited multiple advantages in CO2RR catalyst design, owing to their tunable electronic structures, high SSA, excellent electrical conductivity, favorable chemical stability, and cost-effectiveness[508]. The intrinsic activity and selectivity of active sites within carbon-based catalytic materials can be precisely optimized through atomic-scale structural engineering, such as heteroatom doping, defect construction, and single-atom anchoring[302]. Furthermore, the inherent electrical conductivity and stability of carbon-based materials provide the potential for operation under high current densities. Structures derived from MOFs, such as PCs and CNTs, exhibit rapid electron transfer capabilities, enabling a significant reduction in the reaction energy barrier[509]. Another advantage of SCMs is the potential operation in an acidic electrolyte. Due to the larger solubility of CO2 in neutral or alkaline electrolytes than in acidic electrolytes, the FE for CO2RR can be promoted in high concentrations of OH−. However, the operation of CO2RR in acidic electrolyte is relatively valuable for long-term stability, because the pH of the electrolyte gradually decreases with the continuous injection of CO2 gas. Therefore, the final status for long-term CO2RR tends to be operation in acidic electrolyte regardless of the initial pH of electrolyte, which can highlight the properties of resisting acid for SCMs. Regarding the classification of carbon catalytic materials for CO2RR, these primarily include nonmetal-doped CMs, single-atom-doped CMs, carbon-based composites.
Nitrogen doped carbon
-
Nitrogen-doped carbon is recognized as a typical CO2RR catalyst. The performance of CO2RR by nitrogen-doped catalyst is closely associated with the doping position of nitrogen. Commonly, pyridinic nitrogen is considered as the active site in CO2RR, because the retained lone pair of electrons benefits the CO2 binding and activation[510]. For graphitic nitrogen, electrons occupy the π* antibonding orbitals, thereby increasing the energy barrier for CO2 adsorption (Fig. 21a). Through manipulating the nitrogen defects, nitrogen-doped carbon nanotube (CNT) arrays were developed as CO2RR catalysts with a low overpotential of −0.18 V and a FE of 80% toward CO formation[12]. To further improve the performance of CO2RR, a synthetic method namely high-energy microwave irradiation was developed to achieve gradient nitrogen doping along the radial direction of CNT, yielding a surface N-content of 30.5% that exceeds the upper limit attainable by conventional pyrolysis[511]. The unique temporal characteristics of microwave heating, manifested by precursor residence times shortened to minutes, and relatively low CNT surface temperatures, facilitate maximal retention of surface nitrogen species (Fig. 21b). In an H-cell configuration, this catalyst achieved a FECO of ~100% at −1.16 V vs RHE with a current density of 10 mA/cm2 due to the high nitrogen doping levels and ordered doping structure, thus lowering adsorption energies for CO2.
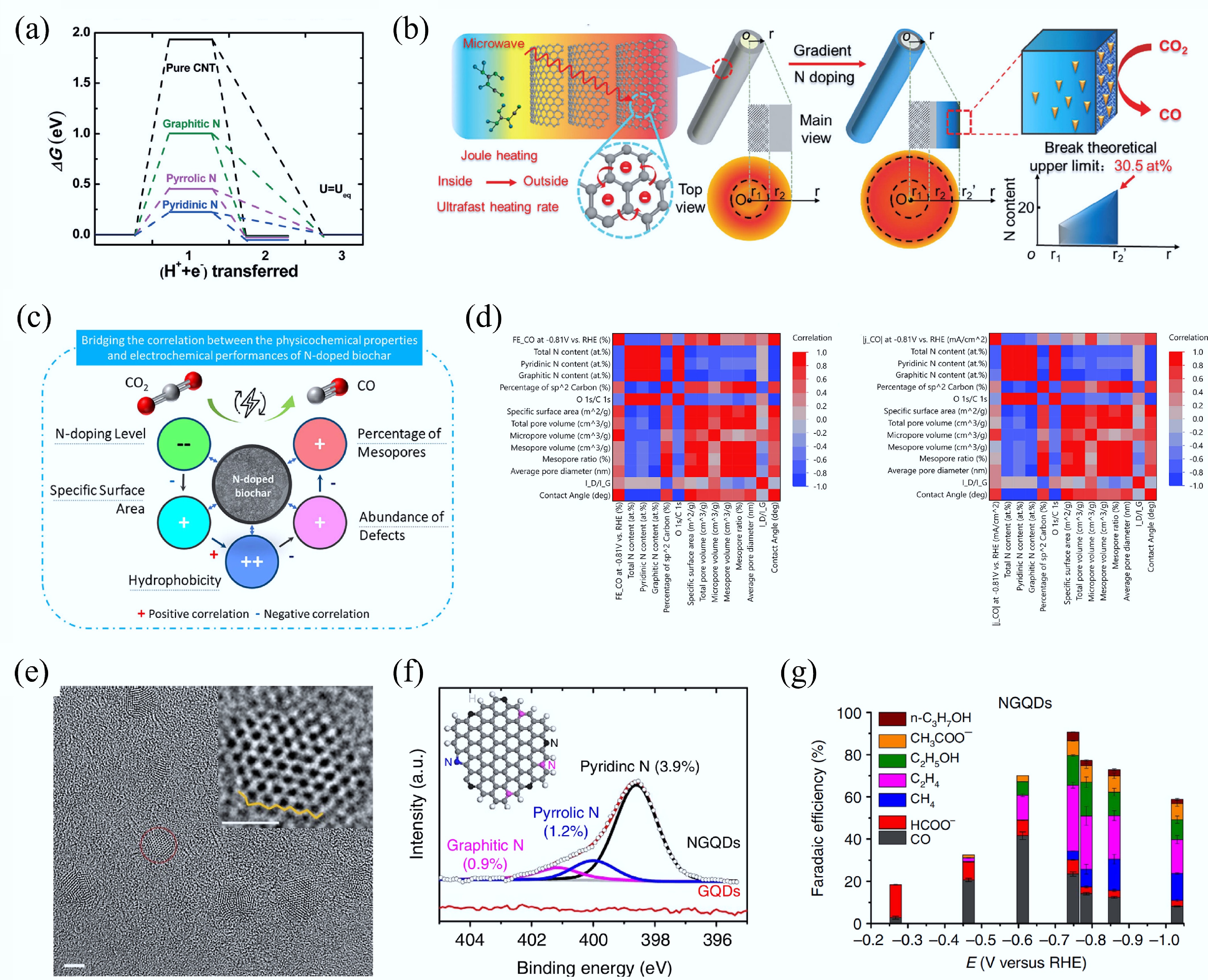
Figure 21.
(a) The Gibbs free energy diagram for CO2RR on different doping positions of N[12]. (b) Microwave irradiation synthesis of gradient nitrogen doping along the radial direction of CNT with ultrahigh N-content[511]. (c), (d) The influence of the structural parameters of N-doped biochar on CO2RR[514]. (e) The high-resolution transmission electron microscopy (HRTEM) image of NGQDs. (f) The N 1s XPS analysis of NGQDs. (g) The CO2RR over NGQDs[515].
Introducing a secondary doping element was also a promising way to tailor the CO2RR selectivity. Although nitrogen can enhance the electrochemical activity of CO2RR through its high spin density and charge delocalization induced by strong electronegativity, theoretical investigations revealed that elevated spin density may concomitantly promote the competing HER during CO2RR processes. To suppress the HER, N, P co-doped CAs were synthesized for CO2RR with the FE for CO formation (FECO) of 99.1% and a partial current density of −143.6 mA/cm2 in the electrolyte of ionic liquid[512]. Phosphorus, as a congeneric element of nitrogen, modulates the local charge distribution and spin density in N-doped carbon due to its lower electronegativity and larger atomic radius. Specifically, the introduction of P atoms could change the valence electron structure of N sites, accelerating the charge transfer of CO2RR against HER. The theoretical calculations also indicated that pyridinic N strengthened both the adsorption of *COOH and *H, whereas the co-doping of P hindered the adsorption of *H and increased the differences between thermodynamic limiting potentials for CO2RR and HER. Moreover, an O-doped C2N catalyst identified the function of C–O–C moieties in nitrogen-doped carbons for CO2RR, proposing a novel "N–C–O–C" synergistic catalytic model[513]. The O-doped C2N catalyst achieved a FECO of up to 94.8% and demonstrated exceptional stability and tunable syngas (H2/CO) ratios.
Apart from the doping content and doping position of nitrogen, the structural parameters of doped carbon were another crucial factor that influenced CO2RR. To elucidate the influence of morphology on CO2RR, a polymer-derived, interconnected, N-doped carbon structure with uniformly sized meso- or macro-pores, and different pore sizes was investigated[516]. The selectivity of CO2RR increased around three times just by introducing the porosity into the carbon structure with an optimal pore size of 27 nm due to the optimized wetting and CO2 adsorption properties. As another example, interconnected nitrogen-doped hollow carbon spheres nanochains forest with high-density sp3 defects were developed to display a superior electrocatalytic performance for CO2RR[517]. The influence of structural determinants on CO2RR performance is particularly pronounced in N-doped biochar. Within the potential window of −0.8 to −0.9 V vs RHE, maximum FE exhibits substantial variations (26.8% to 94.9%) among different N-doped biochar[514]. Crucially, merely increasing SSA and nitrogen doping levels fails to effectively enhance CO2RR catalytic performance. Multivariate correlation heatmap analysis reveals an inverse correlation between N-doping content and electrochemical metrics (Fig. 21c and d). Porous structural characteristics demonstrate significant positive correlation with FECO, yet negligible correlation with partial current density for CO. Notably, augmenting graphitization degree, surface hydrophobicity, defect density, and structural porosity in N-doped biochar catalysts substantially improves their CO2RR performance.
The products of CO2RR over N-doped carbon can be tuned beyond CO. The N-doped graphene quantum dots (NGQDs) with nanoscale dimensions were reported to catalyze the electrochemical CO2 reduction to multi-carbon hydrocarbons and oxygenates with high FE, elevated current densities, and low overpotentials (Fig. 21e–g)[515]. The NGQD catalyst achieves a total FE for CO2 reduction of up to 90%, with ethylene and ethanol conversion selectivity reaching 45%. The C2 and C3 product distribution and production rates mediated by NGQDs rival those of copper nanoparticle-based electrocatalysts. Comprehensive structural characterization demonstrates that the synergistic interplay between the unique nanoarchitecture, maximized density of exposed edge sites, and strategic nitrogen dopant incorporation endows NGQDs with exceptional electrocatalytic activity and C2+ product selectivity. Furthermore, boron and nitrogen co-doped nanodiamond has emerged as a highly efficient and stable electrocatalyst for the selective CO2 reduction to ethanol with a reaction pathway of CO2 → *COOH → *CO → *COCO → *COCH2OH → *CH2OCH2OH → CH3CH2OH[518]. This electrode achieves exceptional ethanol selectivity with a FE of 93.2% at −1.0 V vs RHE. This superior performance stems from synergistic effects of B-N co-doping modulating electronic structure and elevated nitrogen content enhancing *CO dimerization.
Metal-nitrogen doped carbons (M-N-C) single atoms
-
M–N–C catalysts are a kind of popular catalyst for CO2RR, which have the advantages of a high atomic utilization rate, a uniform active center, and tunable coordination structures (Fig. 22a). This unique architecture optimizes electronic structure modulation, enhancing CO2 adsorption and activation kinetics while stabilizing key reaction intermediates. The conductive carbon framework facilitates rapid electron transfer and high current densities. Furthermore, the robust covalent bonding between metal atoms and the N–C substrate confers exceptional electrochemical stability against corrosion under operating conditions. These M–N–C materials were demonstrated to possess exceptional electrocatalytic activity and selectivity for the direct CO2 reduction to CO[519]. The Co–N–C sites exhibited thermodynamically favorable energetics for hydrogen evolution, yet encountered significant kinetic barriers for CO formation (Fig. 22b). Conversely, Ni–N–C single-site configurations feature weakened H* and CO* adsorption energies, necessitating higher overpotentials to overcome the initial activation energy[520]. Upon application of sufficient overpotential, Ni–N–C catalysts transition to a thermodynamically downhill pathway for CO production while simultaneously suppressing the HER through competitive adsorption effects.
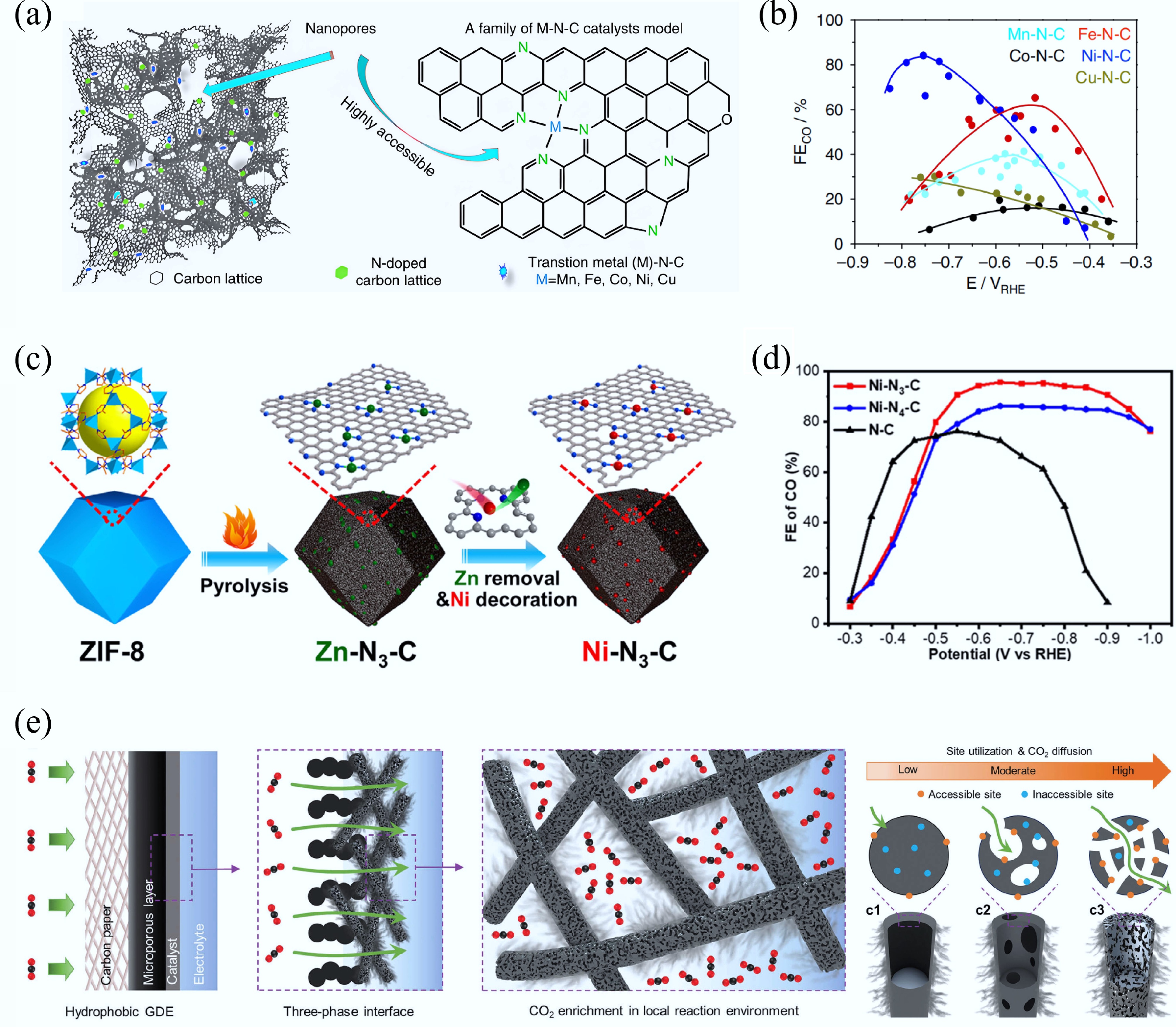
Manipulating the local coordination environment of M–N–C represents a powerful means to tailor the performance of CO2RR. The most common coordination of M–N–C is MN4, whereas the CO2RR is potentially enhanced when tuning the coordination number of M-N bonds. For the Ni center, the Ni–N3–C catalyst on N-doped carbon derived from metal-organic frameworks achieved FECO up to 95.6%, outperforming that of Ni–N4–C[520] (Fig. 22c and d). Theoretical calculations reveal that the lower Ni coordination number in Ni–N3–C can significantly enhance COOH* formation and accelerate CO2RR. In contrast, the CoN4 catalyst exhibits exceptional CO selectivity during CO2RR relative to CoN2 and CoN3 configurations[522]. This performance enhancement originated from the low-spin electronic state of CoN4 sites, which strengthened adsorption of *COOH and CO intermediates while weakening interactions with H2O molecules and H*. Such a distinct adsorption behavior rationalizes the dual optimization of high CO2RR activity and suppressed hydrogen evolution kinetics. Another way to manipulate the coordination structure is by introducing another co-doping element with nitrogen. For example, a series of Co-SxN4-x (x = 0−3) SACs were demonstrated to exhibit a distinct volcano relationship vs coordination number in CO2RR, with Co-S1N3 occupying the apex due to optimized *COOH and *CO binding energetics[523]. This catalyst achieves maximum FECO of (98% ± 1.8%) and a turnover frequency of 4,564 h−1 at 410 mV overpotential, as well as exceptional stability of > 150 h.
