








-
Diseases of the central nervous system (CNS) are principally categorized into brain tumors and neurodegenerative disorders (NDs), including Alzheimer's disease (AD) and Parkinson's disease (PD); the latter may result from the interplay of numerous factors ranging from human genetics and environmental elements to disease pathophysiology. The human brain has a highly sophisticated and complex structure that complicates drug development for treating CNS diseases[1]; the blood-brain barrier (BBB) further hinders effective drug delivery to brain regions[2]. All of the above have created an emerging yet urgent need for designing innovative and intelligent therapeutic preparations specifically fitting for CNS delivery.
Targeting chimera (TAC) technology is regarded as one of the effective approaches to treating CNS disorders by site-specific clearance of disease-associated proteins. Different from small-molecule inhibitors that suppress protein functions in vivo, TAC activates endogenous degradation mechanisms to cleanse pathologic proteins of interest (POI)[3]. Structurally, TAC molecules are divided into types such as proteolysis-targeting chimeras (PROTAC)[4], lysosome-targeting chimeras (LYTAC)[5,6], autophagy-targeting chimeras (AUTOTAC)[7,8], and so on. Advantages of TAC technology may range from expanding the scope of 'druggable' proteomes, enabling event-driven pharmacology, to overcoming cumulative drug tolerance, all of which would help reduce drug dosage and moderate toxicity risks[3,9].
TAC molecules also possess some deficiencies; for example, large molecular weight, low aqueous solubility, low cell permeability, and off-target toxicity induced by widespread expression of functional proteins. More importantly, a high concentration of TAC is capable of readily forming binary complexes, triggering the 'hook effect' and altering therapeutic effects[10,11]. Structural modifications of TAC molecules, such as ligand improvement and linker