










-
Seeds are critical reproductive organs of plants, making them one of the most important stages in the plant life cycle[1]. Seeds also serve as primary reservoirs for nutrient storage, forming the foundation for food yield and quality[2]. Seed dormancy is defined as the inability of intact, viable seeds to germinate under optimal conditions, a mechanism that enhances plant survival by postponing germination until favorable circumstances arise. Conversely, seed germination involves the transformation of quiescent dry seeds from water absorption to embryo axis elongation[3,4]. The transition between dormancy and germination is intricately linked to developmental and environmental signals, dictating both the timing and location of plant growth, thereby rendering this process a highly dynamic field within plant science[5].
Soybean (Glycine max) ranks among the most economically significant oilseed and biodiesel crops globally, serving as a primary source of oil and protein for human consumption and animal feed[6−8]. On one hand, the high protein and oil content in soybean seeds makes them particularly susceptible to stress from elevated temperatures and humidity prior to harvest, leading to seed deterioration that adversely affects grain quality, yield, and seed vigor[9,10]. Conversely, timely germination coupled with uniform seedling emergence under natural field conditions is essential for achieving optimal yields in soybean cultivation[11]. Consequently, maintaining adequate seed dormancy along with a smooth transition from dormancy to germination at the appropriate time is paramount for successful soybean production. Previously, we identified a dormancy locus known as the G gene through genome-wide association studies and demonstrated that this gene underwent parallel selection for dormancy across multiple crop families during their domestication[12]. GmAOC4, which encodes an allene oxide cyclase, has been reported to significantly influence seed germination by promoting jasmonic acid accumulation in soybean[13]. Through deep re-sequencing efforts, Tian et al. uncovered numerous genetic loci and causal genes associated with seed germination in soybean[14]. Additionally, knockouts of abscisic acid (ABA) receptor genes reduced the sensitivity of soybean seeds to ABA during germination[15]. Despite these advances, there remains a need to identify additional genes to achieve the optimal balance between seed dormancy and germination, thereby improving seed yield and quality in soybean.
Sphingolipids, as major constituents of lipid rafts, are indispensable metabolites ubiquitously present in all plant species. These elaborate lipids assume paramount roles in a wide array of cellular processes, including membrane architecture, signaling cascades, and stress response mechanisms[16]. The molecular composition of sphingolipids consists of three principal elements: a long chain base (LCB), a long chain fatty acid (LCFA) or very long chain fatty acid (VLCFA), and a polar head group. The structural diversity of sphingolipids arises from variations in the length and saturation of these fatty acids, as well as modifications to the polar head groups[17,18]. The initial step in sphingolipid biosynthesis is the condensation of serine and palmitoyl-CoA to form 3-ketosphinganine, catalyzed by serine palmitoyl transferases (SPTs)[19]. The intermediate 3-ketosphinganine is then reduced by 3-ketodihydrosphingosine reductase (KSR) to yield sphinganine (d18:0), also recognized as long-chain bases (LCBs). Sphinganine serves not only as a vital building block for subsequent synthesis but also participates in signaling pathways that regulate cell growth and differentiation[20]. Subsequently, LCBs are acylated to fatty acids—typically spanning from 16 to 26 carbons in length—by ceramide synthases (CS), specifically isoforms LOH1, LOH2, and LOH3. This procedure gives rise to the formation of ceramides, which serve as the core scaffold for more complex sphingolipids such as glycosylceramides. Ceramides fulfill multiple functions within plant cells, they contribute to membrane integrity while simultaneously acting as precursors for bioactive compounds involved in stress response mechanisms[21]. Once synthesized, ceramides are translocated to the Golgi apparatus where they undergo further modifications, leading to the generation of complex sphingolipids such as inositol phosphoryl ceramides (IPCs), glucosylceramides (GlcCer), and glucosylinositol phosphoryl ceramides (GIPC), which are the preponderant sphingolipids found within plant lipid extracts. These modifications enhance their functional capabilities within cellular membranes[22]. Additionally, through enzymatic activities involving sphingosine kinase (SPHK) and ceramide kinase, both Sph and Cer can undergo phosphorylation modification into their respective phosphates: Sph-1-P and Cer-1-P. Phosphorylation significantly alters their biological activity. For instance, it can modulate signal transduction pathways related to stress responses or developmental progressions[23]. In plants specifically attuned to diverse ecological niches, hydroxylation at the C-4 position on two-hydroxylated LCBs such as sphinganine leads to the genesis of phytosphingosine (t18:0). This modification confers additional functionality pertinent under specific physiological circumstances[24]. Furthermore, saturated LCBs can be desaturated via enzymes designated as Sphingolipid LCB desaturases at the C-8 position or through Δ4-desaturase at the C-4 position, resulting in d18:1-based or t18:1-based ceramides, among others. Such reactions markedly enhance the structural diversity of plant LCBs, significantly influencing their physical properties such as membrane fluidity and interactions with other biomolecules during various metabolic processes[25].
Although sphingolipids have been reported to play significant roles in plant growth and stress responses, their functions in seed dormancy and germination remain largely unexplored. In the present study, we employed an integrated approach combining transcriptome analysis and sphingolipidomics to demonstrate that saturated sphingolipids—particularly ceramide d18:0/16:0, ceramide d18:0/22:0, and phytoceramide t18:0/24:0—are essential for seed germination in soybean. Additionally, we identified two gene modules associated with seed germination. These modules contain numerous key genes closely linked to seed dormancy and germination, as observed in Arabidopsis and rice. Our findings provide valuable genetic resources for further investigation into soybean seed germination.
-
The soybean cultivars utilized in this study include Zhonghuang13 (ZH13) and Williams 82 (W82), kindly provided by Professor Fanjiang Kong from Guangzhou University (Guangzhou, China). Transgenic lines overexpressing GmSLD1 were developed using the W82 genetic background. Both ZH13 and W82, along with the specified transgenic plants, were cultivated under natural field conditions at the experimental station of the College of Agronomy and Biotechnology, Southwest University, Chongqing, China.
Vector construction and transformation
-
To overexpress GmSLD1, the coding sequence of GmSLD1 was amplified from soybean seed cDNA using SLD-GFP primers (Supplementary Table S1). Homologous recombination was then performed to integrate the amplified sequences into the pTF101 vector using a Ligation-Free Cloning Kit (Applied Biological Materials, USA), resulting in the construction of the SLD1OE vector.
The resultant vector was introduced into soybean plants via Agrobacterium tumefaciens-mediated transformation[26].
Germination assays
-
Freshly harvested mature and elite seeds of ZH13 were immersed in 20 mL of water, either with or without the addition of 5 μM C24 Phytoceramide t18:0/24:0 (CAS No. 34437-74-6, Avanti, USA), in Petri dishes measuring 10 cm in diameter at room temperature. Each treatment consisted of 30 seeds per dish, with nine replicates prepared for each condition. The seeds were transferred to fresh water daily. Germination was monitored every 12 h until the majority of seeds had germinated. Germination was defined as the emergence of the radicle through the seed coat. Photographs were taken using a stereo microscope (ZEISS, Germany) as shown in Fig. 1, and images for Fig. 6d were captured using a Canon 800D camera after 48 h of imbibition.
Transcriptome analysis
-
Ten seeds were randomly collected at 0, 6, 12, and 24 h post-imbibition from a total of 270 seeds, with three biological replicates. The samples were stored at −80 °C at the College of Agronomy and Biotechnology, Southwest University (Chongqing, China). RNA-Seq analysis was performed by Shanghai Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China) following the previously described protocol[27] with some modifications. Specifically, the raw paired end reads were trimmed and quality controlled by fastp (
https://github.com/OpenGene/fastp ) and aligned to the Glycine max genome Wm82.a2.v1 (https://phytozome-next.jgi.doe.gov/info/Gmax_Wm82_a2_v1 ) using HISAT2 (http://ccb.jhu.edu/software/hisat2/index.shtml ). Transcriptome data were analyzed on the free online platform, Majorbio Cloud Platform (www.majorbio.com ).Extraction and detection of sphingolipids
-
Sphingolipids were extracted from the ten randomly selected seeds at 0, 6, 12, and 24 h post-imbibition, with three biological replicates. The extraction and detection were performed by LipidALL Technologies Co. Ltd. (
www.lipidall.com ) as described by Xu et al.[28], with some modifications. Specifically, the collected soybean seeds were weighed and inactivated with hot isopropanol according to a modified protocol as previously described[29]. Following inactivation, an extraction solvent containing chloroform : methanol : 300 mM ammonium acetate (30:41.5:3.5) (v/v/v) was added to the samples, which were then incubated at room temperature for 24 h at 150 RPM. After incubation, the samples were centrifuged, and the clear supernatant was collected and transferred to fresh tubes. The inactivation and extraction steps were repeated once, and the lipid extracts from both rounds of extraction were pooled and dried in a SpeedVac (Genevac, UK). The lipid extracts were stored at −80 °C until LCMS analysis.LCMS analysis was performed using an Exion UPLC coupled with a Sciex QTRAP 6500 PLUS as described previously[30]. For normal phase analysis of polar lipids, individual species were separated on a Phenomenex Luna 3 μm-silica column (150 mm × 2.0 mm internal diameter) under the following conditions: mobile phase A (chloroform : methanol : ammonium hydroxide, 89.5:10:0.5) and mobile phase B (chloroform : methanol : ammonium hydroxide : water, 55:39:0.5:5.5). Quantification of individual lipid species was carried out with reference to spiked internal standards, including d7-Cer d18:1/15:0, GluCer d18:1/12:0, d17:1 Sph, d17:1 S1P, and D-ribo-phytoSph C17, which were obtained from Avanti Polar Lipids (Alabaster, AL, USA), and calibrated with LIPID MAPS.