The structure of the carbon substrate also plays an important role in the CO2RR. For instance, a block copolymer-based strategy was engineered to fabricate an interconnected mesoporous carbon nanofiber (IPCF) and CS network, forming a highly porous IPCF@CS matrix for supporting Ni–N–C single-atom sites[521]. Within this architecture, the high-porosity IPCF effectively prevents CS stacking, while the CS provides abundant fully exposed active sites (Fig. 22e). Meanwhile, the densely interconnected mesochannels in IPCF facilitate CO2 diffusion toward the fiber@sheet reservoir, which establishes a CO2-enriched local reaction environment at the IPCF@CS interface. In acidic electrolyte, the catalyst achieves FECO exceeding 99.5% across a broad current density range (100–550 mA/cm2). Under operational acidic CO2 reduction conditions, the NiN4 sites undergo dynamic structural distortion from an ex-situ planar configuration to an out-of-plane geometry accompanied by Ni–N bond elongation. This transformation modulates charge redistribution and induces low-valent nickel site formation, ultimately reducing kinetic barriers for CO2 activation and CO desorption while suppressing the competing HER. For IPCF@CS, the highly stable structure against an acidic environment and the remarkable acidic CO2RR performance highlighted the potential long-term use. As another example, a series of spherical carbon supports with varying degrees of nanocurvature were used as the substrate of Ni–N–C catalyst[524]. The simulations and in-situ Raman spectroscopy indicated that a higher nanocurvature leads to a larger interfacial electric field. In acidic electrolyte, Ni–N–C exhibited an optimal CO partial current density of ~400 mA/cm2 with FECO > 99% toward CO2RR.
Further extending the M–N–C single-atom into di-atomic system provides a new chance for CO2RR. For example, a series of nitrogen-doped PCs incorporating ultrahigh-loading, atomically dispersed dual-copper sites with Cu2N6 configuration and an adjacent Cu–Cu distance of approximately 2.88 Å was synthesized for CO2RR[525]. The dual-copper site catalysts, with ultrahigh mass loading of atomically dispersed dual-copper sites, achieved a 52% FE for ethylene (C2H4) with a total current density of 180 mA/cm2 at −1.4 V vs RHE in 0.1 M KHCO3 electrolyte. The in situ infrared spectroscopy combined with theoretical calculations revealed the key intermediate pathway *CO → *CHO → *COCHO, confirming that the dual-copper sites significantly enhance C2 product selectivity by lowering the C-C coupling energy barrier. Additionally, the two neighboring single-atom active sites were reported to possess a new reaction pathway, namely inner tandem catalysis for CO2RR[38]. This mechanism enabled directional migration of adsorbed intermediates between adjacent binding centers through a feasible configuration transition, driven by differential affinities of the C and O atoms in intermediates toward the dual binding centers of the inner tandem active sites (Fig. 23). Guided by this principle, a neighboring Co–N–Cr system was developed for efficient CH4 production via CO2RR, exhibiting an exceptionally low overall thermodynamic barrier of 0.35 eV and a kinetic activation energy of merely 0.09 eV for intermediate migration. This integrated tandem catalysis demonstrates superior activity and product selectivity compared to conventional tandem systems, thereby expanding the design paradigm for advanced tandem catalysts across diverse catalytic applications.
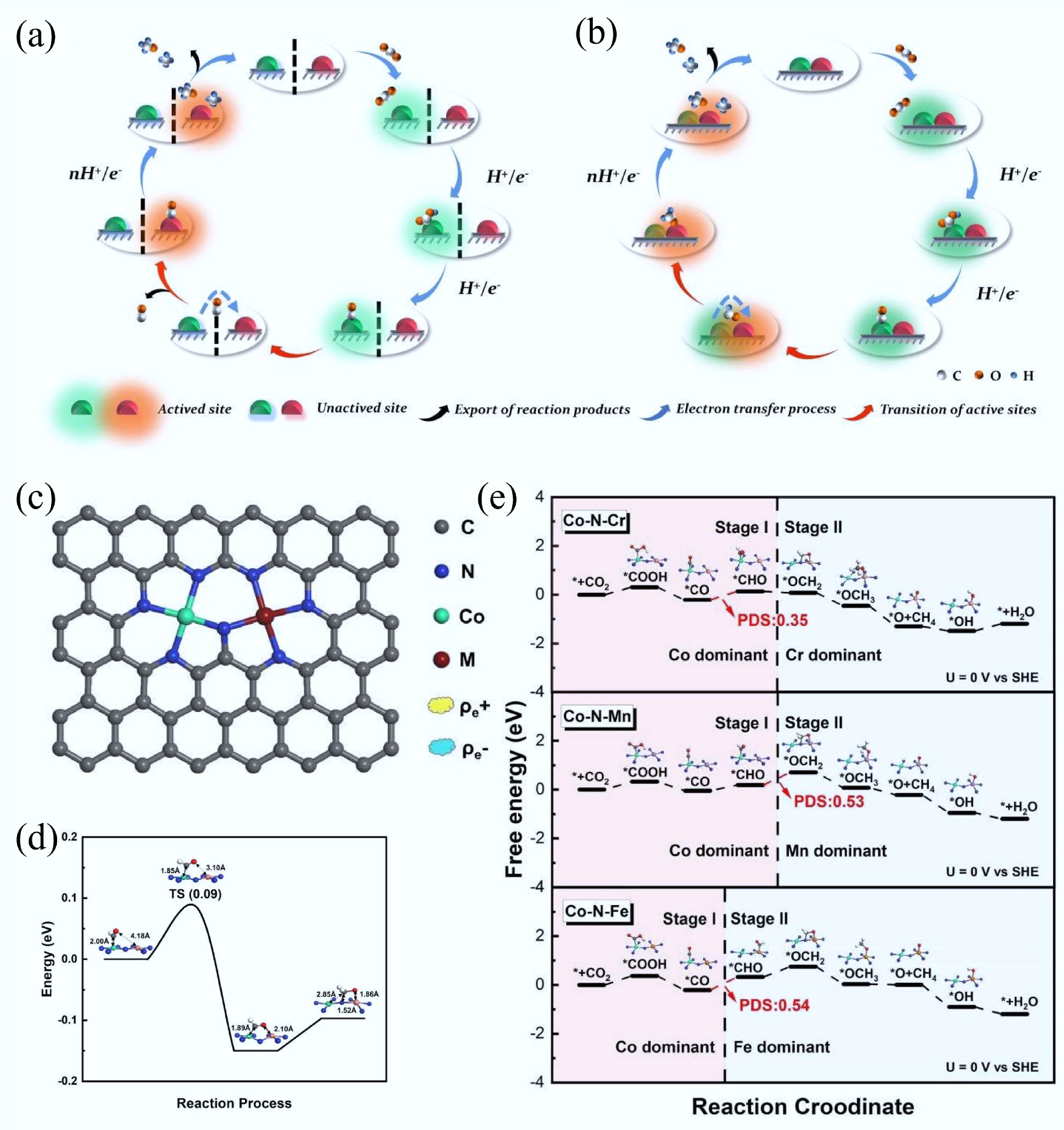
Figure 23.
(a), (b) Schematic diagram of the dynamic catalytic mechanism for the CO2RR on traditional tandem catalysts and inner tandem catalysts. (c) Model of Co–N–M inner tandem catalyst. (d) The activate barrier for the migration of adsorbed intermediates on Co–N–Cr inner tandem catalyst towards CO2RR. (e) The inner tandem reaction path for Co–N–M catalysts[38].
Although much research ascribed the superior CO2RR performance over M–N–C to the intrinsic coordination structure, the mechanism study remains a concern, especially under real reaction conditions. A recent operando X-ray absorption spectroscopy study revealed the structural evolution dynamics of M-N-C catalysts during CO2RR[526]. Comprehensive investigations reveal that even when commencing from structurally well-defined M–N–C systems, the coexistence of single atomic sites and metallic clusters/ NPs during CO2RR complicates experimental identification of active sites. This work unequivocally demonstrates that the in-situ formation of metallic clusters/NPs from single-atom sites during CO2RR occurs at M–N–C sites. Consequently, these metallic species actively participate in CO2 reduction alongside residual cationic single-atom sites. Moreover, a theoretical analysis indicated that Ni–N–C in its ground state adopted dsp2 hybridization, exhibiting intrinsic inertness toward CO2 activation and reduction[527]. Under operational potentials, applied electric fields and adsorbed reaction intermediates triggered dynamic hybridization state transitions to the catalytically active d2sp3 configuration, which was the origin of the exceptional CO2 reduction performance in Ni–N–C catalyst. Such operational hybridization transitions represent a universal phenomenon across diverse single-atom systems, governing catalytic dynamics in multiple reactions through continuous electronic restructuring. To elucidate the fundamental mechanisms, further operando analyses and theoretical computations are appreciated in the future.
CMs hybridized with other active catalysts
-
CMs (such as graphene, CNTs, and PC) can serve as components within composite materials, which are combined with other active catalysts (such as molecular catalysts, metal catalysts, etc.) for efficient CO2 capture and CO2RR[528,529]. This design offers the advantage of synergistically combining the intrinsic merits of CMs—including their excellent electrical conductivity, high SSA, tunable pore structure, and chemical stability—with the high intrinsic catalytic activity of the active centers. The carbon skeleton not only provides an efficient electron transport network and diffusion pathways for reactants/products, but also enables the highly dispersed and stable anchoring of active centers. This maximizes the exposure of active sites while preventing their agglomeration or leaching. Furthermore, interactions such as coordination and electronic coupling between the carbon material and the active centers can effectively modulate the electronic structure of the active sites. This modulation optimizes the adsorption energy of reaction intermediates, thereby synergistically enhancing the catalyst's activity, selectivity, and long-term stability, achieving dual optimization of performance and cost. This composite strategy ingeniously leverages the physicochemical properties of CMs and the catalytic nature of the active centers to cooperatively optimize the electrocatalytic performance.
Combining CNTs with metal phthalocyanine through π–π stacking is a typical design for CO2RR. This system benefits from the well-defined active sites and accurately tailorable structures of molecular catalysts and can overcome the difficulty in CO2RR beyond the two-electron process to generate more valuable products. For example, when immobilized on CNTs, cobalt phthalocyanine (CoPc) catalyzed the six-electron reduction of CO2 to methanol with substantial activity and selectivity via a distinct cascade mechanism wherein CO acted as a key intermediate[41] (Fig. 24a). Under near-neutral electrolyte conditions and at −0.94 V vs RHE, the system achieved a FE exceeding 40% for methanol production and a partial current density > 10 mA/cm2 (Fig. 24b and c). The resulting molecularly tailored electrocatalyst converts CO2 to methanol with high activity, selectivity, and operational stability maintained for over 12 h. As another example, the highly dispersed nickel phthalocyanine (NiPc) with molecularly engineered structure on multi-wall CNT achieved the FECO of > 99.5% at high current densities of −300 mA/cm2 with stable operation at −150 mA/cm2 for 40 h[530]. The CNT can also provide strain engineering of metal phthalocyanine for enhanced CO2RR (Fig. 24d). For instance, the curved CoPc on a single-wall CNT exhibited a methanol partial current density of > 90 mA/cm2 with > 60% selectivity[531]. Mechanistic studies revealed that curvature-enhanced CO binding facilitated its further reduction, while larger-diameter multiwalled CNT favored CO desorption (Fig. 24e).
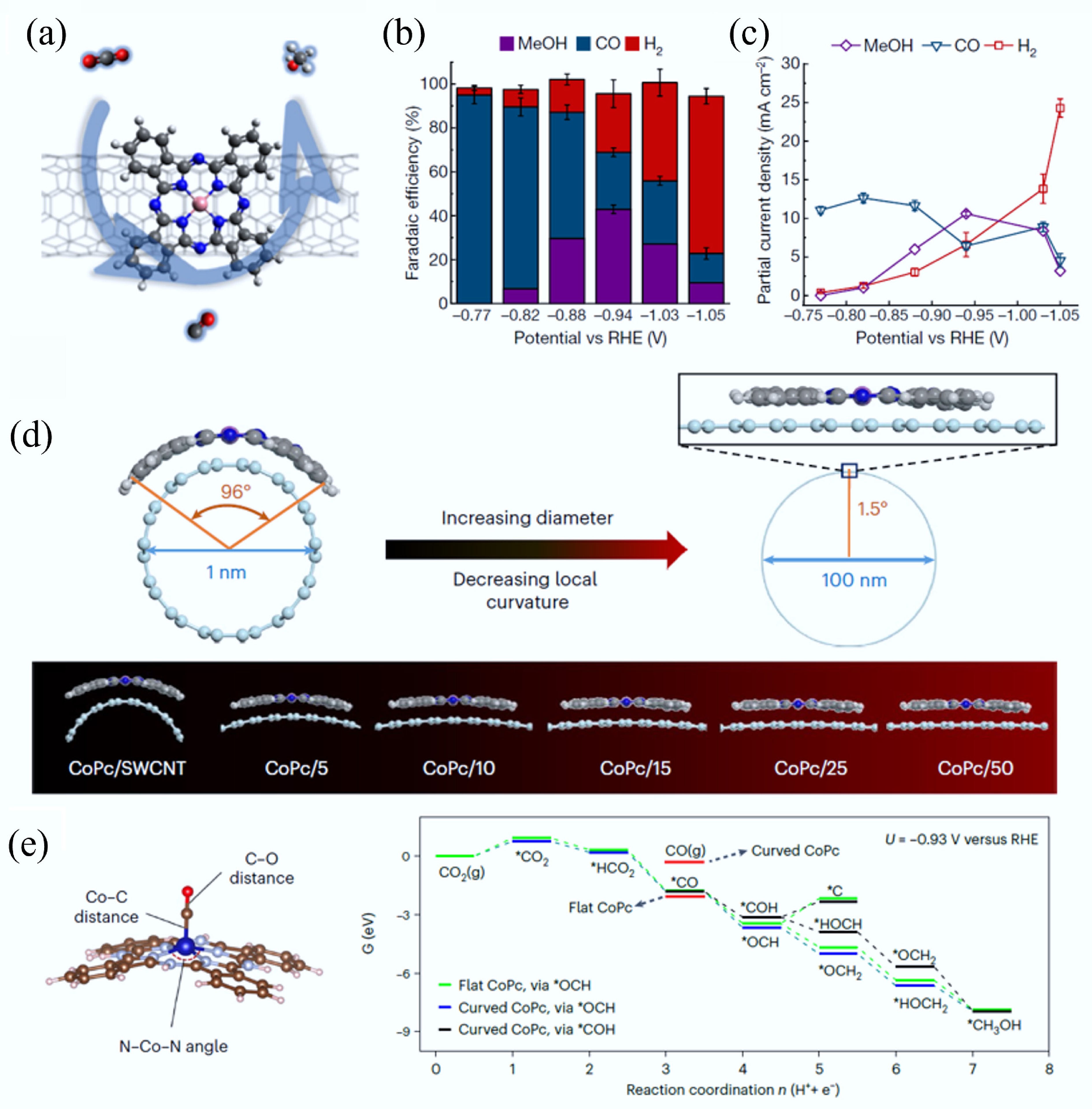
The integration of CMs with metal NPs offers another candidate for CO2RR. The carbon matrix provides exceptional electrical conductivity, ensuring efficient electron transport to the active sites, and a high SSA with tunable porosity that facilitates reactant/product diffusion and maximizes the dispersion of metal NPs, preventing their agglomeration and enhancing the exposure of active sites. For instance, the controllable dispersion of ultrafine Bi NPs within hollow nitrogen-doped carbon shells was demonstrated to be a promising CO2RR catalyst for formate production[532]. The nitrogen-doped carbon matrix induces a confinement effect that lowers the surface energy of bismuth NPs, effectively mitigating their aggregation. The Bi/N-doped carbon interface promotes CO2 activation and facilitates the formation of the key *OCHO intermediate. Consequently, the catalyst achieves 94.8% FE for formate at −0.9 V vs RHE with prolonged operational stability. The interface engineering of metal and CMs gives a new way to get complex products toward CO2RR. As an example, the nitrogen-doped nanodiamonds/Cu interface was reported to synergistically catalyze CO2RR to C2 oxygenates[533]. The catalyst achieves ~63% FE for C2 oxygenates at just −0.5 V vs RHE, while demonstrating exceptional 120-h stability with steady current density and merely 19% activity decay. Mechanism study revealed that the strengthened CO binding at the copper/nanodiamond interface suppressed CO desorption and promoted C–C coupling by lowering the activation barrier for CO dimerization. Moreover, the electronegativity of CMs can also tune the performance of CO2RR. Through constructing Cu NPs on a series of carbon supports with different heteroatom dopants, the high electronegativity of CMs reduced the electron density of Cu and induced a selectivity shift toward multicarbon products[534]. Based on this principle, a composite Cu and F-doped carbon catalyst achieved the FE for multicarbon products of 82.5% at 400 mA/cm2.