adjustment, may subsequently reduce molecular weight, improve solubility, and facilitate efficient ternary complex formation. However, there are still technical challenges in vivo regarding precise targeting of CNS lesions using TAC molecules[12]. Combining TAC candidates with advanced drug delivery systems has become a key strategy to improve their efficacy in vivo. For instance, encapsulation inside functionalized nanocarriers and surface modification with targeting ligands would achieve targeted delivery of TAC molecules to CNS lesions, thereby minimizing interactions with healthy tissues[10]. Moreover, nanocarrier platforms protect TACs from immune clearance, improving their pharmacokinetic properties as well as overall stability in vivo.
This review is to present the modular architecture of TAC molecules by detailing the combination of functional modules such as POI ligands, degradation-inducing ligands, and linkers, which will collectively lead to the specificity and programmability of TACs. The focus of the subject will further expand to TAC applications in managing CNS diseases and the role of TAC molecules within the CNS pathophysiological environment. Performance of novel delivery systems in aiding TAC targeting will also be discussed and evaluated. By combining perspectives of both molecular designs and delivery strategies, we hope to identify key application challenges and provide recommendations for developing innovative TAC therapeutics specifically targeting CNS disorders (Fig. 1).
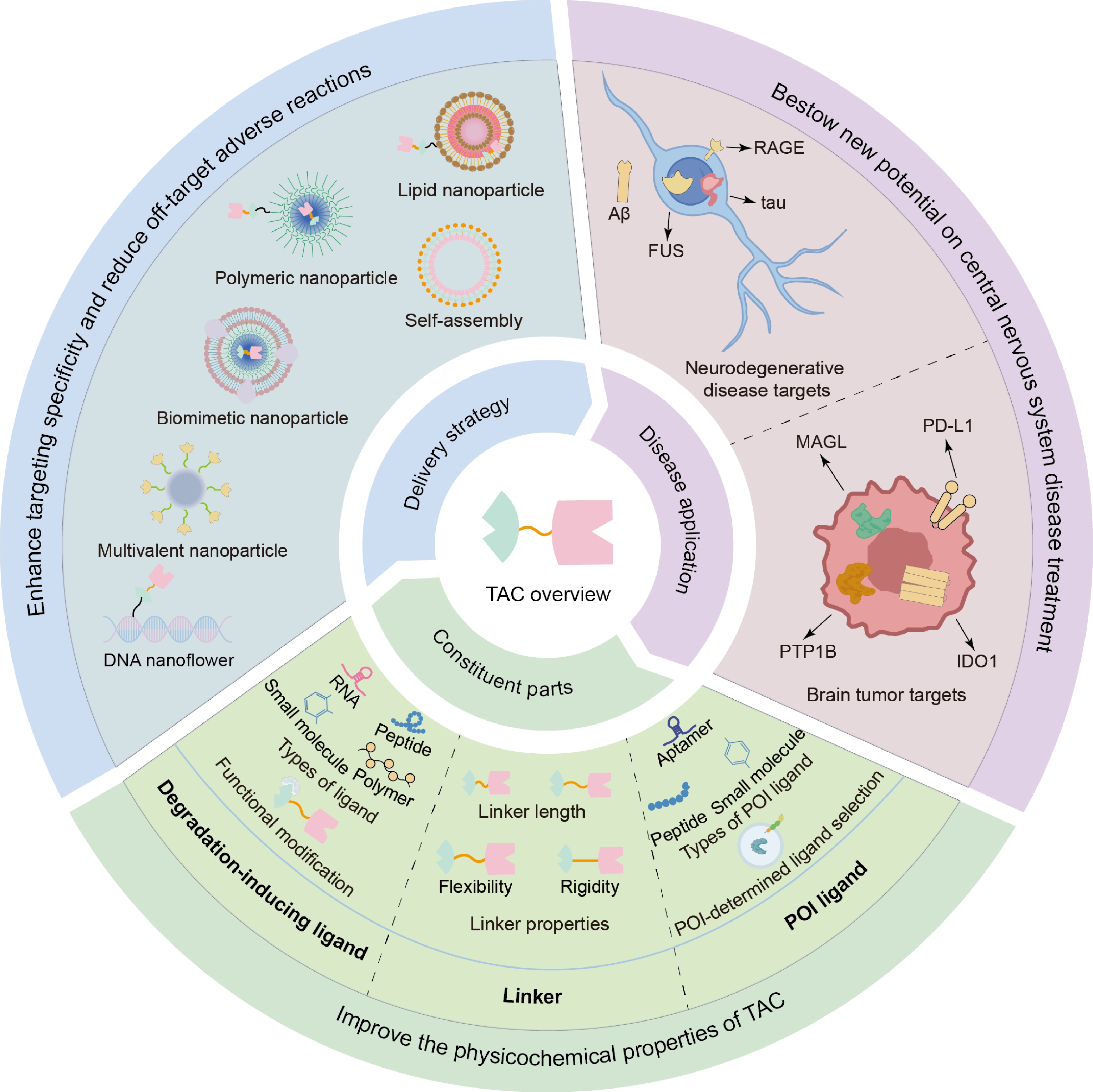
Figure 1.
Schematic descriptions of TAC technology, from molecular components, therapeutic applications, to delivery strategies.
-
A TAC molecule is structurally composed of three components, i.e., a ligand that binds the POI, a ligand that recruits the degradation machinery, and a linker that connects the two ligands[13−15]. TAC induces POI degradation by simultaneously recruiting the POI and intracellular degradation machinery component, hence forming a stable ternary complex (Fig. 2)[6,13−16]. In designing TAC molecules, any structural and compositional alteration to modules could impact the degradation efficiency of a TAC candidate.