RNA extraction and qRT-PCR
-
Total RNA was extracted from ten randomly selected seeds at 0, 6, 12, and 24 h post-imbibition using TRIzol Reagent (Invitrogen, USA) according to the manufacturer's instructions. First-strand cDNA was synthesized from 2 μg of total RNA using an All-in-one 5× RT MasterMix (abm, China) following the manufacturer's protocol. Gene-specific primers were designed as detailed in Supplementary Table S1. Quantitative real-time PCR was performed on a CFX96™ Optical Reaction Module (Bio-Rad, USA) using 2× ChamQ SYBR qPCR Master Mix (Vazyme, China) according to the manufacturer's instructions. GmActin11 and GmTUB4 were used as internal controls. Each analysis was conducted with three biological replicates.
Weighted gene co-expression network analysis
-
Weighted gene co-expression network analysis (WGCNA) was conducted on the Majorbio Cloud Platform, a free online platform. A total of 5,946 genes were selected from 12 time-course germinated seed samples after filtering out the genes with a mean TPM value less than 1 and a coefficient of variation less than 0.1. These genes were further utilized for co-expression network construction using WGCNA software (version 1.63,
https://cran.r-project.org/web/packages/WGCNA/index.html ). Modules were identified by setting the following parameters: a soft power β of 8, a minimum model size of 30, and a merge cut height of 0.25. Gene significance (GS) value was calculated using Spearman's correlation to evaluate the correlation between genes and germination percentage, as well as the amounts of Cer d18:0/16:0, Cer d18:0/22:0, and PhytoCer t18:0/24:0.GA and ABA content detection
-
GA and ABA were extracted from ten randomly selected seeds of W82 and the noted transgenic lines using 2 mL methanol overnight at −20 °C. The concentrations of GA and ABA were determined by UPLC-MS/MS (ultra-high performance liquid chromatography–triple quadrupole mass spectrometry) at Nanjing Convinced-test Technology Co., Ltd. as described by Xu et al.[31]. Each analysis was conducted in triplicate.
-
The plasma membrane, one of the three major systems of a eukaryotic cell, serves as an active communication interface between the cell, neighboring cells, and ultimately the entire organism, which has critical roles in various biological processes essential for development and homeostasis within organisms[32]. Sphingolipids not only serve as key structural elements within membranes but also function as important signaling molecules that mediate responses to physiological cues and stressors encountered by the plant[33]. On another note, soybean seeds are particularly rich in oil content. The high lipid concentration contributes significantly to energy storage during seed germination[7]. To investigate the specific functions of sphingolipids in soybean seed germination more closely, we applied 5 μM C24 phytoceramide t18:0/24:0 (C24 phytoCer) during the germination process. Interestingly, our findings revealed that C24 phytoCer could accelerate soybean seed germination significantly compared to control treatments (Fig. 1a; Supplementary Fig. S1). Specifically, after 24 h of imbibition with C24 phytoCer treatment, we observed a germination percentage of approximately 48.3%. This figure increased dramatically over time; by 36 h post-imbibition, it reached an impressive 85.8%. In contrast, seeds subjected to mock treatment exhibited lower rates of germination, the germination percentage was 29.2% and 54.2% after 24 and 36 h imbibition, respectively, and was only 80.8% after 48 h imbibition (Fig. 1b). These results indicated that exogenous application of C24 phytoCer can indeed accelerate seed germination effectively within soybean.
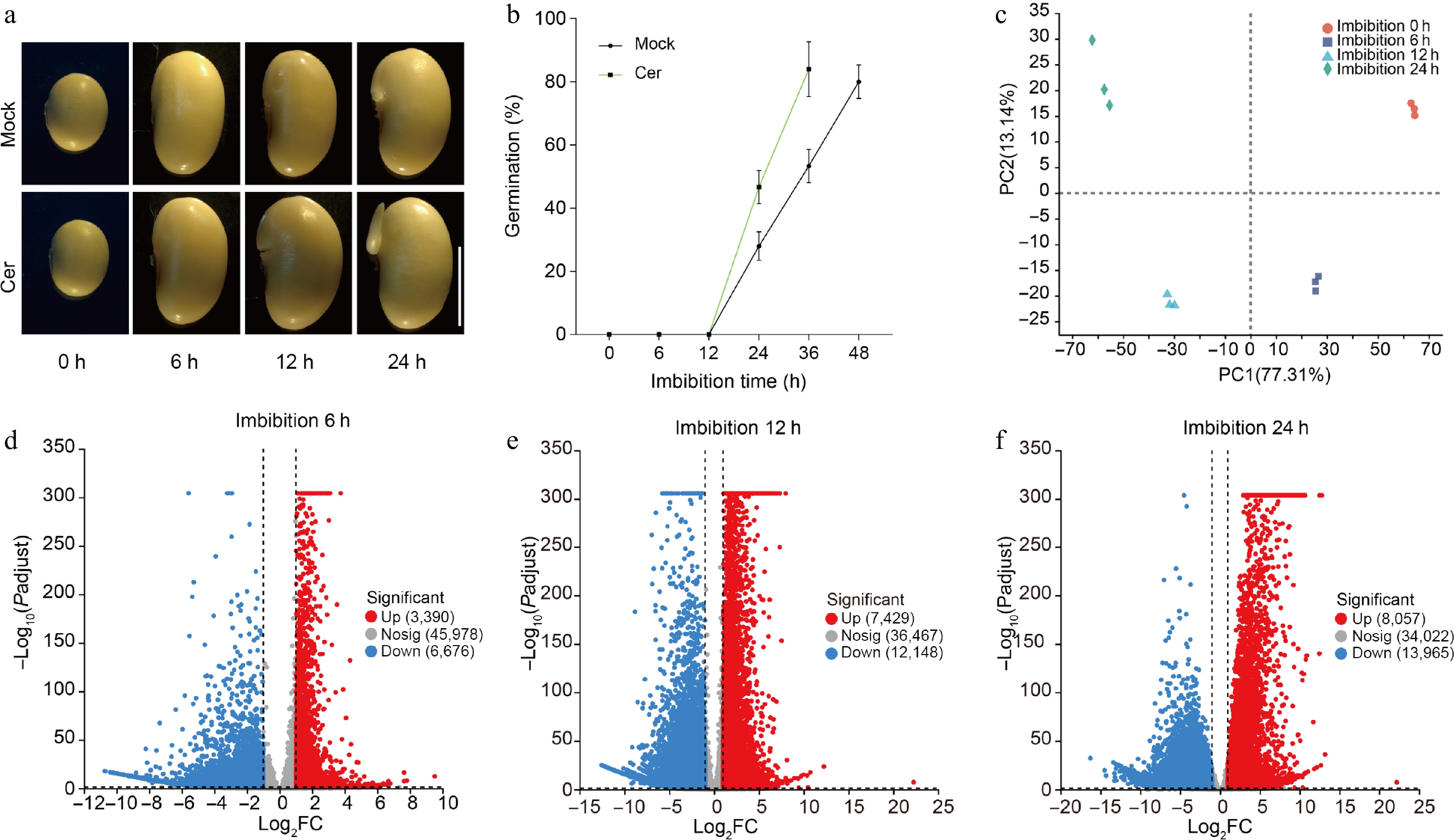
Figure 1.
Germination phenotypes of soybean seeds. (a) Time-course germination phenotypes of freshly harvest soybean seeds with or without C24 phytoceramide t18:0/24:0 (Cer) treatment. (b) Germination percentages of soybean seeds with or without C24 phytoceramide t18:0/24:0 (Cer) treatment. Germination test was repeated with five biological replicates, and 90 seeds were used for each replicate. Photos were taken as indicated. (c) Principal component analysis of the time-course RNA-Seq data. The volcano diagram of up- and down-regulated genes in germination seeds after (d) 6 h, (e) 12 h, and (f) 24 h imbibition.
Identification of different expressed genes during seed germination
-
To explore the molecular basis of soybean seed germination, we conducted a comprehensive RNA-Seq analysis on time-course germinated soybean seeds to generate detailed transcriptome profiles. To ensure robust data collection, we meticulously collected ten random seeds at four distinct time points: 0, 6, 12, and 24 h post-imbibition. Each time point was represented by three biological replicates. The RNA-Seq analysis for the collected samples was carried out at Shanghai Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China). In total, 12 libraries were constructed from these samples and subsequently analyzed. The average number of clean reads per library exceeded 40 million, indicating a substantial depth of coverage that is essential for accurate quantification of gene expression levels. Furthermore, quality control metrics revealed that the Q20 and Q30 values were greater than 97% and 94%, respectively (Supplementary Table S2), signifying high-quality sequencing data with minimal error rates. Following this initial processing phase, we aligned the clean reads from each library against the reference map derived from the assembled V2.0 soybean genome[34]. The alignment results demonstrated impressive mapping efficiency; specifically, both the total mapping rate and unique mapping rate surpassed thresholds of 93% and 88%, respectively (Supplementary Table S3). Through this rigorous analytical process, we identified a total of detectable genes across different imbibition times: specifically, there were found to be 21,053 genes after 0 h; increasing to 26,662 after 6 h; further rising to 31,135 after 12 h; culminating in an impressive count of 32,005 genes detected following 24 h of imbibition with the TPM (Transcript per Kilobase per Million mapped reads) more than 0.5. Principal component analysis clearly illustrated that all 12 samples could be distinctly assigned into four groups corresponding precisely with their respective collection times: namely those taken at intervals representing 0, 6, 12, and 24 h (Fig. 1c). All these data emphasized the reproducibility and reliability of our experimental samples.