Summary and perspectives
-
CMs are an important family in CO2RR catalysis, which not only provides potential metal-free active sites but can serve as a synergistic enhancement for other active catalysts. The intrinsic active center of CMs lies in the heteroatom doping, such as pyridinic nitrogen. Apart from the metal-free design, the CMs are an ideal substrate for single atoms, which construct M–N–C active phase for CO2RR. Moreover, when combined with metal catalysts, CMs exhibit a promoted effect on the catalytic activity and selectivity by the improvement of conductivity and the interfacial effect for the optimized reaction path.
A table is provided which lists the partial current density and FE of SCMs. The SCMs can achieve high FE and current densities for various products, such as CO, CH3OH, and multi-carbon products. The two performance metrics respectively correspond to activity and selectivity, and the intrinsic performance of SCMs is comparable to that of metal-based catalysts (> 90% for a certain product, > 100 mA/cm2 for partial current density, Table 5)
Table 5. A comparison of FE and partial current density of SCMs for CO2RR
Catalyst Main product FE (%) Current density (mA/cm2) Ref. N, P co-doped CAs CO 99.1 143.6 (partial for CO) [512] O-doped C2N CO 94.8 2.75 (partial for CO) [513] Co-S1N3 single-atom CO 98 200 [523] IPCF@CS CO 99.5 550 [521] Ni-N-C with high nanocurvature CO > 99% > 400 (partial for CO) [524] NiPc on multi-wall CNT CO 99.5 300 [530] NGQDs C2H4 and C2H5OH 45 46 (partial for C2H4); 21 (partial for C2H5OH) [515] B, N co-doped nanodiamond C2H5OH 93.2 Not mentioned [518] Cu2N6 C2H4 52 180 [525] Curved CoPc on single-wall CNT CH3OH > 60 > 90 (partial for CH3OH) [531] Nitrogen-doped nanodiamonds/Cu interface C2 oxygenates 63 Not mentioned [533] Composite Cu and F-doped carbon Multicarbon products 82.5 400 [534] For CO2RR, the design of carbon-based catalysts requires consideration of both performance and scalable/sustainable synthesis. From both perspectives, catalytic systems integrating metal single atoms or clusters with carbon matrices represent an ideal system. On one hand, the synthesis of such systems aligns with established methodologies for CMs, requiring only minimal introduction of exogenous transition metals. This results in marginal cost increments while preserving significant structural tunability. On the other hand, the sparse metallic sites can substantially enhance CO2RR performance, exhibiting catalytic activity and selectivity comparable to those of noble metal benchmarks.
In the future, the carbon-based catalysts are expected to achieve the following breakthroughs: (1) machine learning (ML)-guided design of active centers and high-throughput synthesis of CMs for CO2RR; (2) the combined application of multiple in-situ spectroscopies to elucidate the transformation of active sites under applied potentials; and (3) the comprehensive design of carbon-based catalysts for C3+ products in CO2RR.
OER
-
ORR refers to the electrochemical process in which oxygen molecules gain electrons and are reduced to form water (in acidic conditions) or hydroxide ions (in alkaline conditions). Meanwhile, the electrochemical process where water or hydroxide ions lose electrons and are oxidized to produce oxygen is called the OER. ORR consumes oxygen (reduction) and releases energy, while OER generates oxygen (oxidation) and stores energy.
OER plays a crucial role in energy technologies, including water electrolysis, rechargeable metal-air batteries, and energy storage systems[535,536]. However, despite its significance, OER encounters certain challenges in specific applications. For example, OER is a bottleneck for the splitting of water due to its slow kinetics and high overpotential, leading to reduced energy conversion efficiency in water electrolysis[537,538]. CMs can serve as catalysts or catalyst carriers due to their tunable electronic structure, high electrical conductivity, and abundant surface active sites[539,540]. Applications of CMs in OER mainly focus on their use as catalysts or catalyst carriers to improve reaction efficiency and reduce cost[541].
Nitrogen-doped carbon nanotubes (N-CNTs) exhibit good adaptability due to their structural characteristics. Nitrogen doping introduces defect sites and active centers, promoting oxygen adsorption and electron transfer. The tubular structure provides efficient mass transport channels, which are suitable for loading catalysts such as IrO2 to enhance OER activity. 3D porous CAs also have their unique application prospects. The three-dimensional network structure has both high SSA and conductivity, which can uniformly disperse metal oxides such as Co3O4, enhance catalyst stability and mass transport efficiency, and is suitable for high current density scenarios.
However, CMs are prone to oxidative corrosion in acidic OER (generating CO2 at high potentials), while the stability of carbon in acidic HER is relatively better (low potential environment). This difference limits the application of carbon-based catalysts in acidic full water electrolysis systems. The following are specific applications of CMs in OER-related energy fields:
Photocatalytic hydrogen production
-
Hydrogen is regarded as an ideal secondary energy carrier due to its high energy density[542]. Photocatalytic hydrogen production is a method of generating hydrogen by decomposing water using solar energy, involving the separation and transfer of photogenerated carriers in photocatalysts[543]. OER is the anodic reaction in the water electrolysis process, with the reaction equation:
$\rm 4O{H}^-\to {O}_{2}+2{H}_{2}O + 4{e}^- $ CMs serve as effective co-catalysts in photocatalysis by enhancing the separation and transfer efficiency of photogenerated charge carriers, leading to significant performance improvements. Yang et al. developed a carbon-encapsulated Ni/NiOx photothermal co-catalyst featuring high-density frustrated Lewis pair (FLP) sites, as illustrated in Fig. 25[544]. A frustrated Lewis pair (FLP) is a sterically hindered Lewis acid-base system that cannot form a traditional adduct but can cooperatively activate small molecules. The synergistic interaction between FLP–VO–C (basic sites), and FLP–VNi–C (acidic sites) effectively lowers the energy barriers for water adsorption and dissociation. As a result, the Ni/NiOx@C/g-C3N4 (NOCC) composite photocatalyst achieves an exceptional hydrogen evolution rate of 10.7 mmol/g/h, surpassing that of the Pt cocatalyst by 1.76 times. However, the long-term stability of FLP sites under photocatalytic conditions are unclear; the steric hindrance that defines FLPs may be vulnerable to structural rearrangement under prolonged light irradiation or ROS generated during OER, potentially deactivating the sites.
Figure 25.
(a) Schematic diagram of the preparation process of Ni/NiOx@C cocatalyst and Ni/NiOx@C/g-C3N4 photocatalyst. (b) XRD patterns of Ni/NiOx@C structures at different temperatures. (c), (d) Transmission electron microscope (TEM) images of Ni/NiOx@C samples. (e) Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy of Ni/NiOx@C. (f) XRD patterns of Ni/NiOx@C/g-C3N4 with different loading amounts. (g), (h) TEM images, and (i) TEM/energy-dispersive spectroscopy mapping of NOCC5[544].
Hydrogen production via water electrolysis
-
Hydrogen production via water electrolysis is a clean method of hydrogen generation, based on the principle of producing hydrogen and oxygen through the electrolysis of water[545,546]. Traditional OER catalysts are mainly noble metals (such as IrO2 and RuO2), which are costly and have limited stability. CMs, especially those doped with heteroatoms (such as nitrogen, phosphorus, sulfur, etc.), exhibit good OER catalytic performance due to their unique electronic structure and high SSA[547]. Metal-free COFs and POPs-based materials with comparable activity to IrO2/RuO2 have been found. For example, Ghosh et al. strategically designed a noble metal-free thiadiazole (TDA) and triazine (Trz) linked porous organic polymer (TDA-Trz-POP) with N- and S-rich surface[548]. This TDA-Trz-POP-based heterogeneous electrocatalyst shows good activity toward OER in 1 M KOH solutions (Table 6). It has an η10 value (means overpotential for 10 mA/cm2 current density) of 410 mV, and a lower Tafel slope of 104.5 mV/deg for OER in 1.0 M KOH.
Table 6. Comparative OER activity with metal free carbon based materials and TDA-Trz-POP
Metal-free 2D CMs like g-C3N4, graphene, and rGO show promise as OER electrocatalysts (Table 6). Many biomass-derived CMs offer sustainable, high-performance alternatives to precious metal OER catalysts, with the added benefits of low cost and environmental friendliness. The N-doping and porous structure are key to their catalytic effectiveness. In summary, while metal-free heteroatom-doped CMs like TDA-Trz-POP represent a promising low-cost alternative to noble metals for OER, their current performance metrics (overpotential, kinetics), and stability under operational conditions requires significant improvement. Addressing these challenges through rational design of surface active sites and robust framework structures will be essential to realizing their potential in water electrolysis.
OER in fuel cells
-
Fuel cells are devices that directly convert chemical energy into electrical energy. According to the type of electrolyte used, fuel cells are divided into polymer electrolyte membrane fuel cell (PEMFC), phosphoric acid fuel cell, molten carbonate fuel cell, and alkaline fuel cell. In fuel cells, OER is the anodic reaction, with the reaction equation:
$ \rm {O}_{2}+4{H}^++4{e}^-\to 2{H}_{2}O $ PEMFC has attracted much attention due to its high efficiency and low pollutant emission. The gas diffusion layer (GDL) is an important part of PEMFC, which has several essential functions supporting catalytic layers, facilitating gas transport, expelling reaction product water, and providing pathways for proton and electron transfer. Therefore, how to prepare GDL with uniform hydrophobicity in a cost-effective and scalable manner remains a challenge. Recently, the development of cost-effective, environmentally friendly, and scalable fabrication method of fluorine-free bulk self-hydrophobic GDL was demonstrated by Tang et al.[549]. The synthesis and electrode fabrication protocols are illustrated in Fig. 26. The GDL with an interpenetrating network is composed of graphene, cellulose fiber, and modified polypropylene fiber, with impressive mechanical strength (22 MPa) and enhanced hydrophobicity without the use of harmful fluorinated agents through the scalable papermaking. The resulting GDL offers significantly improved PEMFC performance by enhancing water management, achieving a current density of 1.25 A/cm2 at 0.6 V and a maximum power density of 0.746 W/cm2.
Figure 26.
(a) Preparation of GCP composite paper through wet papermaking process. (b) GDL based on GCP paper has uniform bulk hydrophobicity and better water management ability compared to traditional carbon paper GDL. Reprinted with permission from Tang et al.[549].
The high SSA and good electrical conductivity of CMs make them ideal catalyst supports, effectively improving the performance and lifespan of fuel cells while reducing the amount of noble metal catalysts and lowering costs. Addressing long-term stability of hydrophobicity, scaling network uniformity, and optimizing the hydrophobicity-permeability balance will be essential to translating this innovation into commercial PEMFC systems.
OER in metal-air batteries
-
Metal-air batteries are high-energy-density batteries, with their positive electrode reactions typically involving OER[550]. A flexible and free-standing CF membrane immobilized with atomically dispersed Fe-N4/C catalysts (Fe/SNCFs-NH3) were synthesized by Yang et al.[40]. By recombination of MOF materials and CFs, followed by carbonization and activation, the pore structure of the CF membrane and the coordination structures of Fe single-atom catalytic sites were regulated, which greatly improved the ORR/OER electrocatalytic activity. It shows excellent performance in both liquid and flexible solid zinc-air cells (Fig. 27). Modulation of local atomic configurations by sulfur-doping in Fe/SNCFs-NH3 catalyst leads to excellent ORR and enhanced OER activities.
Figure 27.
Zn–air batteries (ZABs) performance of Fe/SNCFs-NH3 catalyst as an air cathode. (a) Discharge polarization curves and corresponding power densities of liquid-state ZABs. (b) Charge-discharge curves of the liquid-state ZAB at different current densities ranging from 1 to 5 mA/cm2. (c) Histogram of the voltage gaps at different current densities. (d) Long-term cycling stability at 1 mA/cm2 of the liquid-state ZAB, and inset is the enlarged charge and discharge curves. (e) Simplified schematic of the solid-state ZAB using a sodium polyacrylate (PANa)-KOH-Zn(CH3COO)2 hydrogel as the electrolyte. (f) Photograph of a wristband with a series of LED lamps lightened by two series-connected solid-state ZABs. (g) Stability of the solid-state ZAB at 1 mA/cm2, and inset are the photographs of the solid-state ZAB at various flat/bent/flat states[40].
In summary, the Fe/SNCFs-NH3 catalyst represents a promising step forward for flexible metal-air batteries, leveraging MOF-derived design and sulfur doping to enhance bifunctional ORR/OER activity. Addressing stability issues of single-atom sites, scaling synthesis, and optimizing the porosity-conductivity balance will be critical to realizing its potential in commercial metal-air battery systems.
HER
-
Hydrogen, characterized by its high energy density and absence of carbon emissions, has emerged as a main candidate for a clean energy carrier. The process of water splitting for hydrogen production has attracted considerable interest due to its efficiency and environmental sustainability, with HER serving as the fundamental mechanism of this process[551,552]. As illustrated in Fig. 28, in acidic conditions, HER is initiated by the Volmer process, resulting in the formation of hydrogen intermediates (H*) on the surface of the catalyst. This is followed by either the Tafel or the Heyrovsky pathways for the desorption of hydrogen molecules[39]. In contrast, under alkaline conditions, HER must first undergo a water dissociation step, which limits proton supply and significantly slows reaction kinetics[553]. Thus, the activity of alkaline HER is often constrained by water dissociation efficiency. Whether in acidic or alkaline environments, the binding energy (ΔGH*) between H* and the catalyst surface must be moderate—too strong binding hinders H desorption, poisoning active sites, while too weak binding impedes initial adsorption. An ideal catalyst should exhibit a ΔGH* value that is close to thermoneutral (~0 eV), thereby achieving a balance between proton adsorption and hydrogen molecule desorption, which is essential for efficient hydrogen production[554].