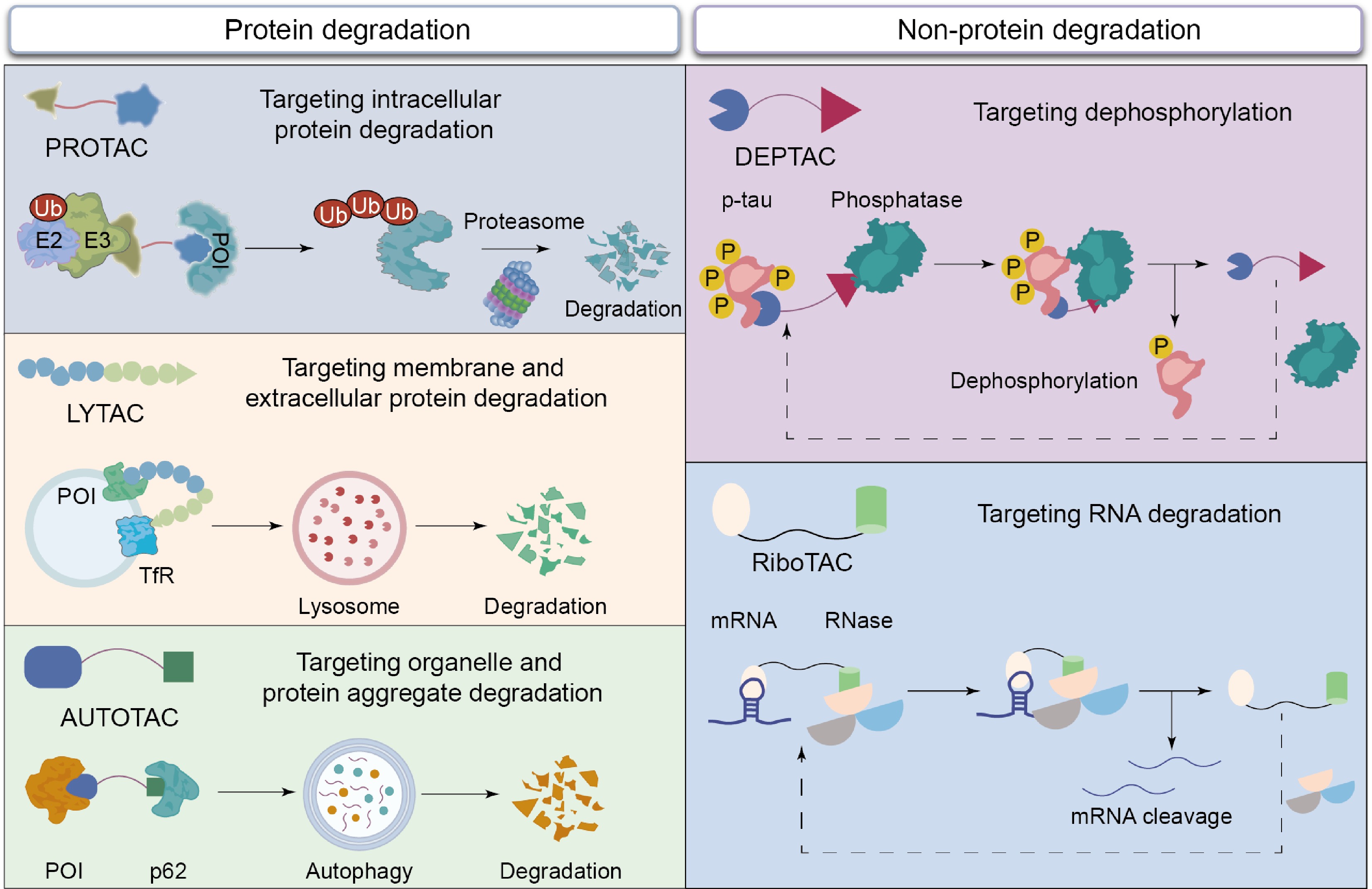
Figure 2.
The action mechanisms of different TAC molecules, including PROTAC, LYTAC, AUTOTAC, RiboTAC, and DEPTAC, for targeting degradation of intracellular protein, membrane and extracellular protein, protein aggregate, RNA, and dephosphorylation.
POI ligands, the anchor for targeting pathogenic proteins
-
In the early design process of TAC, natural products were used as POI ligands, but their high molecular weight severely hindered cellular uptake[17]. Later, small molecule inhibitors were used as POI ligands, showing advantages in terms of affinity, cell permeability, and ease of synthesis, thereby significantly improving drug-like properties[7,10,18]. The complex structure of small molecule inhibitors often leads to poor water solubility and low bioavailability in vivo; many POIs lack binding sites for small molecule drugs. Therefore, peptide ligands have garnered significant attention; the large surface area of the peptide can overcome the limitation of shallow binding pockets and circumvent mutation-induced resistance[19−21]. However, the characteristic of peptide ligands being easily degraded by enzymes weakens their stability in the bloodstream and affects their ability to penetrate cell membranes[22]. Aptamers, as another type of ligand targeting POI, can be screened through SELEX technology to achieve higher specificity[23]. Its advantages include ease of synthesis, structural stability, low toxicity, and limited immune response[24]. Currently, small molecule ligands are still the primary choice when designing TACs. However, based on targeting requirements, peptides and nucleic acid aptamers can serve as good alternatives. Moreover, the structure of the POI also influences the selection of the aptamer.