To identify the genes associated with seed germination, we conducted a systematic pairwise comparison of each sampling time point against the initial period. Through this rigorous analysis, we obtained 10,066, 19,577, and 22,022 differentially expressed genes (DEGs) for 6, 12, and 24 h imbibition, respectively. The criteria for defining DEGs included a two-fold expression difference coupled with a false discovery rate (FDR) value of less than 0.001. Among these identified DEGs after specific imbibition durations: at the 6-h post-imbibition, there were found to be 3,390 up-regulated genes alongside 6,676 down-regulated genes; following 12 h of imbibition revealed an increase to 7,429 up-regulated and an even greater count of down-regulated DEGs totaling at 12,148; finally by 24 h post-imbibition, 8,057 up-regulated and an impressive count of down-regulated DEGs reaching as high as 13,965 were recorded (Fig. 1d−f). The data clearly indicate that the number of DEGs increased significantly in correlation with seed germination progress over time. This trend is consistent with our understanding regarding the physiological transition seeds undergo, from quiescent dormancy characterized by minimal metabolic activity towards rapid germination accompanied by extensive cellular reprogramming and growth initiation.
GO and KEGG analysis for DEGs
-
Among all the DEGs identified, we discovered a total of 7,581 common DEGs that were consistently detected across all time points following imbibition (Fig. 2a). To further analyze these common DEGs, we generated an expression heat-map which visually represents their expression patterns over time. The analysis revealed that these DEGs could be categorized into ten distinct sub-clusters based on their expression profiles. Notably, within sub-cluster 1, there were 1,591 DEGs that exhibited a continuous down-regulation as seed germination progressed. This persistent decrease in expression levels indicates that these genes may have functions negatively associated with seed germination. In contrast, sub-cluster 2 comprised a substantial group of 4,976 DEGs that displayed continuous up-regulation throughout the germination period. The consistent increase in expression for these genes suggests a positive correlation with seed germination (Fig. 2b).
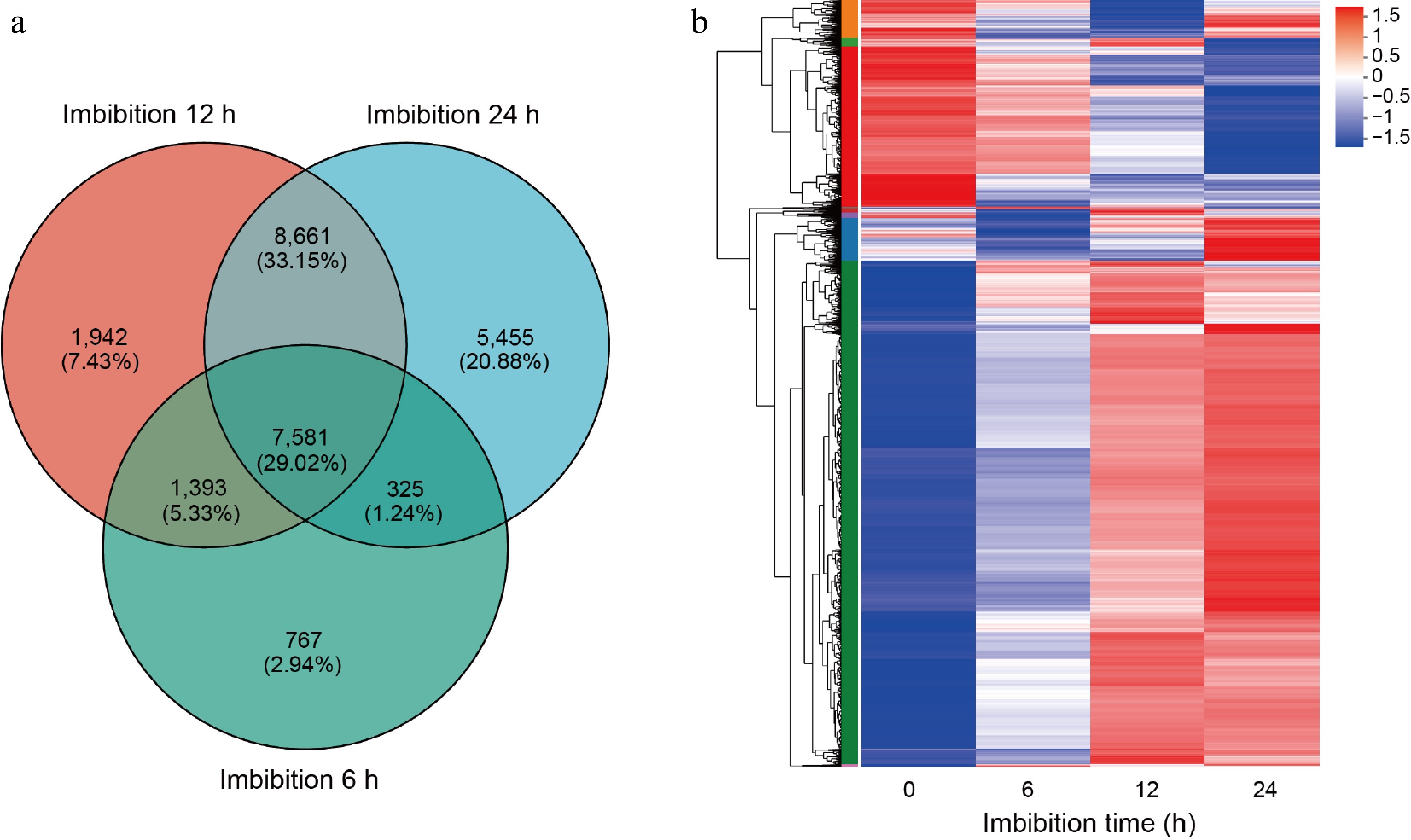
Figure 2.
Cluster analysis of differentially expressed genes. (a) Venn diagrams of differentially expressed genes (DEGs) in germination seeds after 6, 12, and 24 h imbibition. (b) Heat-map hierarchical clustering the common 7,581 DEGs identified in (a). Each column in the chart represents a sample, each row represents a gene, and the color in the chart represents the expression value of the gene after standardized treatment in each sample. Red represents the higher expression level of the gene in the sample, and blue represents the lower expression level. For the specific change trend of the expression level, please see the digital annotation under the color bar on the upper left. On the left are the tree diagrams of gene clusters and the module diagram of sub-clusters.
To elucidate the functions of DEGs in seed germination, we conducted comprehensive Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses on the common set of 7,581 DEGs by using the free online tools of the Majorbio Cloud Platform (
www.majorbio.com ). The results from GO enrichment analysis indicated that these DEGs were predominantly enriched in several biological processes and molecular functions. Notably, they were significantly associated with photosynthesis, which is a critical process for energy production, and regulation of ARF protein signal transduction, which is essential for various cellular signaling pathways. Other enriched terms included thioester biosynthetic processes and acyl-CoA biosynthetic processes, both vital for lipid metabolism; protein dephosphorylation; responses to oxygen-containing compounds; pectin biosynthetic processes; intra-Golgi vesicle-mediated transport; as well as multiple regulatory mechanisms involving small GTPases and Ras proteins (Fig. 3a). Among these findings, four specific GO terms related to the Golgi apparatus particularly captured our attention, since that the Golgi apparatus serves as a central organelle responsible for synthesizing, processing, and sorting proteins, lipids, and carbohydrates within cells[35].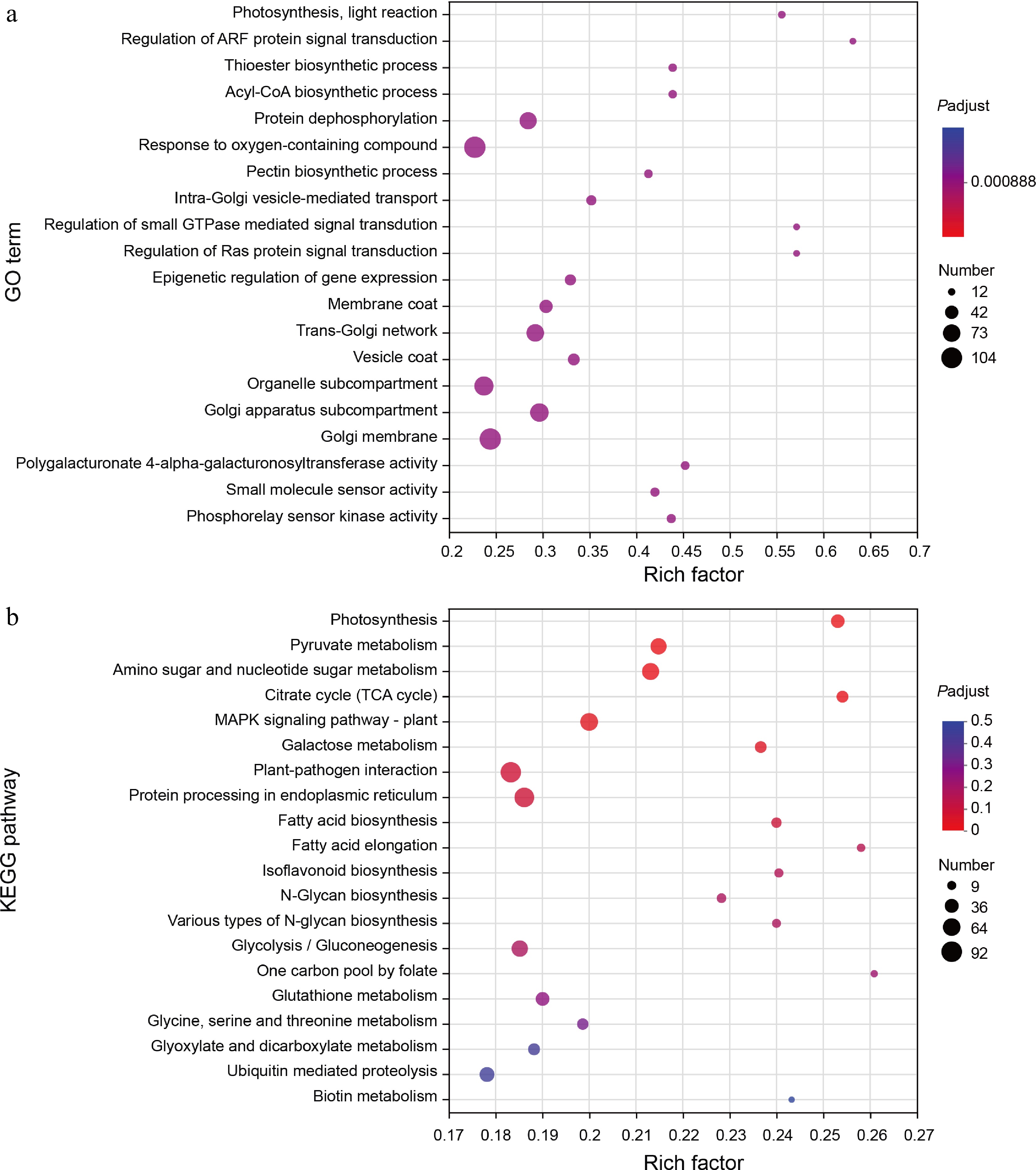
Figure 3.