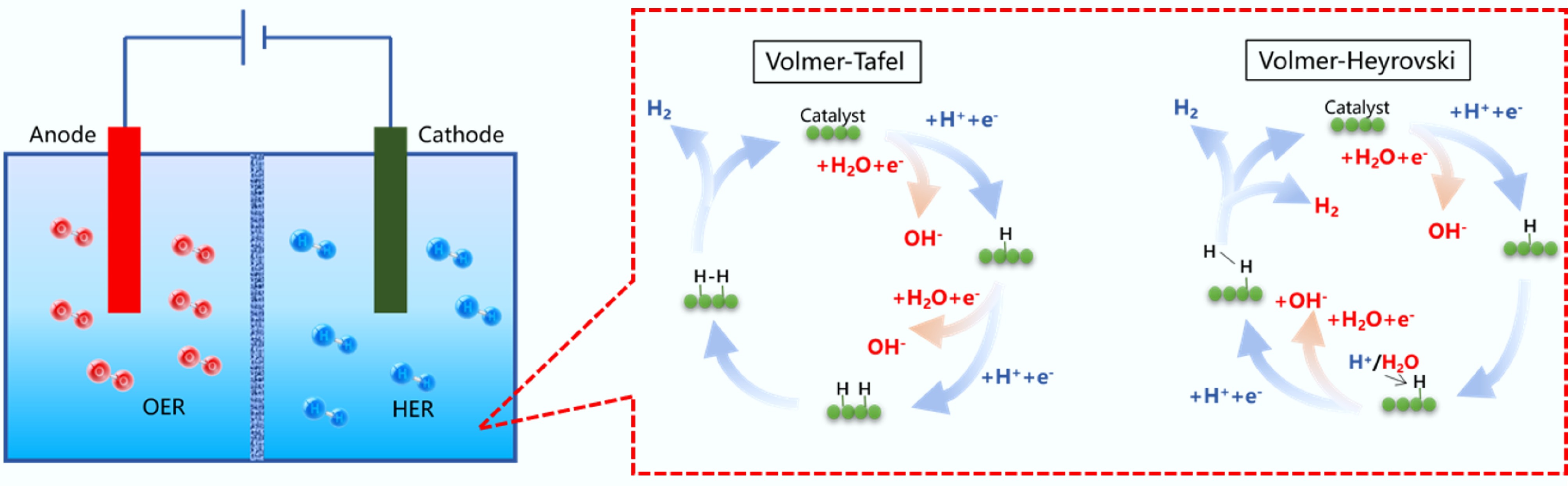
Figure 28.
The reaction steps of HER in acidic and alkaline electrolytes.
The catalytic efficiency of HER is predominantly influenced by catalyst's ability to adsorb hydrogen atoms. Currently, HER catalysts are classified into two principal categories: noble metal-based and transition metal-based catalysts. Noble metals (e.g., Pt, Ir, and Ru) are characterized by their favorable Gibbs free energy change for hydrogen adsorption (ΔGH*), low overpotential, and rapid reaction kinetics, but their scarcity and high costs limit large-scale applications[555,556]. Conversely, transition metal catalysts, while more economically viable, frequently exhibit inadequate conductivity and a tendency to agglomerate at elevated temperatures[557,558].
In recent years, carbon-based materials have emerged as a focal point of research due to their adjustable pore structures, high electrical conductivity, and remarkable chemical stability. The porous architecture of these materials facilitates the exposure of active sites and enhances mass transfer, while their superior conductivity promotes efficient electron transfer. Additionally, their resistance to corrosion ensures durability in both acidic and alkaline environments[559]. Nonetheless, a significant challenge associated with CMs is their inherent ΔGH* values, which often deviate from the optimal range, leading to inadequate hydrogen adsorption capacity. Consequently, improving the hydrogen adsorption capability of CMs through strategies is essential for enhancing their performance in HER.
Fullerene-based catalysts
-
Fullerene, classified as 0D CMs characterized by a distinct structure, low onset potential, coupled with a unique electronic configuration, positions them as an optimal foundation for the development of high-efficiency catalysts. In a study, Wu & Peng constructed a 2D C24 network using [5,6]-fullerene units, which, due to its extensive SSA, offered a plethora of active sites and demonstrated exceptional performance in photocatalytic water splitting (Fig. 29). The C24 monolayer can spontaneously drive reactions through multiple catalytic pathways while maintaining high stability[560]. Additionally, the sp2-hybridized carbon surface of C60 is characterized by the presence of electron-deficient olefinic C=C bonds, which exhibit enhanced π-electron delocalization. This property facilitates the binding of metal atoms in an η2-C60 π-mode, thereby effectively anchoring individual atoms and mitigating the risk of agglomeration. Recent investigations have employed C60 as a carrier for the anchoring of Pt single atoms, achieving a substantial loading of 21 wt% Pt/C60[561]. Further studies have indicated that the strong electron affinity of fullerene (2.7 eV) optimizes the electronic structure of the metal active centers. For instance, the charge redistribution effect between Mn and C60 significantly enhanced the HER activity of Ru sites. The optimized MnRu/C60 achieved an overpotential as low as 8 mV under alkaline conditions, with a mass activity of 2.39 A/mgRu, showcasing the unique advantages of fullerene in electronic structure modulation[562]. 3D crystalline fullerene networks (CFN) can stabilize metal NPs and single atoms through lattice confinement effects. Luo et al. used CFN to in situ capture Ru species, preparing a RuNP-RuSA@CFN-800 with an overpotential of 33 mV at 10 mA/cm2 and long-term stability of 1,400 h[563].
Figure 29.
Top and side view of the C24 monolayer crystal structure[560].
CDs-based catalysts
-
CDs, a novel class of carbon-based materials, have garnered significant interest in the field of electrocatalysis due to their high SSA and superior electrical conductivity. When integrated with metals or their derivatives, CDs will introduce numerous structural defects and active sites. Excellent HER performance was achieved by combining CDs rich in vacancies with Ru. In Liu's work, the vacancies and functional groups present in CDs not only contributed to the stabilization of the Ru-CDs structure but also mitigated the agglomeration of Ru NPs. Under alkaline conditions, the catalyst exhibited a low overpotential of 30 mV (at 10 mA/cm2)[564]. Additional investigations revealed that CDs can also enhance the HER performance of metal carbides. Hollow Mo2C nanoreactors were synthesized through carburization, resulting in the in-situ generation of CDs on their surfaces. This configuration effectively addressed the issue of strong hydrogen adsorption on Mo sites, exhibiting superior activity and stability compared to most Mo2C-based electrocatalysts in alkaline environments[565].
Among the various types of CDs, CQDs are particularly notable due to their pronounced quantum confinement effect. The quantum confinement of CQDs can reduce the recombination of e− and h+, enhance charge separation, and improve the utilization rate of photogenerated carriers. When CQD acts as a carrier, its discrete energy levels hybridize with the d orbitals of the metal active center, which can regulate the position of the d band center of the metal, weaken the excessive adsorption of H*, and promote the Volmer-Heyrovsky step. CQDs possess high crystallinity, which facilitates efficient electron transport, while their uniformly distributed surface functional groups allow for targeted modifications via covalent bonding. For instance, Li et al. synthesized a novel carbon-loaded nano-ruthenium electrocatalyst, which also demonstrated excellent HER performance in alkaline environments[566]. Recently, the application of ML in the design of CQD catalysts has emerged. Baeck et al. employed ML techniques to predict and experimentally confirm the outstanding HER performance of nickel-doped CQDs supported on 3D graphene, achieving an overpotential of 151 mV at 10 mA/cm2 (Fig. 30)[567].
Figure 30.
(a) Flowchart of the proposed BGA and VGA. (b) Performance comparison between conventional GA optimization and BGA optimization for a generic convex-shaped cost function. (c) Score evaluation of important variables for electrochemical measurement. (d) Ranking of TM dopant in CQD toward catalytic performance[567].
Graphitic carbon nitride-based catalysts
-
Among semiconductor nanomaterials, 2D carbon nitride materials have emerged as highly promising metal-free catalysts due to their excellent stability, high SSA, and tunable surface properties. Xie et al. prepared porous graphitic carbon nitride microspheres via a melamine-cyanuric acid intermediate, revealing that the CN-20, obtained after 20 h of calcination, demonstrated remarkable photocatalytic activity for HER under visible light[568].
The most thermodynamically favorable and stable phase of C3N4 is g-C3N4, which provides substantial internal and interlayer space, a large surface area, high stability, and excellent corrosion resistance. Sundriyal et al. illustrated that the incorporation of ZIF-67 into GO resulted in a reduction of electrode conductivity from 4.7 to 3.9 Ω and charge transfer resistance from 1.8 to 1.1 Ω[569]. Similarly, a 3D self-supporting heterostructure comprising vertically aligned CoS nanosheets and rGO networks was fabricated on the carbon cloth. This catalyst required only 76 mV overpotential to achieve 10 mA/cm2 in HER. The porous rGO network facilitated the anchoring and vertical growth of CoS nanosheets, providing numerous catalytic sites[570].
The performance of catalysts can be further enhanced through modification with noble or transition metals. Mai et al. developed a Co/CoxSy@NC-750 structure, wherein the Co framework promoted the formation of highly ordered graphite layers, improving conductivity and exhibiting good HER activity in both acidic and alkaline conditions[571]. Zhang et al. utilized a novel synthesis method, the laser solid phase synthesis method, to fabricate a dual-atom alloy catalyst (PtRu)1Co DAAs[572]. This innovative structure significantly improved HER catalytic performance in alkaline solutions, achieving an overpotential of 27 mV at −10 mA/cm2 and a mass activity 19.6 times higher than that of commercial Pt/C. As shown in Fig. 31, a Fe-CoP@NC/N-rGO composite was synthesized through transition metal doping, phosphorylation, and reduction of GO, significantly reducing charge transfer resistance to 31 Ω through synergistic effects[573].
Figure 31.
Synthesis of Fe-CoP@NC/N-rGO hybrid composites derived from ZIF-67 MOFs.
Despite its advantages, the practical application of g-C3N4 is still limited by rapid recombination of photogenerated carriers and insufficient active sites. In recent years, novel carbon nitride allotropes (e.g., C3N5, C3N7) have shown superior performance by adjusting the carbon-to-nitrogen ratio[574]. Among them, nitrogen-rich C3N5 has garnered significant attention due to its unique electronic structure and abundant active sites. The triazole and triazine groups in its molecule enhance π-electron delocalization, significantly improving catalytic activity. These new carbon nitride materials provide fresh research perspectives for catalyst design. Ng et al. introduced boron-doping into C3N5 via substitutional or interstitial doping. Replacing the nitrogen atom with a boron atom (BN3-C3N5) narrowed the bandgap by 0.6 eV. Boron-doping also reduced the reaction energy of the potential-determining step in HER pathways under acidic and alkaline conditions via the Volmer-Tafel and Volmer-Heyrovsky mechanisms[575].
Future research can focus on developing more precise atomic-level doping strategies, optimizing charge transfer at heterojunction interfaces, and addressing stability issues in large-scale preparation. With a deeper understanding of the structure-activity relationship, carbon nitride catalysts are expected to achieve broader applications in clean energy.
CNFs
-
CNFs, with their 1D nanostructure providing fast electron transport channels, tunable SSA, and excellent chemical stability, are regarded as ideal catalyst substrate materials. Combining transition metal compounds with CNFs not only prevents active component agglomeration but also significantly enhances catalytic performance through interfacial electronic effects. Recent studies have developed various efficient CNF-based catalyst systems through innovative design strategies.
Ding et al. prepared a Mo2C-CoNi@CNFs heterostructure via electrospinning-pyrolysis, in which the unique multiphase synergy enhanced catalytic activity and material stability, demonstrating excellent bifunctional (HER/OER) catalytic performance[576]. Further research revealed that metal component nano-sizing effectively increases active sites, which is considered a potential method to modify hydrogen adsorption. For instance, bamboo-like nitrogen-doped CNFs (NCNFs) embedded with Co and MoC NPs, where ultrafine MoC NPs (≈ 5 nm) and Co electronic modulation synergistically achieved outstanding performance across a wide pH range, requiring only an overpotential of 86 mV to reach 10 mA/cm2 in alkaline conditions[577]. Doping heteroatoms (N, P, S, etc.) and constructing heterostructures are important means to regulate the binding force of protons to molybdenum carbide materials. Similar to the above research work, Wang et al. replaced Co with CoN and simultaneously adopted N-doped multi-channel CNFs (NMCNFs) to design optimization of the MoC/CoN/NMCNFs. MoC/CoN NPs embedded in carbon fibers, with their unique 1D multichannel structure, prevented nanoparticle agglomeration while increasing active site exposure[578]. The highest performance breakthrough came from Zhang et al., who developed a Ru/TiO2/NC system where the TiO2/NC carrier optimized H* adsorption strength, enabling the catalyst to achieve an overpotential of only 18 mV (at 10 mA/cm2) in alkaline conditions, surpassing commercial Pt/C[579].
In summary, by precisely tuning the size and composition of transition metal active centers and their interfacial interactions with CNFs, researchers have successfully developed various high-performance catalyst systems. Future research should focus on: (1) developing more precise atomic-level dispersion preparation methods; and (2) addressing structural uniformity issues in large-scale preparation.
CNTs and other shapes
-
Currently, widely used HER catalysts (noble and transition metal catalysts) require support from 3D substrates with high SSA and conductivity. This support is essential for maximizing the exposure of active sites and facilitating rapid electron transfer rates. Carbon-based materials are renowned for their tunable pore structures, which amplify the exposure of catalytically active sites and accelerate reactant transfer. The pioneering work of Jiao et al. demonstrated that heteroatom co-doped CMs not only augment the number of surface active sites but also foster a synergistic coupling effect, thereby significantly improving the electrochemical performance of carbon-based catalysts[580].
CNTs, with their large SSA and strong charge extraction capability, can improve the conductivity of metal materials when combined with metals. CNTs include SWCNTs and multi-walled carbon nanotubes (MWCNTs). The single-layer structure of SWCNTs offers a more direct electron transmission path and lower resistance. At the same time, their high SSA and uniform surface are more conducive to the dispersion of metal monatomic or NPs, exposing more active sites. While the multi-layer structure of MWCNTs may cause electrons to need to transfer through interlayer jumps and increase resistance, and may shield some active sites, their encapsulation and protection effect on metal particles can effectively prevent agglomeration and corrosion, and the multi-layer structure endows it with stronger corrosion resistance, making it more stable in harsh environments. The encapsulation interaction also protects active particles from corrosion or peeling. For example, Liu et al. developed a low-Pt-content (1 wt.%) Pt/CNT45, which exhibited mass activities far exceeding commercial Pt/C across a wide pH range (acidic: 18.76 A/mgPt, neutral: 3.92 A/mgPt, alkaline: 3.88 A/mgPt). This performance was attributed to the Ptδ+ promoting H2O dissociation and OH desorption[581]. Except for Pt, Yan et al. prepared C@OV-RuO2/CNTs-325, where the CNT carrier enhanced the stability of Ru-based catalysts. The catalyst showed excellent HER activity in acidic, neutral, and alkaline media (η10 = 36.1–19.3 mV), benefiting from the regulation of Ru d-band centers by carbon layers and oxygen vacancies[582]. CNT-supported SACs exhibited curvature-switchable HER activity, with higher curvature leading to greater upward shifts in the d-band center and improved HER activity[583]. Among transition metals, molybdenum stands out as a promising non-noble HER catalyst due to its near-optimal ΔGH*. MS-Mo2C@NCNS provided a large number of active sites and optimized the adsorption energy of H*. Another work synthesized Co,Ni-MoB2@CNT/CC heterostructures through the synergy of CNTs and MoB2, achieving low overpotential (10 mA/cm2) of 98.6, 113.0, and 73.9 mV, respectively, under acidic, medium, and alkaline conditions through the synergistic effect of CNTs and MoB2, breaking through the performance limitations of conventional MoB2 in high pH environment[584,585].
The synergistic effect of CNTs and alloys provides new insights for designing efficient HER catalysts. This composite strategy primarily enhances catalytic performance through electronic structure modulation and spatial confinement effects. A novel synthesis method, nanosecond laser ultrafast restricted alloying, can break the transition limitation from non-miscible to miscible in synthesis (Fig. 32). This method utilizes CNTs as black light absorbers. After absorbing light, the temperature rise triggers the heating of the mixed-phase alloy to synthesize RuM (M = Cu, Rh, and Pd) alloys loaded with CNTs. After synthesis, the Tafel slope of Ru95Cu5/CNTs is only 28.4 mV/dec[586]. Encapsulating alloy species in various carbon substrates has proven to be a viable strategy for manufacturing high-performance OER/HER bifunctional electrocatalysts. Combining CNTs with transition metal alloys can solve the problem of easy agglomeration of metals. An N-doped CNT-encapsulated FeNi alloy catalyst effectively addresses the problem of agglomeration and dissolution of conventional FeNi catalysts. The unique rambutan-like porous structure significantly improves mass transfer efficiency, exhibiting excellent bifunctional catalytic performance (HER and OER overpotentials of 279 and 278 mV at 10 mA/cm2, respectively) in 1.0 M KOH[587].