The function of TAC is also greatly influenced by the mode of ligand binding, which can be mainly divided into three categories: non-covalent binding, irreversible covalent binding, and reversible covalent binding. PROTACs that use non-covalent ligands retain the event-driven mechanism; their dependence on sustained target occupancy, in combination with a tendency for rapid dissociation and systemic clearance, often results in reduced in vivo bioavailability[10]. Irreversible covalent ligands are capable of promoting stable anchoring of PROTAC to the POI, consequently improving intracellular retention and increasing target binding duration[25]. On the other hand, permanent bonding would hinder catalytic turnover[26], and covalent modification of POI may also interfere with proteasome recognition. Reversible covalent coupling forms a dynamic reversible covalent bond with the POI, not only enhancing intracellular accumulation and extending target binding time, but also retaining the ability to dissociate and catalyze cycles, allowing a single PROTAC molecule to promote multiple degradation events[10] (Fig. 3). Although reversible covalent binding has demonstrated specific advantages, non-covalent and irreversible covalent binding is of value too; their applicability would depend on the specific target and the environment.
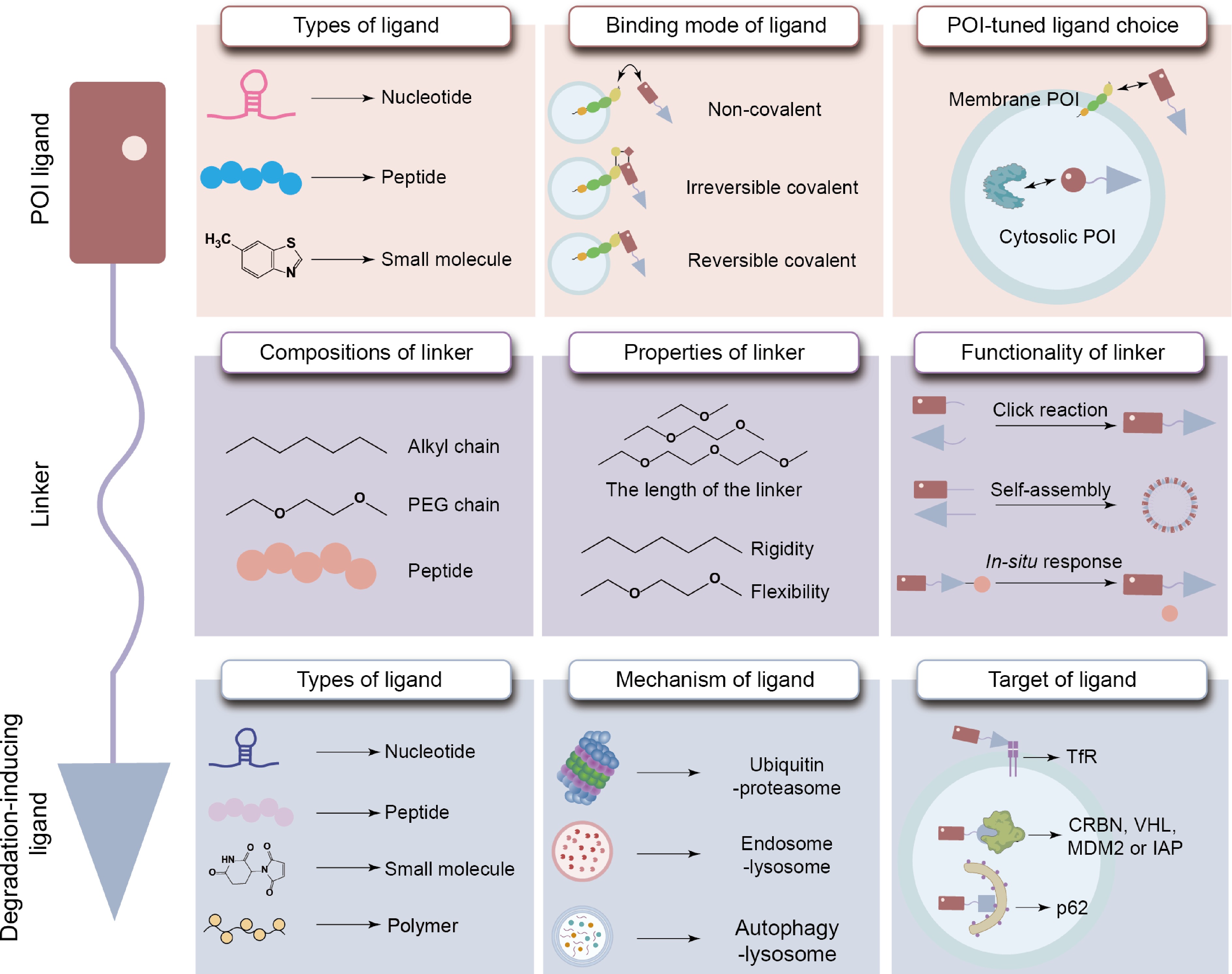
Figure 3.