GO and KEGG enrichment analysis of DEGs. (a) The scatter plot shows detailed descriptions of the pathway within the top 20 GO enrichment analysis of the common DEGs. GO, gene ontology. (b) The scatter plot shows detailed descriptions of the pathway within the top 20 KEGG enrichment analysis of the common DEGs. KEGG, Kyoto Encyclopedia of Genes and Genomes. Analyses of the GO and KEGG enrichments were based on the condition (log2FC > 1, FDR < 0.05). The vertical axis represents the GO term or KEGG pathway, and the horizontal axis represents the ratio of the Rich factor (sample number of genes enriched in the GO term or KEGG pathway) to the annotated gene number. The larger the Rich factor, the greater the degree of enrichment, the size of the dots indicates the number of genes in this GO term or KEGG pathway, and the color of the dots corresponds to different padjust ranges.
In addition to GO analysis, KEGG enrichment analysis revealed that the common DEGs were primarily enriched in pathways such as Photosynthesis, Pyruvate metabolism, Amino sugar, and nucleotide sugar metabolism, Citrate cycle (TCA cycle), MAPK signaling pathway–plant, Galactose metabolism, Plant-pathogen interaction, Protein processing in endoplasmic reticulum, Fatty acid biosynthesis, Fatty acid elongation, Isoflavonoid biosynthesis, N-Glycan biosynthesis, Various types of N-glycan biosynthesis, Glycolysis/Gluconeogenesis, One carbon pool by folate, Glutathione metabolism, Glycine, serine and threonine metabolism, Glyoxylate and dicarboxylate metabolism, Ubiquitin mediated proteolysis, and Biotin metabolism pathways (Fig. 3b). Particularly noteworthy are the pathways related to fatty acid biosynthesis and fatty acid elongation since these biochemical processes likely occur within or involve interactions with components of the Golgi apparatus. Given that soybean seeds are exceptionally rich in lipids, it stands to reason that lipids play significant roles throughout various stages of seed germination.
Sphingolipidomics analysis during seed germination
-
Through the comprehensive GO and KEGG enrichment analyses, we have demonstrated that lipids may play a significant role in soybean seed germination. To further investigate this hypothesis, we conducted a time-course sphingolipidomics analysis on germinated soybean seeds, which were collected simultaneously with transcriptome samples to ensure an integrated understanding of both lipid profiles and gene expression dynamics. The results indicated that these samples could be distinctly categorized into four groups corresponding to their respective collection times: 0, 6, 12, and 24 h post-imbibition (Fig. 4a). This clear separation reinforces not only the reproducibility but also the reliability of our sphingolipidomics analysis as it aligns well with temporal changes observed during seed germination.
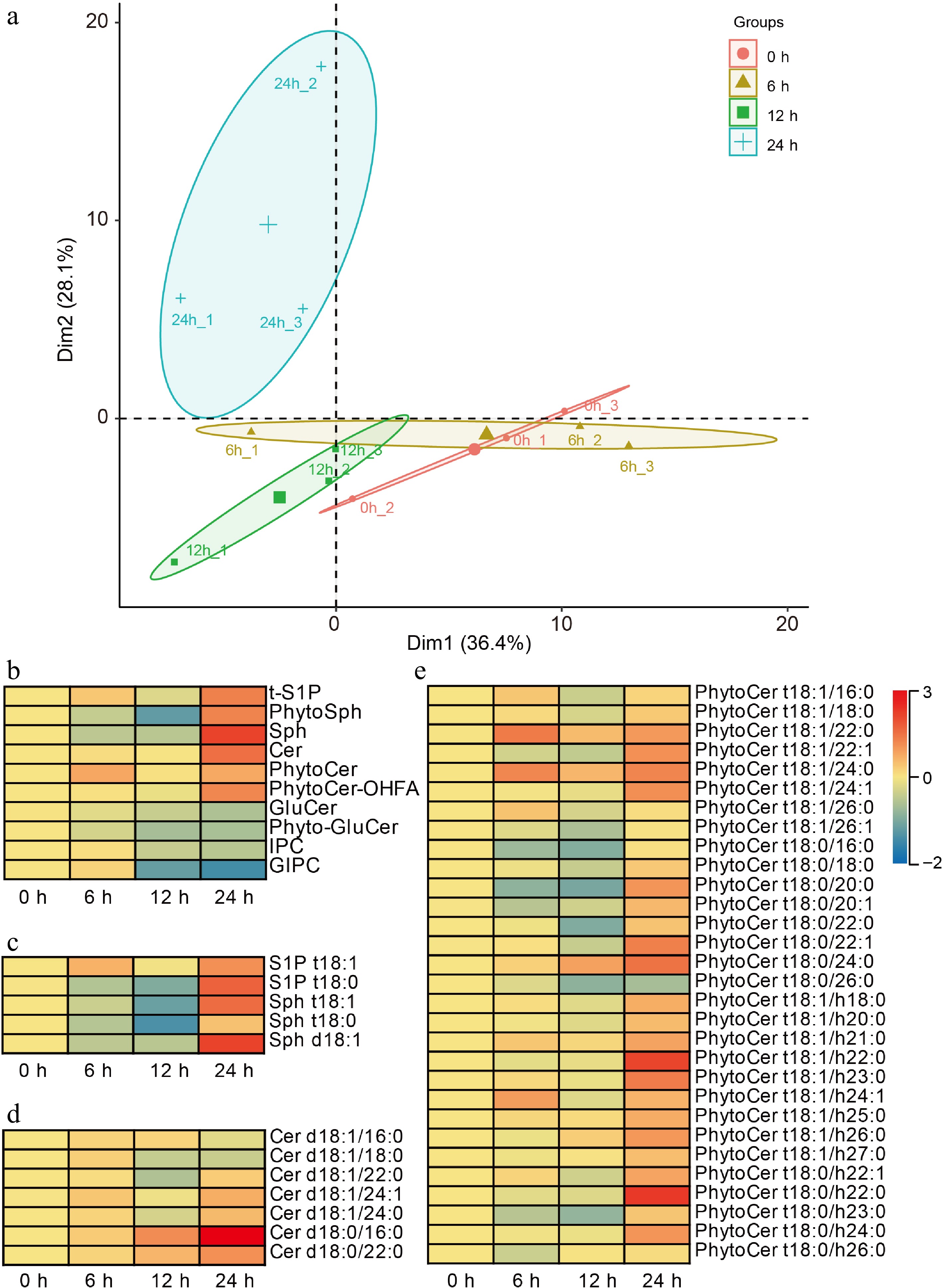
Figure 4.
Sphingolipidomics analysis during soybean seed germination. (a) Principal component analysis of the time-course sphingolipidomics data. (b) Heat-map indicated the amounts of ten-species sphingolipids during soybean seed germination. tS1P, phytosphingosine-1-phosphate; PhytoSph, phytosphingosines; Sph, sphingosines; Cer, ceramides, PhytoCer, phytoceramides; PhytoCer-OHFA, phytoceramides with hydroxylated fatty acyls; GluCer, glucosylceramides; Phyto-GluCer, phyto-glucosylceramides; IPC, inositol phosphorylceramide; and GIPC, glycosyl inositol phosphorylceramide. (c), (d) Heat-maps indicate the amounts of each detected molecules in the ten species. The scale on the right represents the amounts of indicated sphingolipids, and the higher the value, the higher the contents. 'd18:0' and 't18:0' indicates that the long-chain bases (LCB) of sphingolipids has two hydroxyl groups and 18 carbon atoms and no double bonds, or three hydroxyl groups and 18 carbon atoms and no double bonds, respectively. '16:0' and 'h16:0' indicates that the long-chain fatty acid (LCFA) of sphingolipids has 16 to 26 carbon atoms and no double bonds, or is hydroxylated fatty acyls and has 16 to 26 carbon atoms and no double bonds, respectively. Each analysis was repeated with three biological replicates. Heat-maps were generated by applying a base 2 logarithmic transformation to the original dataset.