Figure 32.
(a) RuCu alloy composition modulation by LUCA. (b) TEM images of RuCu/CNTs and their size distributions[586].
Beyond CNTs, CMs of different dimensions can significantly enhance the HER performance of metal catalysts through structural modulation. Yang et al. developed a simple method for preparing nitrogen-phosphorus co-doped CS, constructing a 2D conductive network through the crosslinking reaction of amino acids and phytic acid. The material exhibited an ultra-high active surface area of 93.5 mF/cm2 and an ultrathin thickness of < 10 nm, with charge polarization induced by graphitic N and C3PO groups significantly optimizing ΔGH*[588]. In the 1D material system, Liu et al. innovatively introduced carbon into WO2 nanorods, forming a hybrid structure that not only inhibited nanoparticle sintering (sintering temperature increased by 200 °C) but also improved electron mobility by three orders of magnitude. DFT calculations confirmed that carbon modification reduced the reaction energy barrier by 0.15 eV[589]. By constructing bimetallic alloys on superhydrophilic N-doped carbon nanocages, the abundant OH groups reconstructed the interfacial hydrogen bond network, enabling the RuNi/NC to achieve a record-low overpotential of 12 mV (at 10 mA/cm2) in alkaline conditions, with a mass activity 13.6 times higher than that of commercial Pt/C[590].
Biomass carbon-based catalysts
-
Biomass carbon, with its diverse natural structures and multichannel architecture, can enhance electron transport efficiency. Simultaneously, its rich surface functional groups provide vast space for surface modification, making it a focus of recent attention in HER electrocatalysis.
The multichannel structure and rich surface chemistry of biomass CMs offer unique advantages for HER catalysis. For instance, García-Dalí et al. innovatively used phytic acid as a single precursor to prepare P-doped CMs through heat treatment. Phytic acid, a bio-derived molecule containing carbon and phosphorus, is an excellent precursor for producing high-P-content CMs. Studies showed that the formation of P-C bonds in CMs not only altered the electronic structure of carbon but also significantly enhanced chemical stability, exhibiting an onset potential of −0.27 V in HER (Fig. 33)[591]. In metal-biomass composite systems, lignin-metal supramolecular framework complexes were developed as HER catalysts, and a nitrogen-doped carbon-encapsulated CoRu nanocatalyst was obtained after carbonization at a current density of 10 mA/cm2, with the overpotentials of HER and OER only being 90 and 200 mV, respectively[592]. Research revealed that active sites were mainly located within defects of lignin-derived carbon, displaying a unique 'self-healing' phenomenon within the carbon layers. Oxygen intermediates (*OH, *O, and *OOH) facilitated defect reconstruction, while H* aided the reappearance of defect-rich structures. Further studies found that modulating the activity of carbon atoms on carbon layers through metal clusters has become a promising strategy for achieving efficient and stable HER catalysis. Zhang et al. used Mo-rich reed seedlings as raw materials to successfully encapsulate Mo2C clusters in N-doped biochar via a one-step pyrolysis method. This approach significantly increased active site density, with the prepared BCMo900-1 exhibiting Pt-like activity (η10 = 30 mV, Tafel slope = 33 mV/dec) in acidic conditions, attributed to the synergy between the biochar's large SSA and heteroatom doping[593].
Figure 33.
Schematic of the synthesis process of P-doped carbons from phytic acid.
Current research confirms that by rationally selecting biomass precursors and optimizing pyrolysis processes, precise modulation of electronic structures and active sites can be achieved. Future research should focus on: (1) systematic studies of the biomass precursor-catalyst performance relationship; and (2) dynamic evolution mechanisms of material structures during catalysis.
Other CMs
-
Other CMs, especially those possessing customized nanostructures, have been recognized as exceptionally effective supports for electrocatalysts, providing improved conductivity, stability, and metal utilization. Mesoporous carbons, characterized by their adjustable pore architectures, enhance catalytic performance by facilitating better mass and electron transfer, as well as optimizing interfacial electronic interactions. Concurrently, ultrathin carbon coatings and nitrogen-doped carbon matrices contribute to increased durability in challenging electrochemical environments. Recent developments in intermetallic NPs and heterostructured carbon hybrids exhibit outstanding catalytic efficiency, exceeding the performance of conventional noble-metal catalysts.
Mesoporous CMs, with their tunable pore structures and excellent conductivity, are ideal carriers for reducing noble/transition metal usage. Confining active components within mesopores not only improves metal utilization but also enhances catalytic activity through interfacial electronic effects. In metal compound-mesoporous carbon systems, Liu et al. embedded MoP, Mo2C, and other Mo-based NPs (~5 nm) into CMK-5 mesoporous carbon via a confined growth strategy. The hierarchical structure and strong electronic interaction between the carbon substrate and Mo-based NPs allowed efficient mass/electron transfer, enabling MoP/CMK-5 to exhibit exceptional HER activity in alkaline (η10 = 65 mV), acidic (η10 = 123 mV), and seawater (η10 = 103 mV) conditions[594]. Single-atom mesoporous carbon catalysts offer a new approach to reducing metal usage. For example, Park et al. developed a Ni single-atom/mesoporous carbon catalyst that achieved atomic-level metal utilization, with selectivity three times higher than that of traditional nanoparticle catalysts while reducing metal usage by 90%[595]. Similarly, Li et al. prepared the α-MoC/N-C/RuNSA composite material using a simple electrodeposition technique (Fig. 34). Ru SAs and Ru NCs were incorporated onto the N-doped carbon nanowire arrays modified by cubic α-MoC NPs (α-MoC/N-C). This material exhibited significant bifunctional activity for both HER and hydrazine oxidation (HzOR) under alkaline conditions. The current densities for HzOR and HER reached 1,000 mA/cm2 at 77 and −150 mV, respectively[596].
Figure 34.
(a) The schematic illustration of the formation of α-MoC/N−C/RuNSA. (b) Evaluation of the overall hydrazine splitting performance. (c) The overall hydrazine splitting in simulated alkaline sewage with different N2H4 concentration[596].
The stability of electrode materials under harsh electrochemical conditions is a critical indicator of their application potential. Studies confirm that <10 nm carbon layers significantly improve the stability of Fe-based catalysts. NiFe2O4@Carbonx/Cd0.9Zn0.1S-y heterojunction photocatalysts were synthesized, achieving a hydrogen production rate of 446.99 mmol/g in 4 h of photocatalysis[597]. Adam et al. synthesized mesoporous core-shell FeP/Fe3O4 coated with an ultrathin carbon layer. Carbon coating significantly improved FeP durability, maintaining a stable current density (20 mA/cm2), and these works highlight the importance of thin carbon coatings on catalyst surfaces[598].
Hu et al. developed an N-functionalized carbon carrier (NC) confined growth strategy, directly preparing ultrafine (3.1 ± 0.7 nm) intermetallic Pt3Fe NPs with core-shell or intermetallic structures on NC. This material exhibits excellent HER activity while maintaining atomic-level dispersion[599]. Xiao et al. constructed a MoO2/Mo3P/Mo2C tri-interface heterojunction@N-doped carbon sea urchin structure via polyoxometalate-induced carbonization. The unique interfacial synergy enabled an overpotential of only 69 mV at 10 mA/cm2, and stable operation for 125 h[600].
Summary and perspectives
-
CMs possess distinct advantages in HER, including highly tunable electronic structures and surface chemistry, which facilitate the precise optimization of active sites through heteroatom doping and defect engineering. Graphene is the most suitable carbon material for HER due to its abundant layered space within, the sp2 hybridized carbon network that enhances electron transmission, and its corrosion resistance in acidic and alkaline environments. In contrast, fullerenes have excellent electronic control capabilities, but the preparation process is complex and difficult to scale up for large-scale production. Biomass CMs conform to the concept of circular economy. Compared with materials that require complex chemical precursors (such as CQDs), they better meet the requirements of sustainable development.
CMs continue to encounter obstacles in HER, including insufficient stability under acidic and alkaline conditions, mass transfer limitations at elevated current densities, and issues related to cost and consistency in large-scale production. With an enhanced understanding of structure-activity relationships, it is anticipated that carbon-based catalysts will achieve significant breakthroughs in the future. These include the development of catalyst coatings specifically designed for kilowatt-scale electrolyzers, the establishment of environmentally friendly preparation methods utilizing waste biomass to produce high-performance catalysts, and the potential replacement of platinum-based catalysts in specialized applications, such as near-seawater electrolysis. Such advancements are expected to substantially lower the costs associated with green hydrogen production, thereby facilitating the global transition to sustainable energy sources.
-
The ability to tailor the properties and structure of CMs and their low cost have made their use desirable in energy storage systems. Batteries, such as sodium-ion batteries (SIBs), vanadium redox flow batteries (VRFBs), lithium-ion batteries (LIBs), lithium-sulfur batteries (LSBs), potassium-ion batteries (KIBs), etc., are embedded with CMs to enhance productivity and efficiency. Various prominent features of CMs, such as high electrical productivity and structural stability, have made them the center of attraction for the research community to deal with energy storage systems and develop materials with great electrochemical properties (batteries and superconductors)[601]. Furthermore, this electrochemical energy could be stored via two fundamental approaches: faradaic and non-faradaic processes; that is, the energy storage process in faradic devices takes place through electrochemical redox reactions in the presence of active reagents (examples are batteries and pseudo-capacitors). Whereas, the energy storage in non-faradaic devices takes place electrostatically (examples are supercapacitors)[602].
Moreover, Yu & Manthiram promulgated that the working properties and efficiencies of the electrochemical batteries could be enhanced by incorporating CMs like graphene, graphite, CNFs, and CNTs. CMs generally serve as anodes and electrodes in batteries due to their conductive polymers and characteristics such as structural stability, structure adaptation guarantees, high inertness, and efficient performance[603]. For instance, after 200 cycles of capacity loss, the graphene-doped anode in LIBs was found to maintain over a 40% retention ratio by outperforming the graphite one. In addition to this, CMs, mainly CNFs, have the potential to entrap poly-sulfide material that is present in LIBs as an anode or cathode. Moreover, the CNFs exhibit an annulled capacity of around 300 mAh/g when incorporated into LIBs. This composite makes them a good alternative to graphite, a low-cost option, and a sustainable employment approach. Additionally, a comparison was made by Xie et al. to explore the retention capacity of a battery that was made by a conventional graphite anode with CNF-LIBs[604]. This exploration concluded that the LIB has five times more retention capacity than conventional battery systems. CMs like graphene and CNTs play a crucial role in the battery system in terms of enhancing the electrical conductivity. For example, an examination of sulfur-loaded PC structures maintained 70% of the initial capacity even after 200 cycles, with an elevation in the performance.
Moreover, discharge capacities as high as 1,200 mAh/g have been reported under optimized conditions. For example, in SIBs, the relatively large ionic radius of sodium favors the use of amorphous or disordered hard carbon over conventional graphene as it mitigates challenges associated with inefficient intercalation[605]. According to Saju et al., the cyclic capacities of the carbon anode were discovered to be 320 mAh/g, which showed no capacity fade even after 100 cycles[606]. Another study promoted the resource and cost-efficiency of KIBs by revealing that nano-carbon sheets, if employed as electrodes in KIBs, have enhanced stability and a greater ion diffusion rate[607]. Likewise, CMs are found to exhibit electrocatalyst properties when mounted on electrode surfaces, enhancing redox flow batteries. Eifert et al. highlighted the efficacy of AC-electrodes (with unbound carbon) in improving the vanadium's redox kinetics up to 30% because of AC-electrode's dense power[608]. These multifaceted CMs applications have paved the path to overcome the challenges in the field of battery technology and to promote the discovery of manageable and controllable high-performance systems[609].
CMs' importance for LIBs
-
LIBs continue to hold significant promise in the field of energy storage, primarily due to their superior energy and power densities. The integration of CMs into anodes and electrodes has become widespread, largely owing to their favorable characteristics, such as high electrical conductivity, extensive surface area, and robust structural stability. Nonetheless, untreated CMs often exhibit relatively low specific capacitance when used directly as electrodes, while necessitating structural or chemical modifications for an enhanced electrochemical performance[610]. In the context of lithium-based cathodes, layered transition metal oxides, particularly lithium metal oxide compounds, are widely recognized for their effectiveness. Among them, lithium cobalt oxide (LiCoO2) was first reported by Mizushima et al. with high discharge voltage, impressive theoretical volumetric capacity, notable specific capacity, and excellent cycling stability[611].
Furthermore, Aurbach et al. discovered the effect of conductive agents, i.e., carbon black, mainly MWCNTs, and carbon cellulose, on three different LIBs, adding LiCoO2[612]. The examination revealed that LiCoO2/MWCNT LIBs exhibited the maximum conductivity at a rate of 2C because of the highly conductive nature of MWCNTs and the promotion of electron transfer to the electrode by CNTs, thus building a conductive network with no loss. Whereas the other two composites, i.e., LiCoO2/CF-LIBs and LiCoO2/carbon black-LIBS, showed 30% and 10% capacity losses after two-week cycling, respectively. This discovery provided the fact that composite conductive agents hold better conductivity in comparison to single conductive agents[539,613]. Collectively, it is concluded that CMs are commonly used in LIBs, as presented in Fig. 35.
Figure 35.
Applicability of CMs in LIBs.
Importance of CMs for LSBs
-
LSBs are widely used in battery technology because of the promising recognition they have gained in terms of efficient energy storage solutions. The high performance in terms of substantial theoretical capacity, i.e., 1,675 mAh/g1, an energy density of 2,600 Wh/kg1, good cycle performance, and fast capacity attenuation has made their use an optimal option for commercial applications[614]. Even so, some challenges still prevail, like sulfur's electrical insulating feature, the residual discharge product lithium disulfide/sulfide, the 80% volumetric change in sulfur, and the shuttle effect created by liquefied lithium polysulfides[615–617]. Ample efforts have been undertaken to address these issues regarding electrolyte optimization, cathode and separator upgradation, and stabilization and protection of the lithium metal anode[114,618–620].
In addition, inter-layering is documented to regulate the shuttle effect created by polysulfides without causing any disturbance to Li+ transfer. CNTs, because of their outstanding properties (high electrical conductivity, efficient mechanical durability, and chemical stability) have been employed as intermediary layers and have gained constant attention for LSBs' application[621,622]. In addition to this, a study identified the impact of multifunctional layers in enhancing the electrochemical demonstration of LSBs, concluding that MWCNTs modified with polypropylene and manganese oxide efficiently attract poly-sulfides while supporting the ion-electron transfer[623].
Furthermore, microporous CMs provide a high surface area, and mesoporous and macroporous, have drawn a deal of interest due to their diverse utilization range, and distinctive features[21,624]. Microporous CMs exhibit promising applicability regarding different requirements posed by LIBs separators because of super electrical conductivity, large surface area, large pore capacity, effective electrochemical stability, and changeable pore channels[136,625,626]. Lin et al. synthesized N-doped ordered mesoporous carbon by using CMs, i.e., poly-furfuryl alcohol, and a mesoporous SiO2 hard template[627]. The N-doped ordered mesoporous carbon portrayed efficient electrochemical properties due to its fine graphene structure and 3.5–4.0 nm pore size. Li & Dong used sodium nitrate crystals to prepare a carbon material with cross-coupled mesoporous characteristics[196]. The processed material contained a robust and developed (monolithic 3D) interrelated mesoporous structure that had mesopore size of 2–4 nm and a high SSA of 2,872.2 m2/g[628,629].