Properties of the POI ligand, degradation-inducing ligand, and linker within TAC molecules.
Degradation-inducing ligands, recruiters of the protein degradation system
-
There are two important pathways for protein breakdown in cells: the ubiquitin-proteasome system (UPS) and the lysosomal pathway. The latter also includes the endocytic-lysosomal pathway and the autophagic-lysosomal pathway[11]. Therefore, degradation-inducing ligands should be selected to match the specific degradation mechanism.
In the UPS, E3 ligases intervene in substrate-specific ubiquitination to enable recognition and degradation of the POI by the proteasome[27]. E3 ligase ligands have evolved from low-permeability protein ligands[28] to small-molecule ligands, with recent designs incorporating RNA-based ligands[24]. Furthermore, design selections must also consider distribution characteristics of E3 ligases across different tissues[29]; Cereblon (CRBN), von Hippel-Lindau (VHL), and murine double minute 2 (MDM2) are among E3 ligases that are commonly selected and used[30]. These ligases possess distinct distribution patterns. For example, CRBN is expressed across multiple tissue sites[31]; VHL demonstrates high expression in kidney and gallbladder tissues, while MDM2 shows a nearly ubiquitous distribution[29]. It is possible to improve the in vivo degradation efficiency of PROTACs by taking into account these distribution differences during the design process.
LYTACs and their analogs primarily interact with recyclable transporters, such as transferrin receptors, to intervene in LYTAC internalization and subsequently initiate lysosomal degradation[32]. Based on this mechanism, peptide-programmed lysosome-targeting chimeras (SPYTACs) were designed to specifically target low-density lipoprotein receptor-related protein 1 (LRP1). SPYTACs can effectively permeate the BBB via LRP1-mediated trans-endocytosis and endocytosis, activating lysosomal degradation of POIs[33]. Polymeric lysosome-targeting chimeras (PolyTACs) would further increase endocytic uptake by multivalent interactions and facilitate degradation of membrane-associated proteins[34]. In the AUTOTAC, ligands bind to autophagy receptor p62, which subsequently guides the POI into the autophagic-lysosomal pathway for degradation. Because p62 is widespread in vivo, this approach would offer a viable and flexible method for selectively degrading POIs[35].
From the above perspective, it is essential that a balance between cellular permeability and tissue distribution be maintained in selecting E3 ligase ligands so that satisfactory degradation efficiency is achievable. For other TACs, a balance between high affinity and in vivo stability is needed in order to sufficiently induce degradation by the ligands. For LYTAC, ligands should not only bind to lysosomal transport receptors but also maintain functional integrity within lysosomes at the same time; AUTOTAC ligands must activate autophagy while still avoiding self-degradation.
Beyond inducing protein degradation, degradation-inducing ligands within a TAC platform may also be replaced with other functional groups in order to achieve functional regulation of non-protein degradation; these non-protein-degrading TACs have demonstrated great potential in precisely regulating functions of POIs. For example, dephosphorylation-targeting chimeras (DEPTACs) and phosphorylation-targeting chimeras (PhosTACs) were capable of promoting POI dephosphorylation by recruiting phosphatase PP2A[36,37]; similarly, ribonuclease-targeting chimeras (RiboTACs) facilitated mRNA cleavage by recruiting ribonuclease L, hence influencing protein translation[38].
Linkers, the bridge connecting ligands
-
Linkers are generally composed of alkyl chains, PEG chains, or peptide segments, each of which possesses variable physicochemical properties and may subsequently dictate the overall characteristics of the resultant TACs. Linkers of structural rigidity would restrict molecular conformation and improve cell permeability; incorporating triazole units into linkers further improves molecule stability[10,39,40]. Novel preparation methods have also used linkers to achieve self-assembly and construct multivalent TAC nanoparticles[41] or enable spatiotemporal control of TAC activation by introducing light-responsive groups via linkers[42]. Linker length and rigidity could also be modified and improved. For instance, excessively long linkers may hinder interactions between the POI and E3 ligase; overly short linkers may result in steric obstruction to prevent ternary complex formation[43,44]. In addition, linker flexibility would ease protein–protein interactions, whereas linker rigidity would improve ternary complex stability[43]. All these findings could provide insights into appropriate linker design and development.