In total, we identified ten major species of sphingolipids present in germinated soybean seeds, including phytosphingosine-1-phosphate (t-S1P), phytosphingosines (PhytoSph), sphingosines (Sph), ceramides (Cer), phytoceramides (PhytoCer), hydroxylated fatty acyl phytoceramides (PhytoCer-OHFA), glucosylceramides (GluCer), phyto-glucosylceramides (Phyto-GluCer), inositol phosphorylceramide (IPC), and glycosylinositol phosphorylceramide (GIPC). Notably, our findings revealed that the amounts of simple sphingolipids, including t-S1P, PhytoSph, Sph, Cer, PhytoCer, and PhytoCer-OHFA, were elevated throughout the process of seed germination. This increase was particularly pronounced after 24 h of imbibition. Conversely, we observed a marked reduction in complex sphingolipids such as GluCer, Phyto-GluCer, IPCs, and GIPC throughout seed germination; this decline was especially sharp following the same period at 24 h post-imbibition (Fig. 4b). These results suggested that simple sphingolipids may have some roles in seed germination.
To further explore the functions of simple sphingolipids in soybean seed germination, we conducted a detailed analysis of the quantities of each molecule within the detected simple species. The contents of S1P t18:1 and S1P t18:0 for tS1P, and Sph t18:1 and Sph t18:0, for PhytoSph, and Sph t18:1 for Sph were all increased in seed germination after 24 h imbibition (Fig. 4c). In addition to these observations, we examined Cer, PhytoCer, and PhytoCer-OHFA. The results indicated that nearly all molecular species within these categories also experienced an elevation following 24 h of imbibition (Fig. 4d & e). More intriguingly, we found that certain specific molecules, namely Cer d18:0/16:0 and Cer d18:0/22:0 from the ceramide category, as well as PhytoCer t18:0/24:0 from the phytoceramide group, demonstrated positive correlations with sample collection time throughout seed germination (Fig. 4d & e), implying that these saturated simple sphingolipids might play positive roles in seed germination.
Weighted gene co-expression network analysis with sphingolipidomics during seed germination
-
To identify the specific genes that are highly associated with seed germination and sphingolipid metabolism, we performed a weighted gene co-expression network analysis (WGCNA) utilizing both time-course transcriptomic data and sphingolipidomics. 5,946 genes were filtered out from the 7,581 common DEGs for WGCNA. The construction of co-expression networks was predicated upon pairwise correlations between gene expression levels and key metrics such as seed germination percentage, as well as the contents of specific sphingolipids: Cer d18:0/16:0, Cer d18:0/22:0, and PhytoCer t18:0/24:0 across all samples analyzed. In this context, modules were defined as clusters comprising highly interconnected genes; those within the same cluster exhibited high correlation coefficients among one another. Through this analytical approach, we identified four distinct modules labeled by different colors, turquoise, blue, brown, and gray. Notably, the turquoise module which encompassed 4,555 genes whose expressions were positively correlated with both time-course seed germination percentages and concentrations of Cer d18:0/16:0, Cer d18:0/22:0, and PhytoCer t18:0/24:0. Conversely, the blue module contained 1,174 genes that displayed negative correlations with these same parameters (Fig. 5a). These results from WGCNA align closely with phenotypic observations noted in C24 PhytoCer-treated seed germination. Furthermore, the biosynthetic genes responsible for gibberellin (GA) production (specifically GmGA3OX1), and brassinosteroid (BR) synthesis via GmCYP85A2 were found to be part of the turquoise module. Both phytohormones are known to play positive roles in promoting processes essential for successful seed germination. In contrast to these findings regarding growth-promoting hormones, the blue module included various biosynthetic and signaling genes related to abscisic acid (ABA), a hormone recognized for its critical role in maintaining seed dormancy under unfavorable conditions (Supplementary Table S4). Additionally noteworthy is that expression levels of DEGs within the turquoise module demonstrated continuous up-regulation throughout seed germination stages (Fig. 5b, Supplementary Figs S2 & S3a). Conversely, genes categorized within the blue module exhibited a consistent down-regulation correlating with increased imbibition time (Fig. 5c; Supplementary Figs S3b & S4).
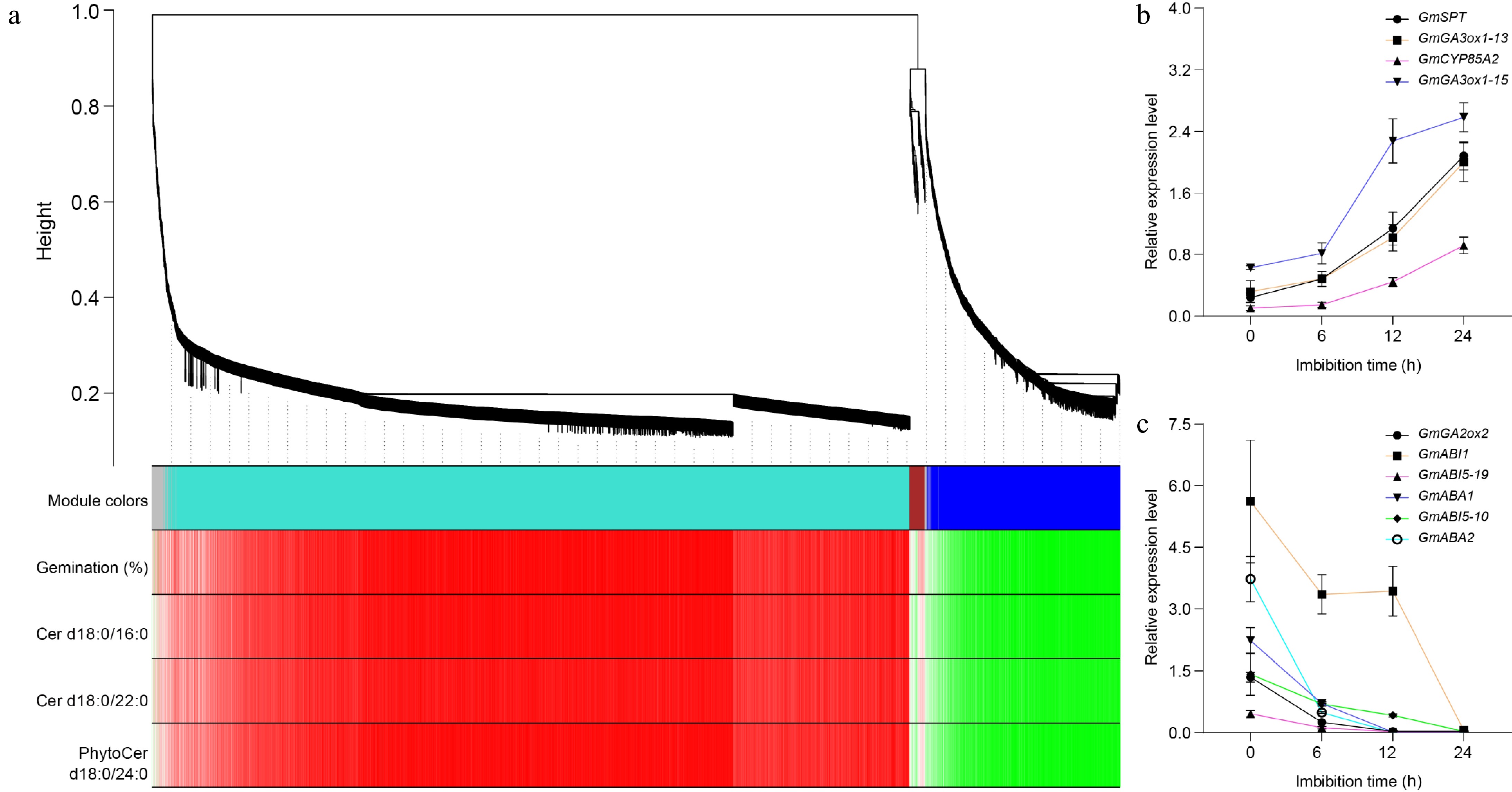
Figure 5.
Weighted gene co-expression network analysis of DEGs. (a) Hierarchical cluster tree showing co-expression modules identified by WGCNA during soybean seed germination. Each leaf in the tree represents one gene. The major tree branches constitute four modules, labeled with different colors, including gray, turquoise, brown, and blue. Red color represents the positive correlation between genes and traits; Green color represents the negative relationships between genes and traits. Traits including germination percentages, the amounts of Cer d18:0/16:0, Cer d18:0/22:0, and PhytoCer d18:0/24:0. (b) The expression of turquoise module genes during seed germination detected by Quantitative real-time PCR. (c) The expression of blue module genes during seed germination detected by Quantitative real-time PCR. GmGA3ox1-13 indicates the GmGA3ox1 gene is on chromosome 13, and so on. GmActin11 was used as an internal control. Each analysis was repeated with three biological replicates for Quantitative real-time PCR. Error bar, ± SEM.