Role of CMs in SIBs
-
SIBs are currently being recognized as a potential substitute for conventional LIBs, largely due to the abundant and geographically diverse availability of sodium, which contributes to their cost-effectiveness and long-term sustainability[630]. This scenario makes SIBs a strategic solution for meeting the demands of grid-scale and large-capacity energy storage applications. Among recent innovations, 2D materials—particularly carbon monolayers—have attracted growing interest in their outstanding electrochemical properties. These monolayers offer exceptionally high theoretical capacities, reportedly reaching up to 2,680 and 1,788 mAh/g, thereby surpassing many current LIB anode materials in terms of potential energy density.
Therefore, the broad spectrum of anode materials for SIBs has been extended to a vast range of precise materials like soft carbon. The flaky structure of soft carbon allows Na+ for insertion and extraction within the layers, which facilitates the retention and discharge of electrical energy. This influences the charge and discharge characteristics of the battery because the migration rate of Na+ elevates due to the scaly/flaky structure of soft carbon that enhances the battery's cycle stability and rate capability[631–633]. Mishra et al. tailored graphite and nitrogen content to produce nitrogen-doped soft carbons via tempering temperatures from 800 to 1,400 °C[634]. The materials displayed prominent cycling stability with a fundamental specific capacity of 201 mAh/g1. Moreover, following 500 cycles, the cycling stability maintained 87% of the initial reversible capacity at around 100 mAh/g[635].
Application of CMs in VRFBs
-
VRFBs have received magnified attention owing to their captivating characteristics and being one of the most favorable electrochemical energy storage systems. Nevertheless, the low energy efficacy and high -yielding cost somehow limit their practicability. One of the deprecating VRFB components is the electrode, which fundamentally determines its strength and ultimate tariff. Thus, highly electrically conductive, cost-effective, chemically stable, ample surface area, and electrochemically reactive electrocatalysts are required for the V2+/V3+ and VO2+/VO2+ redox reactions[636–638]. Furthermore, biomass-derived CMs like N-doped carbon, AC, and PC have been recently documented as precursors to enhance the battery performance. For example, Cheng et al. obtained an N-doped PC electrocatalyst from the kiwifruit and doped it into a graphite felt electrode[639]. This study resulted in peak current densities at both the anode and cathode by utilizing this electrocatalyst. Therefore, the study proved that the addition of a catalyst elevated the electrode's electrical conductivity. Wang & Li used twin cocoons to derive oxygen- and nitrogen-treated CMs[640]. They compared the impacts of both derived carbon species on VRFB performance. The results showed a spike in electrolyte utilization rate, electrolyte accessibility, electrode hydrophilicity, and diffusion. It was observed that by increasing the functional group with oxygen and nitrogen employment, an increase in activated sites and the battery's energy efficiency ranging from 60.7% to 72.5% was witnessed[641]. Additionally, Jiang et al. synthesized PC via fungi (as electrocatalysts), followed by electrode preparation by the immersion method, resulting in a high redox peak potential[637]. The electrocatalyst was used for doping a cloth electrode to reduce the effect of the electrode's spiked electrochemical activity. Doping the electrode reduced the electrocatalytic stability and over-potentials in comparison to the bare electrode, thus increasing and stabilizing the energy efficiency and discharge capacity. Furthermore, fish scales were utilized to derive a carbon-based electrocatalyst, which was further modified by doping the graphite felt electrode with the electrocatalyst. The study concluded that the electrode's polarization in the context of the electrochemical, ohmic, and mass transfer aspects showed a decline. Additionally, with the bare electrode, the battery's discharge capacity yielded 89 mA/h, whereas the doped electrode value was 101 mA/h, even though the current density of the bare electrode was 100 mA/cm2[642].
Challenges and future directions for CMs in energy storage systems
-
The unique aggregation of chemical and physical properties, such as high surface area, high thermal stability, high electrical conductivity, resistance to corrosion and high chemical stability have raised the popularity of CMs in the energy storage system. CMs show compatibility with many other materials to form composites; they have diverse surface chemical attributes, provide various textures and structures, and are easy to process. Still, there lie several challenges, like expensive electrochemical systems, ecologically unfriendly, and a lack of autonomous usage in some cases[643,644]. There may be concerns related to rechargeable batteries, like the environmental impacts of disposing of massive quantities of exploited batteries[645,646]. Moreover, replacement of organic liquid electrolytes with solid-phase electrolytes is a requirement at the current time, as organic solid electrolytes are safe in terms of cell design, are energy-rich, and have fewer safety issues[647,648]. Performance contrasts with non-carbon systems show that traditional inorganic materials (e.g., metal oxides) often give high energy density, CM-enhanced systems uniquely address stability and safety trade-offs. Conventional batteries face rapid degradation under thermal stress, whereas CM composites (e.g., graphene-coated electrodes) leverage high thermal stability to extend cycle life by 40%–60%[647]. Organic liquid electrolytes pose flammability risks during scaling, but CM-solid electrolyte hybrids enable safer cell designs, which are critical for large-format applications[646,648]. Scalability evidence from industrial pilots reveals CM's advantages in real-world contexts. Pilot-scale CM-supercapacitors achieved 98% capacity retention after 10,000 cycles, with higher performance than ceramic alternatives (≤ 80% retention)[643]. This aligns with their structural resilience during rapid charge/discharge. CM-enhanced solid-state batteries demonstrated 30% higher energy density at the module level vs sulfide-based systems[645,647].
Applications of CMs in biomedical sciences
-
Over the past few decades, the synthesis of CMs and their applications in biomedical sciences have gained enough attention. CNTs, MXenes, and graphene are CMs that possess outstanding surface characteristics, chemical stability, targeted delivery potential, and biological acceptance, making them a more promising choice to sort out longstanding challenges both in the field of biomedical sciences and clinical sciences[649–651]. Today, scientists and researchers are expanding the use of CMs in oncology to refine biosensor accuracy and cancer therapy. According to published literature, CMs are used in a variety of domains, including diagnosis and treatment of diseases, brain interfaces, imaging, transfer of genes, engineering of tissues, and antimicrobial applications[652]. When talking about them as a whole, these innovations have provided not only sustainable solutions to complex medical challenges but also tremendous shifts to fulfill the needs of future generations.
Drug delivery
-
Cancer drug delivery is being transformed by CMs because they overcome several challenges to success, such as low drug solubility and off-target effects. As an example, Dash et al. were the first to use a dual-targeting mechanism presented with magnetic GO to increase the Adriamycin tumor-destroying capacity[653]. The technique successfully synergized tumor-specific peptides with the magnetic directing technique and introduced accurate drug delivery without affecting healthy organs. The mGOCG further enhanced targeting via the coating of chitosan on magnetic GO. In addition to graphene, similar versatility can be observed in the case of CNTs. Gul et al. increased drug entry into cells at the level of the cell membrane through a functionalization of carboxylated CNTs using PS-derivatives[654]. These systems modified with polymers suggest the potential for revolution in changing the mode of carriers and cells. Likewise, Moniriyan et al. applied PEGylated magnetic CNTs to transport platinum-based drugs, efficiently inhibiting the in vitro proliferation of cancer cells[655]. Overall, these advances in oncology represent another paradigm shift in the use of CMs in human therapeutics that exploit the tunable nature of CMs to overcome barriers to effectiveness, such as limitations in cellular uptake and control of nonspecific biodistribution to reduce collateral damage to human therapeutics.
Biosensors
-
CMs are excellent for biosensing with special qualities in sensitivity and specificity of detection. The most illustrative example is the study by Elugoke et al., who used CuO nanotubes together with CQDs to measure clinically relevant concentrations of dopamine, paving the way for addressing challenges of pharmaceutical quality control[656]. Subsequently, Gao et al. designed a hybrid sensor of manganese oxide and CDs to detect S. aureus food contaminants, which is an essential improvement in food safety management approaches[657]. Adding even more pertinence, DNA/RNA-conjugated probes reveal indisputable specificity in early disease detection, which equips the stakeholders with practical methods to revise security measures. Mishra et al. have demonstrated that regular CNTs are incapable of detecting SARS-CoV-2, but the hybrid platforms (the combination of fiber Bragg grating and functionalized SWCNT) delved far deeper than the traditional methods[658]. This comparison highlights the necessity of designing CMs based techniques in complicated diagnostics.
Bioimaging
-
The CMs are unique characteristic participants in bioimaging in that they demonstrate superior photoluminescence, biocompatibility, and significantly low toxicity. Sravani et al. applied hydroxylated-SWCNTs that deliver cisplatin to gastric cancer stem cells and kill them by decreasing proliferation via combined cytotoxicity[659]. Molaei demonstrated that nitrogen-doped CQDs, specifically those functionalized with polyethyleneimine, are better cell imaging monitors through photoluminescence than hexamine-doped counterparts[660]. Sharker et al. created hyaluronic acid-based carbon fluorescent NPs, which are ideal for in vivo studies due to their solubility and brightness[661]. Moreover, CDs' high quantum yield makes them broadly useful for cellular imaging[662]. Arjenaki et al. engineered a multifunctional graphene CQD probe labeled with technetium-99m and pembrolizumab, which simultaneously enhances breast cancer imaging and delivers immunotherapy—a dual-utility breakthrough[663].
Tissue engineering
-
Tissue engineering leverages CMs to recreate microenvironments that induce regeneration. In cardiac repair, Adel et al. designed graphene scaffolds that reduce oxidative stress and inflammation in macrophages, accelerating heart recovery in mice[664]. For structural applications, CNTs emerge as ideal bone implant coatings due to their mechanical resilience and elasticity[665–668]. These advancements highlight CMs' capacity to resolve both biological and structural challenges in tissue engineering (Table 7).
Table 7. Application of CMs in biomedical sciences
CMs Applications Purpose Ref. Country (year) BC-MWCNT composite Potential antiviral agents, wound healing Anti-oxidant function, healing of diabetic wound,s and controlled cytokines expression, [751] Pakistan, 2022 BCDs Bio-imaging, biosensor, energy storage Used commonly, Food safety, aureus detection, mode sensing, and radiometric fluorescence [657] China, 2023 CNTs It plays crucial role in drug delivery system Stem cell culture, assisting drug release into cells, biosensing, delivery of doxorubicin to cancer cells [654] Turkey, 2022 CDs with red emission Cancer therapy, bioimaging, biosensing Successful detection of bio thiols, rapid cell internalization [752] India, 2021 MWCNTs Biomedical imaging, tissue engineering, drug delivery system Enhanced bioavailability, targeting tumor site drug delivery, cytotoxicity, HeLa cells [753] Brazil, 2023 Gc-NPs Biosensing, tissue engineering, bioimaging High protein adsorption capacity, imaging of sentinel lymph nodes [97] USA, 2013 Fullerene/Buckyball Photo-sensitizers, wound healing, anti-oxidant, Most commonly used in cancer especially breast cancer [754] China, 2019 GTCEnc Tissue engineering, anti-cancer, wound healing Anticarcinogen, and antimicrobial activity, mostly progressive against COLO205 cell lines [755] Saudi Arabia, 2023 NDs Diagnostics, and wound healer tool Tissue engineering, and neural regeneration and [664] Iran, 2023 Challenges in clinical translation
-
The biomedical applications of CMs show immense promise (e.g., targeted drug delivery, wound healing, and bioimaging). However, clinical adoption of CMs faces significant challenges:
• The CMs (e.g., CNTs or graphene derivatives) can exhibit biocompatibility variability based on synthesis methods, surface functionalization, size, or impurities. For instance, residual metal catalysts in CNTs may trigger cytotoxicity or immunogenicity[669].
• Long-term in vivo safety data remain sparse for most CMs. For example, studies like Gul et al. (Table 7) demonstrate a decline in toxicity of surface-modified CNTs[654].
• There is a lack of tailored guidelines for CMs by regulatory bodies (e.g., FDA, EMA). For example, nano-diamonds (NDs) show excellent neural regeneration potential, but their clinical translation is slowed by undefined clearance mechanisms and potential nanoparticle accumulation in organs[664].
• Multifaceted CMs' interactions with biological systems (e.g., protein corona formation, immune activation) pose a challenge for their application.
• Carbon-based nanocomposites exhibit promising anticancer activity, and their inflammatory potential in humans requires rigorous assessment[670].
• The gap between in-vitro efficacy and in-vivo safety is the biggest barrier for carbon nanomaterial translation[671].
Application of CMs in agriculture
-
Population growth, climate change shifts, and reduced rainfall have accelerated land degradation and diminished irrigation water availability with serious threats to global food security[672]. Concurrently, these issues amplify risks of crop disease proliferation, accidental introduction of harmful plant pathogens, and dependency on synthetic fertilizers and pesticides[673]. Global annual consumption of fertilizers (170–180 million tons) and pesticides continues to rise sharply, reaching approximately 4.6 million tons[674]. These chemicals inflate production costs while causing widespread environmental contamination and degradation[675]. This reality compels policymakers and scientists to urgently explore sustainable agricultural practices that minimize chemical inputs. Consequently, developing alternative approaches, including nanomaterial-based strategies, is essential for achieving environmental resilience and sustainable food systems[676].
The NPs and nanocomposites, particularly CMs, have emerged as promising tools in agriculture. Their applications have improved plant physiology, thereby enhancing nutrition and productivity to relieve biological/physical stresses[677,678]. Hegde et al. emphasize applications in plant growth, proper fertilizer supply, and pest control[679]. Kamle et al. emphasized more applications of nanomaterials as nano-biosensors, soil amendments, and stimulants to plant growth; for example, CNTs can serve as stress-sensing chemical probes to detect stress-indicated molecules such as H2O2 or Ca2+ in plants[680,681]. The flexibility of CMs, such as biocompatibility, low-level toxicity, adjustable surface characteristics, and water solubility, allows using them to improve internal responses and plant yields[682,683].
At the molecular level, CMs reduce the fertilizer dependency by using in-situ nutrient delivery in soil. They have a large surface area and porosity to adsorb and release nutrients (e.g., nitrogen, phosphorus), which have a direct effect on the root uptake capacity and decrease leaching[682]. Some CMs even have pH-buffering, which maintains soil conditions to ensure maximum nutrient bioavailability. Besides, they can be used as nano-sensors which make it possible to monitor pesticide stress signals in real-time thereby lowering the use of pesticides[679,680]. However, these properties that make CMs effective necessitate rigorous biosafety and environmental risk assessments. Potential toxicity, long-term bioaccumulation in soil organisms, and impacts on microbial communities remain critical concerns. Some studies have revealed the low toxicity of CMs, and their environmental persistence and interaction with existing pollutants require thorough investigation[682]. Regulatory frameworks for CMs in agriculture are still evolving, underscoring an urgent need for standardized toxicity testing, fate studies in complex soil ecosystems, and lifecycle analyses to ensure sustainable distribution.
Beyond farming, CMs enhance food packaging and safety through antimicrobial/antioxidant properties, extending shelf-life and enabling pathogen detection[684,685]. Biochar demonstrates scalable environmental benefits. Yadav et al. confirmed its role in sequestering soil organic carbon (20%–50% more than controls at > 10 t/ha), potentially locking in 0.5–3t CO2/ha annually[686]. Lee & Park noted its nutrient delivery efficiency but warned that high application rates (> 8 t/ha) can alter soil microbiota[687]. Bhattacharya et al. further emphasized biochar's value in upcycling agri-waste (1 tonne ≈ 3.7 m3 of saved landfill space)[688]. Collectively, CMs offer diverse solutions across agriculture and food systems (Table 8), showing significant potential to address food security challenges. Nevertheless, responsible implementation demands parallel research into their ecological footprint and long-term biosafety.