The type of TAC determines the application scenario
-
TAC molecules have diverse mechanisms of action; they can achieve therapeutic effects by inducing protein degradation or by inhibiting disease progression through non-proteolytic regulatory mechanisms. Each TAC molecule has its own unique characteristics and limitations (Table 1)[45]; therefore, appropriate selection must be based on the specific disease target and therapeutic objectives.
Table 1. The characteristics and limitations of different TACs, as well as the challenges encountered when applying them to CNS disorders.
Types of TAC Characteristics Key challenges in the application of CNS diseases Future design strategies PROTAC Inducing the efficient degradation of intracellular soluble POI Proteasome pore size is only 13 Å, restricting entry of protein aggregates[46];
Neuronal proteasome impairment in AD affects UPS pathway activity[47];
Neurons express lower VHL and related E3 ligases than peripheral tissues, increasing susceptibility to off-target toxicityIntervening on protein monomers or oligomers in the early stages of the disease;
Incorporate additional proteasome repair elements during design to restore proteasome activity;
Development of PROTACs targeting E3 ligases specifically expressed in neurons, such as TRIM28LYTAC Inducing lysosomal degradation of membrane-bound and extracellular POIs via the endocytic system Structures frequently contain antibodies or glycopeptides, readily causing strong immune responses[45];
Impaired lysosomal acidification in AD severely impairs protein degradation efficiency[48];
Lysosomal transport receptors such as CI-M6PR are widely expressed in various tissues, posing a risk of nonspecific uptake[49]Replace antibodies or peptide ligands with small chemical molecules to mitigate immune responses;
Co-delivery with drugs used to treat lysosomal acidification disorders;
Identification of endocytic receptors overexpressed in CNS lesion sites via single-cell sequencing and subsequent design of targeting ligandsAUTOTAC Driving the autophagic degradation of cytoplasmic protein aggregates It may affect the cellular autophagy pathway, leading to excessive autophagy and damaging healthy neurons[45];
p62 levels are upregulated or depleted under pathological conditions in ND[50]Administer the drug in pulses to prevent excessive autophagy;
Design of an autophagy ligand that is independent of p62 levelsDEPTAC Using a phosphatase to promote the dephosphorylation of the overphosphorylated POI PP2A expression is downregulated in the affected brain regions[51];
It can only remove the phosphorylated groups from the toxic protein, but cannot eliminate the protein backboneDesigning ligands to recruit phosphatases specifically enriched in neurons;
Combining dephosphorylation and protein degradation functions to develop DEPTAC-degradation chimerasRiboTAC Eliminate POI at the source by using ribonuclease L to catalyze mRNA cleavage Widespread expression of RNA-binding proteins carries a high risk of off-target degradation By employing a bispecific antisense oligonucleotide approach, target specificity is further enhanced Abbreviation: CI-M6PR, cation-independent mannose-6-phosphate receptor. Solutions have been proposed to address the specific challenges faced in the application of TAC molecules for CNS diseases. Combining these strategies with the screening and optimization methods for TAC molecular building blocks mentioned earlier can provide concrete guidance for the design of TAC molecules for future CNS disease treatments.
Clinical progress in TAC molecules
-
Positive results from preclinical research have demonstrated TAC molecules' potential as effective therapeutic agents; ongoing clinical trials, a majority in Phase I studies and small numbers in Phase II or III trials, are evaluating more than 50 TAC candidates for their therapeutic efficacy. PROTACs account for the vast majority of study candidates, reflecting strong momentum in the development and application of the technology[52]. In contrast, other types of TACs are making slow yet steady progress into clinical translation. At present, no LYTAC or endoTAC candidate has entered clinical trial; two AUTOTAC molecules are in clinical trial phases (Table 2).
Table 2. TAC therapies in clinical disease treatment, with degradation-induced targets, candidate molecules, clinical phases, POI targets, and indications.