The KEGG enrichment analysis revealed that the DEGs associated with the turquoise module were predominantly enriched in a variety of metabolic and signaling pathways, including various types of N-glycan biosynthesis, Citrate cycle (TCA cycle), N-Glycan biosynthesis, Amino sugar and nucleotide sugar metabolism, Isoflavonoid biosynthesis, MAPK signaling pathway–plant, Fatty acid elongation, Glycine, serine, and threonine metabolism, Phagosome, One carbon pool by folate, Fatty acid biosynthesis, Steroid biosynthesis, Glycolysis/Gluconeogenesis, Pyruvate metabolism, Plant-pathogen interaction, Circadian rhythm–plant, Glyoxylate and Dicarboxylate metabolism, Glycosylphosphatidylinositol (GPI)-anchor biosynthesis, Aflatoxin biosynthesis, and Flavonoid biosynthesis pathways (Supplementary Fig. S5). In contrast to these findings regarding the turquoise module, the DEGs linked with the blue module exhibited predominant enrichment within several other key biological functions. These included Photosynthesis, Galactose metabolism, Protein processing in endoplasmic reticulum, Glycerolipid metabolism, Ascorbate and aldarate metabolism, Glutathione metabolism, Pyruvate metabolism, Cutin, suberine and wax biosynthesis, Fatty acid biosynthesis, Glucosinolate biosynthesis, Cysteine and methionine metabolism, Oxidative phosphorylation, Glycosphingolipid biosynthesis-globo and isoglobo series, Peroxisome, Valine, leucine and isoleucine biosynthesis, alpha-Linolenic acid metabolism, Glycolysis/Gluconeogenesis, Fatty acid degradation, Pantothenate and CoA biosynthesis, and Alanine, aspartate, and glutamate metabolism pathways (Supplementary Fig. S6). Collectively analyzing these results implies that distinct sets of biochemical pathways might exhibit either positive or negative relationships concerning soybean seed germination outcomes depending on their regulatory influences throughout developmental transitions.
Saturated ceramide is required for soybean seed germination
-
Through a combination of chemicobiological methods, sphingolipidomics analysis, and weighted gene co-expression network analysis (WGCNA), we have established that saturated simple sphingolipids play a positive role in soybean seed germination. To further confirm the specific roles of these sphingolipids during this critical developmental phase, we investigated the expression levels of key biosynthetic genes involved in sphingolipid production. Fortunately, our examination revealed that the expression levels of several important genes, specifically Gm8DES, GmSBH2, and GmSPHK, were predominantly down-regulated throughout the process of seed germination (Supplementary Table S5). To validate our findings regarding gene expression patterns, we then examined the expression of these genes by qRT-PCR, and the results were consistent with the transcriptome data (Fig. 6a−c; Supplementary Fig. S7). Notably, Δ8DES is responsible for catalyzing the c8 desaturation of LCBs. Our data indicated that all detected Gm8DES genes exhibited continuous down-regulation correlating with increased imbibition time during seed germination. This trend aligns well with our findings showing that concentrations of ceramide species such as Cer d18:0/16:0 and Cer d18:0/22:0, as well as PhytoCer t18:0/24:0, were consistently elevated throughout soybean seed germination. The observed relationship between decreased expression levels of biosynthetic genes and increased amounts of saturated simple sphingolipids provides compelling evidence supporting their functional importance during soybean seed germination.
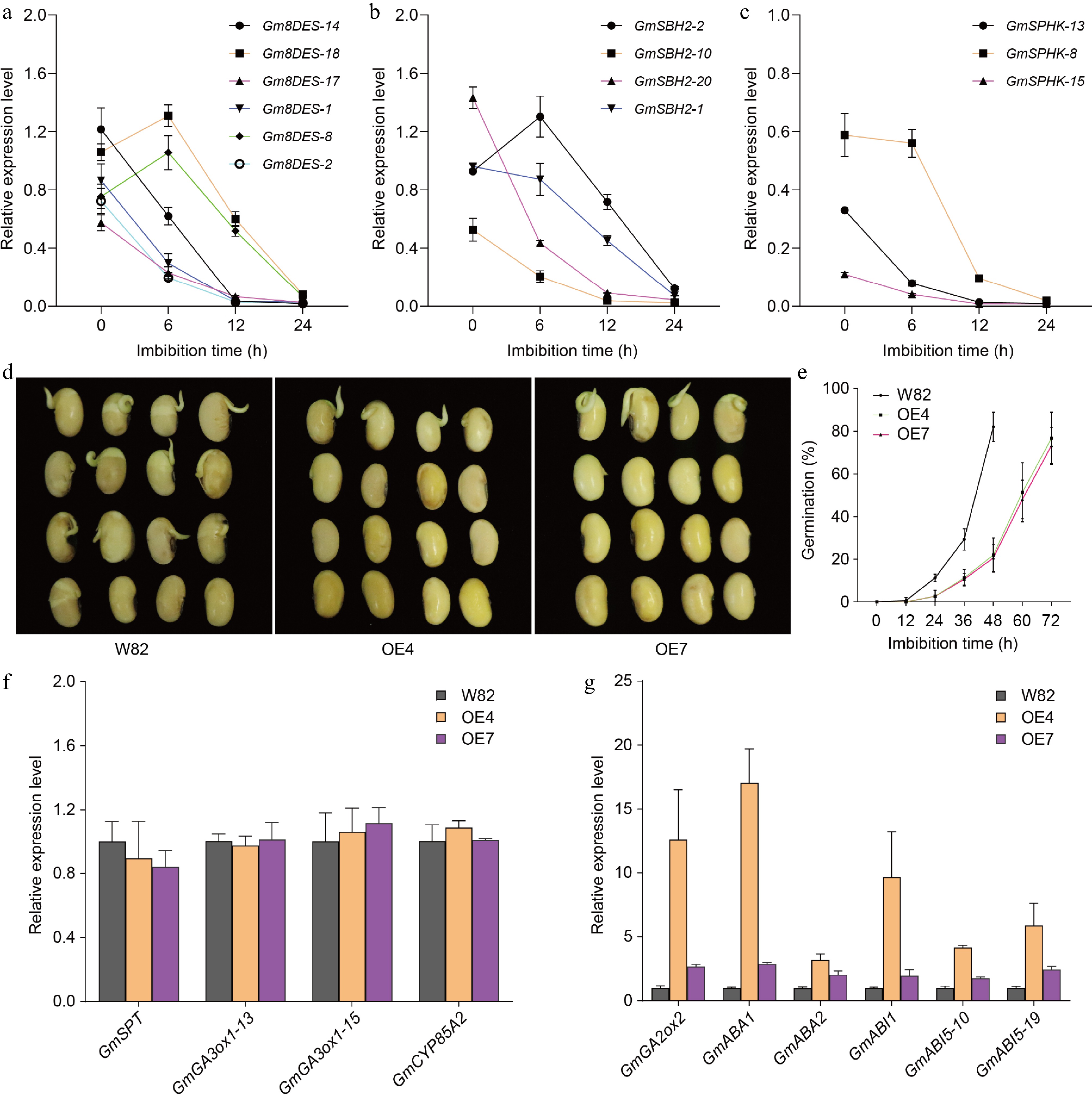
Figure 6.
The expression levels of sphingolipid biosynthetic genes. (a) The expression of Gm8DES genes during seed germination detected by Quantitative real-time PCR. (b) The expression of GmSBH2 genes during seed germination detected by Quantitative real-time PCR. (c) The expression of GmSPHK genes during seed germination detected by Quantitative real-time PCR. Gm8DES-14 indicates the Gm8DES gene is on chromosome 14. GmActin11 was used as an internal control. Each analysis was repeated with three biological replicates for Quantitative real-time PCR. Error bar, ± SEM. (d) Germination performance of freshly harvest seeds of GmSLD1 overexpression lines after 48 h imbibition. (e) Time-course germination percentage of GmSLD1 overexpression lines. Each analysis was repeated with four biological replicates. Error bar, ± SEM. (f) The expression of turquoise module genes in GmSLD1 overexpression lines detected by Quantitative real-time PCR. (g) The expression of blue module genes in GmSLD1 overexpression lines detected by Quantitative real-time PCR. GmGA3ox1-13 indicates the GmGA3ox1 gene is on chromosome 13, and so on. GmActin11 was used as an internal control. Each analysis was repeated with three biological replicates for Quantitative real-time PCR. Error bar, ± SEM.
To further investigate the roles of saturated simple sphingolipids in seed germination, we generated overexpression lines for GmSLD1 (Glyma.01G046000, one of the Gm8DES genes). From this effort, we successfully obtained eight positive transgenic lines that exhibited enhanced expression levels of the GmSLD1 gene (Supplementary Fig. S8). Subsequent germination assays were conducted to assess how these overexpression lines influenced seed dormancy and germination. The results revealed that seeds from the GmSLD1 overexpression lines displayed a strong dormancy phenotype compared to wild-type seeds (Fig. 6d). Specifically, after 24 h of imbibition, wild-type seeds achieved a germination percentage of approximately 11.3%. This figure demonstrated significant improvement as time progressed; by 48 h post-imbibition, the germination rate surged to an impressive 82%. In stark contrast, seeds subjected to GmSLD1 overexpression showed markedly lower rates of germination: only 2.7% and 20.7% after 24 h and 48 h of imbibition respectively. Even at the extended duration of 72 h imbibition, their maximum germination percentage reached just 76.7% (Fig. 6e). To elucidate the pathways through which the GmSLD1 influences seed dormancy mechanisms, we examined the expression levels of genes within both turquoise and blue modules in seeds derived from our GmSLD1 overexpressing lines. Our analysis indicated that expression levels for four specific genes within the turquoise module did not exhibit significant differences when compared to those found in wild-type plants (Fig. 6f). Conversely, six genes located within the blue module were significantly up-regulated in response to increased expression of GmSLD1 in these transgenic lines; this finding aligns well with their observed strong dormancy phenotype (Fig. 6g). These results collectively suggest that GmSLD1 may affect seed dormancy primarily by influencing gibberellin metabolism alongside abscisic acid synthesis and its subsequent signal transduction pathways. Overall, our study provides compelling evidence indicating that saturated simple sphingolipids play indeed positive roles during soybean seed germination by modulating critical hormonal pathways involved in regulating both metabolic activities necessary for growth initiation as well as mechanisms governing seed dormancy.