Table 8. Application of CMs in agriculture and the food industry
CMs Promising characteristics Applications Ref. Country Applications of CMs in food the industry F-C60 Biological sensor, antioxidants Food quality, food safety, increase caloric value [756] Germany C-Ar Verstyle stability, host of ingredients, culture media It increases shelf life, and also used in food packing [757,758] Italy BC It is highly porous, with a high surface area. Antimicrobial Sustainable food production, waste management, food preservation [688] Korea AC Surface chemistry, adsorption potential, It plays a key role in decolorization, deodorization, and purification [759] India CDs It acts as a biosensor as well as a chemical sensor Detection of food additives, quality, food safety [760] Pakistan CNTs Antibody, antimicrobial properties, biosensing, immobilization, Ethylene production, food ripening, food packaging [761] Bangladesh G Bio and chemical sensor, antimicrobial properties Food safety and preservation, packaging and quality control [762] Pakistan CNFs It has antimicrobial and antioxidant properties Food packaging, quality control, and preservation of food [763] Hungary Applications of CMs in agriculture BC Carbon-rich material, cation exchange capacity, porous, high surface area Reduce soil acidity, high porosity, sequester carbon, improve soil health [686] Nepal F-C60 Free radical binding, resistance to radiation Antioxidant and Nrf2 activation, plant protection, potential for pest control [764] Thailand CNFs Graphene layer, cylindrical structure Plant protection, nutrients, nutrient delivery, carriers for pesticides [763] Hungary C-Ar Low density and porosity, high surface area Biopesticides and biofertilizers, soil amendment, nutrient delivery [687] Korea AC Surface chemistry, porous, high surface area, adsorption potential Control odors, filter water, nutrient availability, improve soil [765] Malaysia CDs Antioxidant and antimicrobial Stress resistance, accelerating plant growth [312,596] China CNTs Unique structural, special electronic, and mechanical properties Plant growth regulation, gene delivery, seed germination [766,767] Pakistan, India G Single layer of carbon atoms, high binding energy Improving soil health, antifungal, enhancing plant growth, antibacterial agents [232] United States Applications of CMs in other fields of the socioeconomic system
-
CMs have emerged as versatile, innovative solutions in modern science and technology due to their exceptional strength, light weight, flexibility, and conductivity. CMs and their composites now permeate diverse sectors beyond energy, environment, biomedicine, and agriculture, driving advancements in electronics, aerospace, textiles, sports, construction, and computing[689–691].
In electronics, CMs enable flexible, high-performance devices, sensors, and storage systems that boost efficiency while reducing costs[692]. Aerospace leverages their strength-to-weight ratio not only for defense applications, such as protective gear and stealth vehicles, but also for commercial aircraft components[693]. Similarly, sports industries utilize CMs in equipment like golf clubs, bicycles, and athletic wear, enhancing performance through lightweight durability[694]. The construction and automotive sectors are increasingly adopting CMs for enhanced structural efficiency and longevity[695]. Compared to conventional materials, CMs consistently deliver longer product lifespans, cost reductions, and manufacturing efficiencies across these fields.
However, the socioeconomic promise of CMs necessitates parallel consideration of their environmental footprint. While their durability can extend product lifecycles, fabrication processes often involve energy-intensive methods, and end-of-life disposal poses challenges[695]. Certain CM composites may resist recycling or degrade into persistent environmental contaminants. Responsible deployment thus demands developing standardized recycling protocols and assessing long-term ecotoxicological impacts, particularly for high-volume sectors like electronics, textiles, and automotive manufacturing. Collectively, CMs have solidified their role as indispensable materials, reshaping technological progress. Yet, maximizing their socioeconomic contribution requires balancing innovation with sustainable lifecycle management to mitigate unintended ecological consequences.
-
Although CMs have demonstrated broad utility in energy and environmental domains, their structure–property relationships are often highly nonlinear and governed by interdependent microstructural features. These complexities, coupled with long experimental cycles and substantial resource consumption, limit the effectiveness of traditional trial-and-error strategies in rapidly optimizing material performance. To overcome these constraints, AI, particularly ML and deep learning (DL), has emerged as a transformative tool. By mining experimental and computational data, AI models can capture hidden correlations between structure and function, enabling accurate property prediction, rational structural design, and optimization of synthesis conditions[696]. Compared with conventional approaches, AI excels in handling nonlinear, high-dimensional systems and is accelerating a paradigm shift in materials research from empirical workflows to data-driven, predictive methodologies.
As illustrated in Fig. 36, AI is increasingly being embedded into closed-loop workflows that enable intelligent and adaptive carbon material development[697]. These workflows integrate data input, model construction, performance prediction, structure generation, synthesis planning, and experimental feedback, supporting both forward and inverse design strategies across the material development pipeline. Building on this integrated framework, AI offers a suite of specific capabilities that directly address key bottlenecks in CMs research. These include property prediction, where ML models capture nonlinear correlations between structural features (e.g., surface area, porosity, and doping configuration) and performance metrics such as adsorption capacity, catalytic activity, or electrical conductivity[698]. AI also enables inverse design, allowing researchers to define target properties and identify structural candidates most likely to achieve them[699]. In addition, AI contributes to synthesis optimization by revealing the relationships between processing parameters and material quality, and facilitates the automated interpretation of characterization data, such as extracting morphological features from microscopy images or deconvoluting overlapping spectra peaks[700,701]. It supports high-throughput screening, enabling rapid evaluation of material libraries under multi-objective constraints[702]. These capabilities form the basis of next-generation, data-driven, and autonomous materials discovery frameworks.
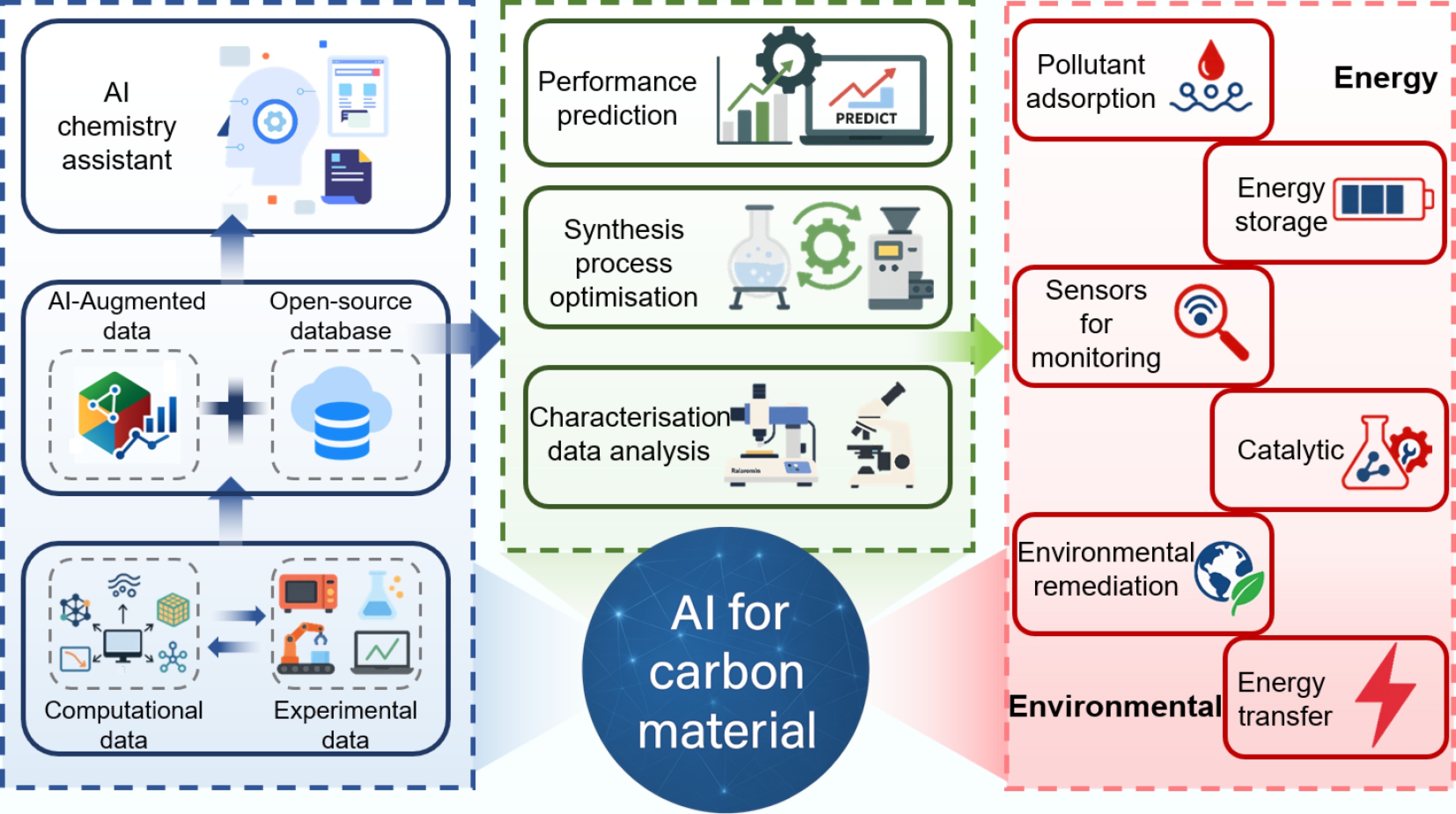
Figure 36.
Schematic overview of several insights into the development of AI approaches in CMs for various practical applications.
Algorithm selection and model matching
-
Given the diversity of research tasks in CMs development, selecting the appropriate ML algorithm is essential for achieving reliable and efficient outcomes. Before diving into model-specific strategies, it is helpful to briefly introduce the overall categorization of ML approaches commonly applied in CMs research. ML algorithms can generally be classified into supervised learning, unsupervised learning, and reinforcement learning[703]. Supervised learning methods such as decision trees, support vector machines (SVM), neural networks, and gradient boosting are widely used for tasks like regression and classification, where the target variable is known. Unsupervised learning algorithms, including K-means clustering and principal component analysis (PCA), are typically used for data preprocessing, feature extraction, and pattern recognition without predefined labels. Reinforcement learning involves an environment where the machine interacts with it to obtain rewards as feedback. Building on this categorization, different tasks such as property prediction, inverse design, synthesis optimization, and data-driven characterization often involves distinct data structures, feature complexities, and computational demands, necessitating tailored algorithmic strategies[704–706]. Table 9 provides a systematic overview of these commonly used algorithms, summarizing their application domains, strengths, and limitations, and serving as a reference point for researchers seeking detailed implementation strategies[708].
Table 9. Representative AI approaches (mainly ML and DL) applied in CMs
Model Application Advantages Disadvantages Ref. K-means clustering Structural clustering, feature selection Easy to implement; fast training; scalable to large datasets. Requires specifying the number of clusters (K); may not capture complex, non-linear feature relationships. [707] PCA Dimensionality reduction, feature extraction Reduces dimensionality; removes feature correlation; reduces overfitting risk; improves visualization; few hyperparameters. Requires data standardization; may be hard to interpret; sensitive to outliers; assumes linearity; potential information loss. [707] GAN Image generation, data augmentation, style transfer Can generate high-quality, diverse samples; does not require explicit likelihood modeling. Training instability; can be computationally intensive; evaluation metrics can be challenging. [708] KNN Classification, regression Simple and intuitive; no explicit training phase (lazy learner); adapts naturally to new data; robust to outliers/noise (depending on K); supports multi-class. Computationally expensive prediction (slow for large datasets); sensitive to irrelevant features and the curse of dimensionality; requires feature scaling/normalization; sensitive to K choice; missing data needs handling. [768,769] Decision tree (DT) Classification, regression Easy to understand and interpret (white box); requires little data preprocessing (handles mixed types, missing values, no scaling needed); handles non-linear relationships. Prone to overfitting (especially deep trees); unstable (small data changes can alter tree); biased towards features with more levels; poor extrapolation for regression; can create complex trees. [768,770−772] Extremely randomized trees Classification, regression Very fast training; reduces variance compared to single DT; introduces extra randomness for robustness. Less interpretable than single DT; potential slight increase in bias; may ignore some feature interactions; still requires ensemble for best results (like RF). [773] Gradient boosting regressor Regression High prediction accuracy; naturally handles non-linear relationships and interactions; robust to outliers (via loss functions). Computationally intensive training; requires careful hyperparameter tuning; can overfit if not regularized; less interpretable than linear models or single decision trees; sequential training limits parallelism. [769, 774−777] SVM Classification, regression Effective in high-dimensional spaces; robust to overfitting via margin maximization; versatile (different kernels for non-linearity); memory efficient (uses support vectors). Computationally expensive training for large datasets; choice of kernel and hyperparameters (C, gamma) is crucial; less effective on very noisy datasets; poor scalability; 'black box' nature for kernels. [768−772,
777−780]Partial least squares Regression (especially with multicollinearity), dimensionality reduction Handles multicollinearity well; allow small numbers of samples, even less than that of variables; provides interpretable components (latent variables); model is interpretable. Performance can degrade with very high dimensionality or complex non-linearities; components may be harder to interpret than PCA; less common than PCA/RF for screening. [779] Gradient boosted decision tree Classification, regression Often achieves state-of-the-art accuracy; handles mixed data types; robust to outliers and missing data; provides feature importance. Highly sensitive to hyperparameters; prone to overfitting without careful regularization; sequential training is slow; harder to tune than RF; more complex. [781] RF Classification, regression High accuracy; robust to overfitting and noise; handles high dimensionality well; provides feature importance; parallelizable training; handles missing data. Less interpretable than single DT; can be computationally expensive for very large forests; prediction speed slower than linear models; may not extrapolate well for regression; biased towards categoricals. [704, 768,769,
772,776−779,
783]XGBoost Classification, regression Very high performance; fast and efficient training; built-in regularization; handles missing data; parallel computing; often top choice in competitions. Many hyperparameters to tune; prone to overfitting if not regularized; less interpretable than RF; prediction can be slower than RF with large forests; 'black box'. [675, 705, 776] MLP Classification and regression Can approximate complex non-linear functions; learns feature representations automatically; universal function approximator. Requires careful hyperparameter tuning (layers, neurons, LR); prone to overfitting (needs regularization); sensitive to feature scaling; 'black box'; training can be slow; local minimal risk. [776, 783] ANN Classification; regression; clustering (similar to MLP) Can model complex non-linear relationships. Requires extensive tuning; prone to overfitting; sensitive to feature scaling; 'black-box' nature; slow training. [724, 778, 784] Back-propagation neural network Classification; regression Can approximate complex non-linear functions; learns feature representations. Requires careful tuning (architecture, learning rate); prone to overfitting; sensitive to feature scaling; 'black box'; slow training. [768, 771] Multiple linear regression (MLR) Regression, forecasting, trend analysis Simple, interpretable model (coefficients); computationally efficient; well-understood statistical properties. Assumes linearity, independence, homoscedasticity, normality; poor performance with non-linear relationships or high-dimensional data; sensitive to outliers and multicollinearity. [772, 778] Linear regression Regression Simplicity and interpretability; computationally very efficient; statistical inference possible. Same strict assumptions as MLR; limited to modeling linear relationships; only handles a single predictor. [777, 779] Least absolute shrinkage and selection operator Regression, feature selection Performs feature selection (shrinks coefficients to zero); helps mitigate multicollinearity; produces sparse models. Can be unstable with highly correlated features; selects at most n features; biased estimators; choice of lambda critical. [785] RR Regression Reduces overfitting by shrinking coefficients; handles multicollinearity well; always solvable. Does not perform feature selection (all coefficients remain non-zero); less interpretable than models with feature selection; biased estimators. [773] RNN Sequential data: Time series, NLP Designed for sequential/temporal data; maintains memory/hidden state; can process variable-length inputs. Suffers from vanishing/exploding gradients; struggles with long-term dependencies; relatively slow to train; training instability. [786] Long short-term memory Sequential data: Time series, NLP Solves vanishing gradient problem of basic RNNs; excels at capturing long-term dependencies in sequences. More complex and computationally expensive than basic RNNs; still challenging for extremely long sequences; many parameters to tune; slow training. [783] CNN Grid data: Image recognition, Computer Vision Highly effective for image/audio data; exploits spatial/temporal locality; translation invariance; parameter sharing reduces complexity. Requires large datasets; computationally intensive (especially training); less intuitive interpretation; architecture design requires expertise; primarily for grid-like data. [787] GNN Graph-based tasks (e.g., social network analysis, molecular modeling) Specialized for graph-structured data; captures node and edge relationships. Requires graph-structured data (specific input format); model design and implementation can be complex; computationally expensive for large graphs; may not generalize well to non-graph data. [712, 788,789] Deep neural networks Complex tasks: classification, regression (using architectures like MLP, CNN, RNN) Can model highly complex non-linear relationships and hierarchies in data; state-of-the-art performance for many tasks. Requires very large datasets; computationally very expensive to train; severe overfitting risk (needs strong regularization); extreme 'black box'; sensitive to hyperparameters and initialization; debugging hard. [790] Structure–property modeling
-
Understanding the structure–property relationships of CMs is crucial for their application in energy storage and environmental remediation. These relationships describe how the structural features (e.g., porosity, SSA, functional group distribution, and crystal defects) influence their performance metrics, including adsorption capacity, electrochemical performance, and mechanical properties. However, the multiscale complexity and nonlinear coupling of these materials often exceed the scope of traditional empirical or physical models[709,710]. In recent years, AI methods have been introduced as complementary tools to investigate such relationships, drawing on their strengths for data mining and pattern recognition[711,712].