Degradation-induced targets TAC Clinical phases (date) POI targets Indications CRBN Dezandrodeg[53,54] III (2025, 3) AR Prostate cancer ARV-766[55] II (2025, 10) AR Prostate cancer KT-474[56,57] II (2023, 11) IRAK4 Atopic dermatitis, hidradenitis suppurativa BMS-986458[58] II (2023, 10) BCL6 Relapsed/refractory non-Hodgkin lymphoma, BCL Zelebrudomide[59] I (2021, 4) BTK
IKZF1
IKZF3BCL, MZL, WM, LPL/IC, FL, CLL, DLBCL, SMZL, MCL, SLL, central nervous system tumor Zaloblideg I (2024, 9) BCL6 NHL, AITL, BCL, DLBCL CFT8919[60] I (2025, 2) EGFR NSCLC UBE3 Vepdegestrant III (2023, 3) ESR1 Breast cancer, MBC, HR+/HER2- breast cancer ARD-266 I (2025, 9) AR CRPC ARV-102 I (2024, 2) LRRK2 PSP, Parkinson's disease, neurodegenerative disease TQB3019[61] I (2025, 1) BTK Advanced malignant cancer VHL PRT3789[52,62] II (2025, 3) SMARCA2 Advanced and metastatic solid tumor, esophageal cancer, NSCLC ASP4396[63] I (2024, 4) KRAS G12D Solid tumor ASP3082[64] I (2025, 4) KRAS G12D Solid tumor SQSTM1 ATC-202 I (2025, 4) TTR Familial amyloid polyneuropathy ATC-104 I (2024, 4) TDP-43 Amyotrophic lateral sclerosis Others (undisclosed) Catadegbrutinib[52,65,66] III (2025, 5) BTK B cell malignancy, NHL, MCL, Chronic spontaneous urticaria, relapsed cancer, refractory cancer, CLL, SLL, MZL, FL GT-20029 II (2024, 3) AR Alopecia, acne vulgaris PT0253 I (2024, 12) KRAS G12D Solid tumor AXT-1003[67] I (2024, 6) EZH2 Relapsed/refractory non-Hodgkin lymphoma, NHL, advanced solid tumor Abbreviations: ESR1, estrogen receptor 1; MBC, metastatic breast cancer; HR+/HER2- breast cancer, hormone receptor positive/human epidermal growth factor receptor 2 negative breast cancer; AR, androgen receptor; BTK, Bruton tyrosine kinase; NHL, non-Hodgkin lymphoma; MCL, mantle cell lymphoma; CLL, chronic lymphocytic leukemia; SLL, small lymphocytic lymphoma; MZL, marginal zone lymphoma; FL, follicular lymphoma; IRAK4, interleukin 1 receptor associated kinase 4; SMARCA2, SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 2; NSCLC, non-small cell lung cancer; BCL6, B-cell lymphoma 6 protein; BCL, B-cell lymphoma; CRPC, castration-resistant prostate cancer; IKZF1, IKAROS family zinc finger 1; IKZF3, IKAROS family zinc finger 3; WM, Waldenström macroglobulinemia; LPL/IC, lymphoplasmacytic lymphoma/immunocytoma; DLBCL, diffuse large B-cell lymphoma; SMZL, splenic marginal zone lymphoma; LRRK2, leucine-rich repeat kinase 2; PSP, progressive supranuclear palsy; AITL, angioimmunoblastic T-cell lymphoma; EGFR, epidermal growth factor receptor; EZH2, enhancer of zeste homolog 2; TTR, transthyretin; SQSTM1, sequestosome 1; TDP-43, TAR DNA-binding protein 43. Clinical translation of PROTAC has significantly accelerated since 2019, when the first PROTAC molecule, ARV-110, entered clinical trials. As of 2025, three PROTAC candidates have entered Phase III clinical trials[68], among which an NDA for vepdegestrant has been submitted to the FDA, and it is expected to become the first PROTAC drug to be launched. Additionally, two AUTOTAC molecules have progressed to Phase I evaluation. To date, only four TAC molecules have advanced to Phase I clinical trials for CNS disorders. ARV-102, which was developed for PD, showed good tolerability in healthy men. It built up in the cerebrospinal fluid (CSF) in a way that depended on the dose. In addition, it reduced LRRK2 levels by more than 50%. For zelebrudomide, ATC-202, and ATC-104, Phase I results remain undisclosed. Serious questions still persist in the clinical development of TAC molecules for CNS applications, including limited BBB permeability and suboptimal metabolic stability in vivo. To solve these problems, several reasonable approaches should be adopted, including: (i) improving the molecular structure to achieve a balance between lipophilicity and polar surface area, thereby promoting the passive diffusion of TAC and improving its metabolic stability; (ii) designing delivery carriers capable of targeting the brain, confirmed using in vitro BBB models and animal models of CNS diseases; and (iii) exploring alternative routes of administration, such as intranasal delivery, which uses the olfactory pathway to bypass the BBB and increase drug accumulation in the brain[69,70]. Currently, all TACs in the clinical stage are based on small molecules, which may be attributed to the susceptibility of peptides and nucleic acids to enzymatic degradation in the systemic circulation; high costs also pose questions for large-scale production. To ease the clinical translation of peptide and nucleic acid drugs, efforts should be made to address stability limitations through structural improvements and delivery technologies, and cost-effective production processes should be actively explored.