Saturated ceramide promotes seed germination by modulating the GA/ABA ratio
-
To further elucidate the relationship between simple saturated sphingolipids promoting seed germination and phytohormones, we quantified hormone levels in seeds of GmSLD1 overexpression lines. The results indicated that the contents of active gibberellins GA1 and GA3 were significantly reduced (Fig. 7a & b), while the GA4 content was slightly decreased (Fig. 7c), whereas the ABA content was significantly elevated in these seeds (Fig. 7e). Given the critical role of the GA/ABA ratio in regulating seed dormancy and germination, we also measured this ratio in the overexpression lines. We found that the GA/ABA ratio was markedly reduced in the seeds of GmSLD1-overexpressing lines (Fig. 7f−h), consistent with their enhanced dormancy phenotype. These findings provide robust evidence that GmSLD1 overexpression decreases the GA/ABA ratio, thereby enhancing soybean seed dormancy.
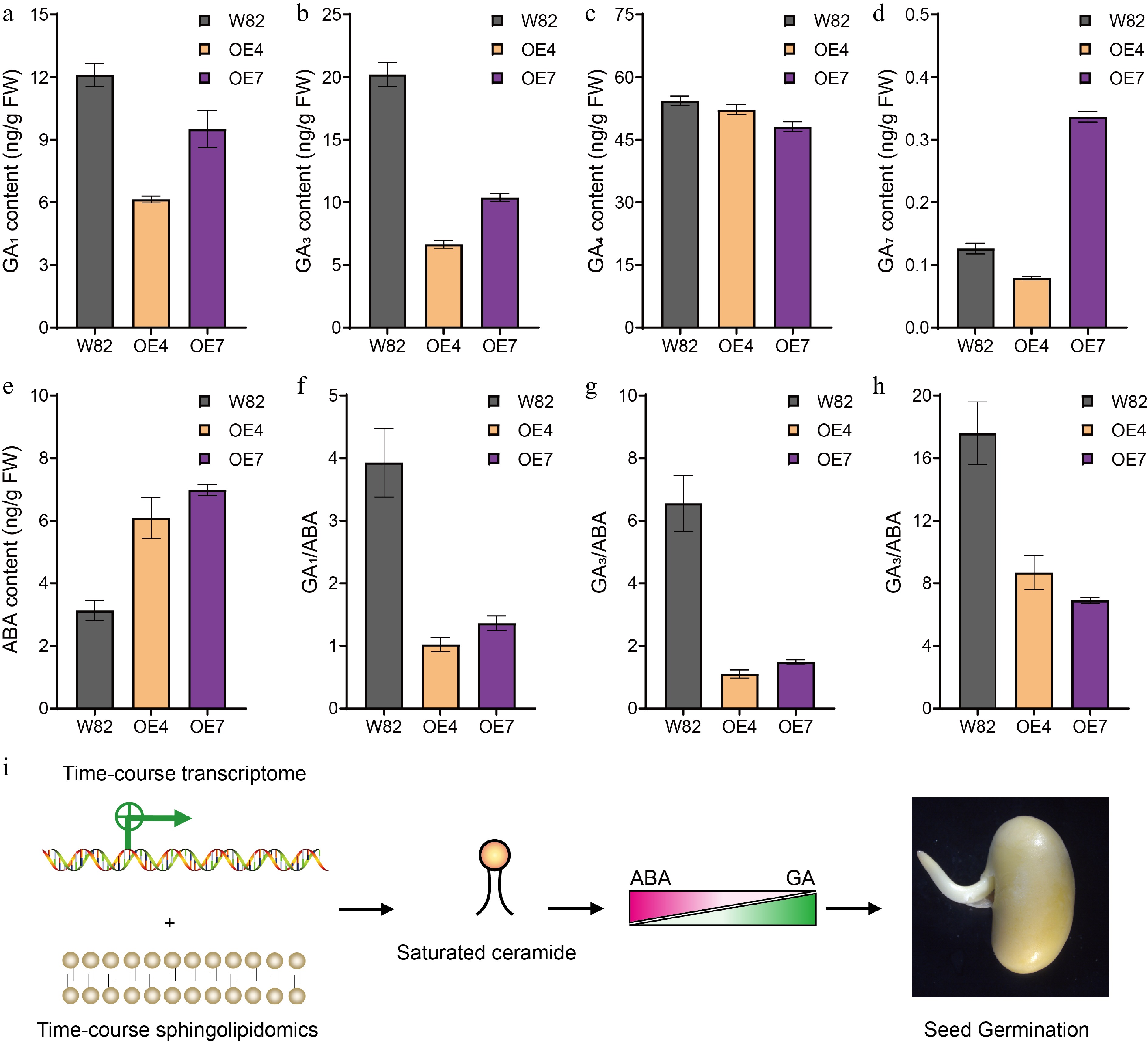
Figure 7.
The GA and ABA contents in seeds of GmSLD1 overexpression lines. The content of (a) GA1, (b) GA3, (c) GA4, and (d) GA7, in seeds of GmSLD1 overexpression lines. (e) The content of ABA in seeds of GmSLD1 overexpression lines. The (f) GA1/ABA, (g) GA3/ABA, and (h) GA4/ABA in seeds of GmSLD1 overexpression lines. Each analysis was repeated with three biological replicates. Error bar, ± SEM. (i) Through the integration of time-course transcriptomic and sphingolipidomic analyses of germinated soybean seeds, we found that saturated ceramides promoted seed germination in soybean. Furthermore, our genetic evidence demonstrated that this promotion was mediated by modulating the GA/ABA ratio within seeds.
-
The transition from dormancy to germination represents a critical initial step in the plant life cycle, ensuring that plants grow at optimal times and subsequently reproduce successfully[1]. To identify additional genes associated with this vital process, we conducted a comprehensive time-course transcriptome analysis during soybean seed germination. This approach allowed us to capture dynamic changes in gene expression as seeds transitioned through various stages of imbibition. Utilizing WGCNA, we identified four distinct modules within our transcriptomic gene set: turquoise, blue, gray, and brown. Notably, the turquoise module was positively associated with soybean seed germination while the blue module exhibited a negative correlation with this process. Within the turquoise module specifically, we screened for two gibberellin (GA) biosynthetic genes known to be positively correlated with seed germination, which aligns well with previous research indicating their importance in promoting seed germination[36,37]. Intriguingly, we also identified SPATULA (SPT), a core gene implicated in both seed dormancy and germination processes. SPT has been shown to couple GA3ox expression with light and temperature signaling pathways, affecting seeds' responses to environmental cues during seed germination[38]. Furthermore, SPT interacts with another important protein called INDEHISCENT which is essential for reproductive tissue development originating from carpel margins within the gynoecium prior to fertilization. Additionally, IND plays a role in promoting effective seed dispersal in Arabidopsis[39]. SPT could repress the expression of ABI4, RGA, and MFT, but induce the expression of ABI5 and RGL3, driving two antagonistic 'dormancy-repressing' and 'dormancy-promoting' routes that operate simultaneously in freshly matured seeds. This explains the opposite effect of SPT on seed dormancy of the in Landsberg erecta and Columbia Arabidopsis ecotypes[40]. Interestingly, in our time-course transcriptome data, the expression trends for these pivotal genes were all found to be positively correlated with increasing percentages of germination over time. Furthermore, we validated these observations using qRT-PCR, confirming that the expression levels of these genes gradually increased alongside imbibition duration as depicted in Fig. 5. In addition to these established factors influencing seed germination, it is plausible that other genes present within the turquoise module may also play positive roles specific to soybean seeds during this process.
From the blue module, we first noted the presence of ABA biosynthetic genes, specifically GmABA1 and GmABA2. These genes are particularly significant because ABA is known to induce and maintain seed dormancy during the maturation process[41]. Previous studies have shown that Arabidopsis mutants that lacking functional ABA1 and ABA2 exhibited reduced dormancy levels[42]. In addition to these biosynthetic genes, we identified a protein phosphatase 2C gene, GmABI1, from the blue module. This gene has been reported to function within a negative feedback regulatory loop of the ABA signaling pathway in Arabidopsis[43]. More intriguingly, we also discovered a core transcription factor involved in ABA signaling: GmABI5. ABI5 acts downstream of ABI3 and plays a crucial role in executing an ABA-dependent growth arrest during germination[44,45]. Furthermore, our analysis revealed a GA deactivation gene, GmGA2ox, which functions by inactivating active GAs[46]. This action suppresses germination rates specifically under conditions where seeds are imbibed without light exposure, a common scenario encountered by many plant species during their life cycles[47]. We observed that expression trends for these key genes were negatively associated with germination percentages; as imbibition time increased, expression levels gradually decreased across all examined samples (Fig. 7). Such findings reinforce our understanding that certain hormonal pathways operate antagonistically when it comes to seed dormancy versus germination. Taken together, our screening efforts have highlighted numerous well-characterized genes related to both seed dormancy and germination processes, particularly those linked with ABA and GA pathways which exhibit opposing roles within this context. Moreover, the relationships established through WGCNA indicate that specific gene modules correlate positively or negatively with seed germination outcomes, respectively. This insight underscores how intricately connected these two modules are concerning soybean seed dormancy and germination. Understanding their interactions will facilitate further analyses aimed at elucidating molecular mechanisms governing soybean seed germination while potentially informing breeding strategies focused on optimizing crop performance under varying environmental conditions.