By processing large experimental and computational datasets, AI can reveal structure–performance correlations that traditional methods often miss, thereby improving prediction accuracy and model generalizability[713]. In mechanical property prediction, Hajilounezhad et al. developed a comprehensive dataset of simulated CNT forests with diverse arrangement densities and morphologies[714]. They trained a convolutional neural network (CNN) classifier to predict the stiffness and buckling load of these structures, achieving an accuracy of over 91%. This study highlights the potential of integrating image analysis with AI algorithms to capture the complex microstructure–mechanical property relationships. In the field of electrochemical performance prediction, Reiser et al. combined graph neural network (GNN) with physics-informed features, such as symmetry constraints, to predict the conductivity and energy storage properties of MOFs and carbon-based materials, thereby improving prediction accuracy[712].
Moreover, Desgranges & Delhommelle employed an ensemble learning approach, integrating multiple weak learners into a unified framework to predict the partition functions of adsorbed fluids in nanoporous materials (Fig. 37a)[715]. This approach allows for rapid estimation of selectivity and desorption free energy, significantly reducing computational costs and demonstrating AI's potential to accelerate the evaluation of porous materials. Zhang et al. further proposed a Monte Carlo tree search combined with a recurrent neural network framework (RNN, Fig. 37b), enabling the inverse design of application-specific MOFs. This approach integrates target applications, metal node types, and organic linker selection, achieving efficient high-throughput screening, and candidate optimization[713]. He et al. employed a transfer learning approach that combined ML and ab initio calculations to identify metallic MOFs from a database of 2,932 structures[711]. Their approach ultimately revealed six MOFs with zero band gap (Fig. 37c−e), including Mn2[Re6S8(CN)6]4, Mn2[Re6Se8(CN)6]4, and Mn2[Re6Te8(CN)6]4, which displayed promising electrical conductivity with large CB dispersions-up to a few hundred meV, suggesting high carrier mobility[711]. These studies illustrate the versatility of AI in structure–property prediction and establish a foundation for efficient screening, rapid evaluation, and performance-driven structural design and synthesis.
Figure 37.
(a) Schematic illustration of an ensemble learning framework integrating ANNs to predict the partition functions of adsorbed fluids in metal–organic and COFs[715]. (b) Representation of the Monte Carlo tree search and recurrent RNN framework for the inverse design of application-specific MOFs[713]. (c)–(e) Optimized crystal structures and calculated electronic band structures of three DFT-confirmed metallic MOFs—Mn9Re24C24S32N24, Mn9Re24C24Te32N24, and Mn9Re24C24Se32N24—highlighting their zero band gaps and potential electrical conductivity[711].
Inverse design and synthesis recommendation
-
AI is moving beyond property prediction toward inverse design and synthesis optimization, shifting from passive evaluation to proactive discovery, which is especially important for carbon materials with complex structures and vast design spaces. At the virtual design level, AI has been widely applied to guide synthesis optimization and accelerate structural generation. For example, Rao et al. employed an ML model to identify optimal synthesis conditions for SWCNTs within a narrow diameter range, completing the task with fewer than 100 experiments and significantly improving experimental efficiency[716]. Similarly, Lin et al. trained a regression model to balance the trade-off between crystallinity and growth efficiency in SWCNT forests, successfully validating the model with fewer than 50 experiments, thereby demonstrating AI's potential in synthesis optimization for carbon-based materials[717].
Meanwhile, generative DL models have demonstrated clear advantages in performance-driven structural design. Yao et al. proposed a supramolecular variational autoencoder framework to map carbon material structures into a continuous latent space, enabling smooth interpolation and targeted generation of new structures, such as porous graphene and defect-engineered carbons, thereby expanding the design space[718]. Song et al. combined GANs with ML to predict material synthesizability and generated over 260,000 potential new materials. They subsequently validated these materials' formation energies, thermal stabilities, and exfoliation energies using DFT calculations, establishing a closed-loop process from generation to screening and verification, which accelerated new material discovery[708].
Recent studies show that AI can be integrated into laboratory workflows to support the development of autonomous and intelligent materials research platforms. Zheng et al. developed the ChatGPT Chemistry Assistant, which combines prompt engineering in chemistry with AI to automatically extract MOF synthesis conditions from various literature formats, achieving an F1 score exceeding 90%. This tool provides valuable support for synthesis planning and data-driven experimental design[719]. Furthermore, the CARCO platform integrates the Carbon_BERT model to screen TiPt bimetallic catalysts with superior performance compared to traditional Fe catalysts. Coupled with an automated CVD system, CARCO leverages digital twin technology to simulate over one million experiments within 20 min, achieving high-density HACNT array synthesis with an accuracy of 56.25%. The entire experimental process was completed in just 43 d, greatly reducing research time and opening new avenues for high-throughput, intelligent CNMs research[720]. Szymanski et al. introduced the A-Lab, which uses computation, historical data from the literature, ML, and active learning to plan and interpret the outcomes of experiments performed using robotics[721]. Over 17 d of continuous operation, the A-Lab realized 41 novel compounds from a set of 58 targets and was able to summarize and suggest failing formulations to further improve the synthesis success rate. Jiang et al. developed an autonomous chemical synthesis robot for the exploration, discovery, and optimization of nanostructures, driven by real-time spectroscopic feedback, theory, and ML algorithms that controlled the reaction conditions and enabled selective templating of reactions[722]. This platform further optimized nanostructures with desired optical properties by combining experiments and extinction spectrum simulations to achieve a yield of up to 95%. These studies demonstrate that AI-driven structural design and synthesis recommendation is becoming a key direction in CM research, establishing the design–synthesis–validation loop and enabling high-throughput, intelligent material discovery.
Advances in electron microscopy, Raman spectroscopy, Fourier-transform infrared spectroscopy, and X-ray diffraction have led to the accumulation of vast amounts of complex, high-dimensional, and often unstructured data, such as microscopic images and spectral signals, which pose major challenges for conventional analysis. The complexity and subjectivity of these datasets make manual interpretation slow, labor-intensive, and inconsistent. To address these challenges, AI techniques particularly DL have been introduced to improve the efficiency and accuracy of data interpretation. For instance, Förster et al. developed a CNN-based DL model that automatically extracted chiral indices of CNTs from HRTEM images, significantly enhancing the throughput and statistical reliability of HRTEM image analysis[700]. Similarly, Zhang et al. applied a deep neural network to analyze Raman spectra, achieving classification accuracies exceeding 90% for CNTs even at a very low signal-to-noise ratio of 0.9 and up to 98% at signal-to-noise ratio of 2.2, demonstrating the potential of DL for robust feature extraction in complex mixtures[701]. Through the application of AI, characterization data processing is gradually shifting from traditional descriptive analysis to predictive understanding, enabling the integration of visual and spectral data streams for intelligent interpretation. This evolution lays the foundation for closed-loop materials development, where insights extracted from images and spectra not only describe structure but also inform design and performance optimization.
Application of AI models in environmental and energy domains
-
In environmental remediation, AI models have been widely applied to predict pollutant adsorption, including gases such as methane and carbon dioxide. For instance, Zhang et al. used an ML model to systematically analyze the influence of pore structure features of porous CMs on methane storage performance. They highlighted the contributions of SSA, micropore volume, mesopore volume, temperature, and pressure to adsorption behavior, effectively complementing and validating experimental data[723]. Similarly, Burner et al. applied a neural network model to predict the CO2 uptake capacity and CO2/N2 selectivity of MOFs under low-pressure conditions[724]. Their model demonstrated excellent predictive performance, achieving R2 values of up to 0.96, and significantly accelerated the screening process-capturing over 99% of the top 1000 high-performing MOFs compared to traditional Grand Canonical Monte Carlo simulations at a speed at least ten times faster. As illustrated in Fig. 38a, the heat map shows the strong correlation between ML-predicted CO2 working capacity and Grand Canonical Monte Carlo-calculated values, highlighting the efficiency and accuracy of the ML model in high-throughput screening. Furthermore, Hasan et al. integrated ML algorithms into a molecularly imprinted polymer-based electronic nose system to tackle the challenges of VOC detection in complex environments. Their approach established a high-sensitivity and high-selectivity platform for multi-component VOC detection, providing a strong foundation for the development of intelligent environmental monitoring systems[725].
Figure 38.
(a) Heat map showing the correlation between ML-predicted CO2 working capacity and Grand Canonical Monte Carlo-simulated CO2 working capacity for MOFs[724]. (b) Heat map of ML-predicted theoretical limiting potentials for Pc DACs; the redder, the better the catalytic activity for the CO2RR. (c) Feature importance analysis highlighting the top 12 descriptors (contribution 41.5%)[726]. (d), (e) Free-energy diagrams and geometries of prominent M@NₓCᵧ SACs for the ORR and OER, at zero electrode potential. The rate-determining steps are highlighted by shades, and the blue and gray balls represent nitrogen and carbon atoms, respectively[727]. (f) Visual comparison of limiting potentials for the NRR and the HER for various SACs and related materials[728].
In the energy sector, AI techniques, particularly GNNs, have shown strong potential in the design and development of carbon-based catalysts and energy storage materials. Wu & Li reviewed the use of ML in predicting the electrochemical performance of SACs, emphasizing its role in activity screening and mechanistic analysis for key reactions such as CO2RR, HER, and OER[726]. Li et al. combined RF algorithms with DFT calculations to investigate the electrochemical properties of 104 graphene-supported SACs across ORR, OER, and HER pathways[373]. Their model effectively identified the importance of various descriptors and accelerated the screening of promising catalyst candidates, thereby supporting the design of high-performance carbon-based electrocatalysts. This approach employed DFT simulations to calculate the structures and limiting potentials of graphene-supported M@NxCy SACs, and ML models (mean square errors of 0.027/0.021/0.035 V for ORR/OER/HER, respectively) to predict catalytic activity. The study confirmed the reliability of the ML models and identified top-performing SACs, such as Ir@pyridine-N3C1 and Ir@pyridine-N2C2, that outperform conventional noble-metal oxides. These results demonstrate how AI can significantly accelerate catalyst screening and performance evaluation, thereby providing robust support for carbon-based catalyst design and development in energy applications (Fig. 38b, c). Additionally, Wan et al. employed a gradient boosting regression algorithm integrated with DFT-based screening to predict the CO2RR activity of phthalocyanine-based dual-atom catalysts (Fig. 38d, e), achieving an R2 score of 0.96 and identifying promising materials with low limiting potentials[727]. Furthermore, Zafari et al. utilized ML models integrated with DFT calculations to systematically evaluate the NRR selectivity and HER competition of various catalysts[728]. They visualized the relationship between the limiting potentials of NRR and HER (Fig. 38f), highlighting the performance distribution and revealing promising catalyst candidates with high selectivity and activity.
These studies demonstrate that the integration of AI into CMs research has accelerated the shift from trial-and-error to data-driven development. By leveraging powerful data processing, pattern recognition, and optimization capabilities, AI has transformed the research paradigm from performance prediction and structural design to synthesis optimization and characterization substantially improving both efficiency and precision. This data-driven approach not only accelerates the discovery of novel materials but also expands the application potential of CMs in energy storage, environmental remediation, and catalysis.
Nevertheless, key challenges persist. Data silos, inconsistent formats, and limited interoperability hinder integration, while scarce and heterogeneous datasets restrict model generalizability. The interpretability and deployment of AI models in complex CM systems remain limited, and gaps between simulations and experiments slow iterative progress. Overcoming these issues will require standardized frameworks, open databases, user-friendly toolkits, interpretable models, and closed-loop systems linking prediction, synthesis, characterization, and feedback.
-
In this review, the synthesis strategies, structural modifications, and multifunctional applications of CMs in the fields of energy, environmental remediation, and health-related technologies are summarized. The discussion covers both traditional and emerging preparation methods, the integration of AI, toxicity concerns, and future potential. While CMs possess advantages such as tunable structures, chemical stability, and cost-effectiveness, several key challenges still hinder their large-scale, sustainable application. These include the difficulty of translating lab-scale performance to practical use, limitations in stability, selectivity, recyclability, synthesis scalability, environmental safety, and the early-stage integration of AI technologies. To address these challenges and accelerate progress, targeted future research directions are proposed.
-
There are still some challenges for the real applications of SCMs: (1) most reported record values are achieved at the laboratory level. From laboratory study to real application is still a big challenge; (2) the stability and reusability of some materials under extreme conditions do not meet the requirements for real conditions; (3) the selectivity of CMs for the special target is still a problem, especially under complex systems in the presence of competing pollutants; (4) the separation of powder CMs from solutions restricts the real application at a large scale; (5) under industrial level conditions, the efficiency and stability after long time operation can not merit the requirement; (6) some carbon nanomaterials with excellent properties can not be synthesized at low cost in large scale, and effective recycling strategies are still lacking; (7) the toxicity of some carbon nanomaterials in environment should not be ignored; (8) AI technique will contribute significantly in the guiding to construct carbon nanomaterials for special properties in near future; (9) last but not least, the excellent properties of carbon nanomaterials are promising materials in energy and environmental pollution management. With the development of science and technology, it is no doubt that carbon-based materials will contribute greatly to industrial development and to the improvement in the quality of human life.
-
All authors contributed to the study conception and design. The first draft of the manuscript was written by Z Sun, Y Liao, Y Zhang, S Sun, Q Kan, Z Wu, L Yu, Z Dong, Z Wang, R He, L Wang, Q Meng, H Wang, Q Wang, L Mao, D Pan, S Wang, Z Zhang, W Zhu, S Liu, M Wakeel, B Hu, T Duan, X Tai, X Wang, and all authors commented on previous versions of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets used or analyzed during the current study are available from the corresponding authors on reasonable requests.
-
This work was supported by National Natural Science Foundation of China (22176077, 22376059, 22406028), and Fundamental Scientific Research Funds for Central Universities (lzujbky-2023-stlt01).
-
All authors declare that there are no competing interests.
-
Different techniques for synthesizing carbon materials are described.
Applications of carbon materials in environmental pollution treatments are summarized.
Applications of carbon materials in OER, HER, and CO2RR areas are reviewed.
AI techniques in carbon material synthesis and application are discussed.
Carbon materials exhibit great potential in environmental and energy applications.
-
Full list of author information is available at the end of the article.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Sun Z, Liao Y, Zhang Y, Sun S, Kan Q, et al. 2025. Sustainable carbon materials in environmental and energy applications. Sustainable Carbon Materials 1: e007 doi: 10.48130/scm-0025-0002 

Sustainable carbon materials in environmental and energy applications
- Received: 08 June 2025
- Revised: 17 July 2025
- Accepted: 10 September 2025
- Published online: 11 November 2025
Abstract: Fast industrial development not only requires a sustainable energy supply but also generates environmental pollutants. Carbon materials, particularly sustainable carbon materials (SCMs), have attracted multidisciplinary interest because of their special physicochemical properties, such as porous structures, high surface areas, sufficient active sites, abundant functional groups, high stability, and post-modification. Herein, the techniques for the synthesis and characterization of SCMs are summarized, along with recent achievements in the removal of environmental pollutants (e.g., heavy metal ions, radionuclides, and organic pollutants), and in energy-related applications (e.g., CO2 reduction reaction, hydrogen evolution reaction, oxygen evolution reaction, and battery systems). The reaction mechanisms are discussed from macroscopic results, advanced spectroscopic characterization, and theoretical calculations. The potential toxicity to the environment and living organisms is described, and artificial intelligence techniques in the carbon material studies are also explored. Finally, the challenges and perspectives regarding the applications of SCMs in energy and environmental areas are presented from the authors' viewpoint.