Significant expression trends during seed germination
-
Seed germination is a complex biological process that can be divided into three distinct phases: the initial phase characterized by substantial water uptake leading to seed swelling, followed by a plateau in water absorption, and finally, the resumption of water uptake culminating in the rupture of the seed coat or testa. This sequence is then succeeded by cell elongation and expansion, ultimately resulting in the protrusion of the embryonic root or radicle through the endosperm. Each of these stages plays a critical role in ensuring successful germination and subsequent plant development[1]. Given the continuous and uninterrupted nature of seed germination, it follows that internal gene expression during this stage should exhibit significant temporal trends reflective of these physiological changes. Indeed, our analysis revealed that gene expression patterns during seed germination displayed clear temporal characteristics. These included various trends such as continuously up-regulated expressions and continuously down-regulated expressions (Supplementary Figs S2 & S4). When considering results from WGCNA, we found compelling associations between specific gene expression patterns and their roles in either promoting or inhibiting germination processes. Genes displaying continuously up-regulated trends were positively correlated with successful germination outcomes; conversely, genes showing consistently down-regulated expressions were linked to maintaining seed dormancy. These temporal expression trends suggest an orchestrated sequence whereby dormancy must first be alleviated for effective seed germination to occur. This release from dormancy is likely accompanied by a marked down-regulation of genes associated with quiescent states, a necessary step before transitioning towards active growth mechanisms facilitated by the up-regulation of genes specifically related to seed germination.
The functions of sphingolipids in seed germination
-
The membrane system, comprising a complex arrangement of proteins and lipids, is one of the three major systems found in eukaryotic cells. This system plays a crucial role in perceiving environmental cues that are essential for cellular responses and adaptations[32]. For instance, Cold1 is a plasma membrane-localized protein that functions as a cold sensor, enabling plants to detect low temperatures and initiate appropriate physiological responses[48]. Additionally, the membrane receptor FLS2 works in conjunction with BAK1 to recognize pathogen-associated molecular patterns, thereby triggering defense mechanisms against potential pathogen threats[49]. In response to low temperatures, there is an observed increase in the unsaturation of membrane lipids. This modification enhances membrane fluidity under stress conditions, allowing for better functionality at lower temperatures[50]. Sphingolipids have traditionally been regarded primarily as structural components within cellular membranes[51], however, recent studies have highlighted their role as signaling molecules involved in various plant growth processes and stress responses. These sphingolipids actively participate in critical cellular processes such as programmed cell death, cell-cell interactions, and hormone signal transduction pathways[23,50,52]. Research has demonstrated that exogenous application of phytosphingosines t18:0 or genetic knockout of specific genes like SLD can elevate the expression levels of disease resistance genes while enhancing overall plant resistance to biotic stresses such as pathogens[53]. Furthermore, GIPC sphingolipids act as Na+ and NLP toxin receptors, and have essential roles in both abiotic and biotic stress responses: Jiang et al. showed that GIPC sphingolipids sense salt and trigger Ca2+ influx to improve salt resistance in plants[54]. Mutant plants with reduced GIPC contents exhibit increased resistance to NLP toxins, while monocot plants' insensitivity to NLP might be attributed to the length of the GIPC head group and the architecture of the NLP sugar-binding site[55]. Collectively, these findings suggest that plasma-membrane lipids are integral not only for maintaining structural integrity but also for facilitating adaptive mechanisms toward diverse environmental stimuli. Various sphingolipid species exhibit distinct functionalities tailored toward these adaptive processes.
For normally dormant seeds, it is imperative that they accurately sense environmental cues conducive to germination to successfully complete their life cycle. However, the specific contributions of sphingolipids during this critical phase remain largely unexplored. In our presented study focusing on soybean seed germination dynamics, time-course transcriptome analysis revealed significant findings regarding gene expression patterns associated with lipid metabolism. We observed a gradual reduction in transcripts from Gm8DES genes throughout the seed germination stages (Fig. 6a). To further elucidate the relationships between lipid profiles and gene expression changes during this process, we conducted comprehensive analyses involving time-course sphingolipidomics on germinated soybean seeds. Our results indicated an increasing trend in the contents of simple saturated sphingolipids, including Cer d18:0/16:0, Cer d18:0/22:0, and PhytoCer t18:0/24:0, as seed germination progressed over the analyzed time periods (Fig. 4d & e). Additionally, we established positive correlations between the amounts of these specific ceramides and both the germination percentage and the expression of turquoise module genes identified by WGCNA (Fig. 5a). Furthermore, genetic transformation demonstrated that overexpression of a sphingolipid Δ8-desaturase encoding gene, GmSLD1, significantly enhanced soybean seed dormancy by modulating GA metabolism alongside ABA biosynthesis and signal transduction. These findings further substantiate the involvement of saturated sphingolipids in seed germination. Taken together, this research provides compelling evidence suggesting that saturated simple sphingolipids possess beneficial roles in promoting successful soybean seed germination by modulating the GA/ABA ratio. These conclusions are supported by integrated approaches combining temporal transcriptomic data with detailed sphingolipidomic profiling methodologies and transgenic genetic evidence (Fig. 7i).
This study was supported by the National Natural Science Foundation of China (Grant No. 32172059), the Fundamental Research Funds for the Central Universities (Grant No. SWU-XDJH202315), the Chongqing Technology Innovation and Application Development Special Key Project, China (Grant No. cstc2021jscx-gksbX0011), and the Collection, Utilization and Innovation of Germplasm Resources by Research Institutes and Enterprises of Chongqing, China (Grant No. cqnyncw-kqlhtxm).
-
The authors confirm contribution to the paper as follows: experiments design: Xu F; experiments performing: Gao Y, Liu F, Zhang JL, Qiao Z, Liu N, Tan P, Hu M, Zhang J, Yan X; project supervision: Yi Z, Luo M, Xu F; writing manuscript: Xu F, Luo M. All authors reviewed the results and approved the final version of the manuscript.
-
The raw sequence data supporting the results of this article are available at the Majorbio Cloud Platform (www.majorbio.com), a free online platform. Sequence and sphingolipidomics raw data were submitted to the CNCB database (www.cncb.ac.cn), and can be found with BioProject PRJCA023939. Further information and requests for data and material should be directed to and will be fulfilled by Fan Xu (xufanfeiren@163.com) and Ming Luo (luo0424@126.com).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Yinghui Gao, Fei Liu
- Supplementary Table S1 List of primers used in this study.
- Supplementary Table S2 Evaluation of sequencing quality.
- Supplementary Table S3 Mapping efficiency statistics.
- Supplementary Table S4 Listed of known genes involved in seed dormancy and germination in turquoise and blue module.
- Supplementary Table S5 List of differentially expressed genes (DEGs) involved in sphingolipid biosynthesis.
- Supplementary Fig. S1 Germination performance of soybean seeds.
- Supplementary Fig. S2 Cluster analysis of turquoise module genes.
- Supplementary Fig. S3 The expression levels of turquoise and blue module genes.
- Supplementary Fig. S4 Cluster analysis of blue module genes.
- Supplementary Fig. S5 KEGG enrichment analysis of turquoise module genes.
- Supplementary Fig. S6 KEGG enrichment analysis of blue module genes.
- Supplementary Fig. S7 The expression levels of sphingolipid biosynthetic genes.
- Supplementary Fig. S8 Identification of GmSLD1 overexpression lines.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gao Y, Liu F, Zhang J, Qiao Z, Tan P, et al. 2025. Saturated ceramide is required for seed germination in soybean. Seed Biology 4: e006 doi: 10.48130/seedbio-0025-0006 

Saturated ceramide is required for seed germination in soybean
- Received: 20 December 2024
- Revised: 18 March 2025
- Accepted: 31 March 2025
- Published online: 17 April 2025
Abstract: Soybean (Glycine max) constitutes one of the paramount oil and forage crops globally. High and uniform seed germination is essential for optimizing soybean yield. Sphingolipids are major constituents of membrane lipid rafts and exert pivotal roles in plant growth and stress responses. Nevertheless, comprehension regarding the functionalities of sphingolipids in seed germination remains elusive. Using chemicobiological approaches, we discovered that exogenous application of C24 phytoceramide t18:0/24:0 could facilitate seed germination in soybean. To elucidate the role of sphingolipids in soybean seed germination, we conducted an integrated time-course transcriptome and sphingolipidomic analysis on germinated soybean seeds. Through time-course transcriptome analysis, we identified two gene modules positively or negatively correlated with seed germination; the expression levels of these genes were successively up- or down-regulated during seed germination, respectively. Notably, numerous genes involved in sphingolipid biosynthesis exhibited down-regulation during seed germination, particularly LCB Δ8 desaturases, which attracted our attention. Furthermore, our time-course sphingolipidomics outcomes indicated that the concentrations of ceramide d18:0/16:0, ceramide d18:0/22:0, and phytoceramide t18:0/24:0 increased during seed germination, suggesting a positive association between saturated ceramides and seed germination in soybean. Additionally, overexpression of GmSLD1 (encodes LCB Δ8 desaturase) gave rise to strong seed dormancy in soybean by reducing the GA/ABA ratio within soybean seeds. In conclusion, through comprehensive time-course transcriptome profiling combined with sphingolipidomics and genetic evidence, we identified two sets of genes related to soybean seed germination and demonstrated that saturated simple ceramides play crucial roles therein.
-
Key words:
- Soybean /
- Seed germination /
- Sphingolipid