-
NDs, brain tumors, brain infections, and inflammations are all CNS disorders[71]. Barrier of the BBB will restrict access of chemicals to the brain, making it extremely difficult for pharmaceuticals to reach effective concentrations within the brain[72]. TACs would only be able to permeate the BBB and accumulate at the site of the lesion and cleanse disease-associated proteins by rational molecule designs; this will also help expand the range of protein targets available for treatment in CNS diseases[73,74].
TAC molecule design for the treatment of brain tumors
-
Glioma is a type of malignant brain tumor and accounts for approximately 45% of all intracranial tumors[75]. Standard treatment procedures for gliomas include surgical resection followed by radiation and chemotherapy. Because tumor cells easily spread along blood vessels and through the white matter in gliomas, it is difficult for surgery to completely remove tumor tissues. Cytokines produced by glioma tumors may also induce M2 polarization of microglia and peripheral macrophages, which creates an immunosuppressive environment that further promotes tumor growth and drug resistance[76]. TAC molecules are capable of degrading specific POIs, subsequently eliminating their catalytic and structural functions and overcoming the aforementioned therapeutic barriers (Fig. 4).
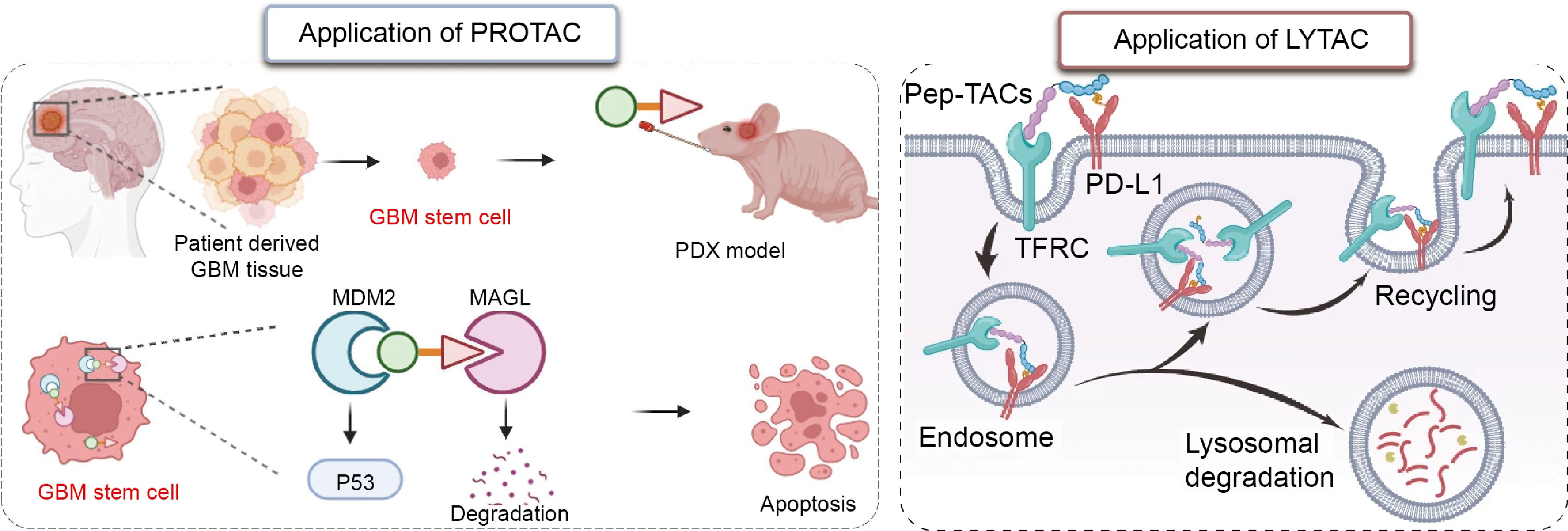
PROTACs have already been applied to multiple pathological targets in glioblastoma (GBM). A PROTAC targeting monoacylglycerol lipase (MAGL), a key factor in the development and progression of GBM, was combined via an esterification reaction[78]. The piperidine moiety in the MAGL inhibitor was modified via ring-opening to serve as the POI ligand, and given the high expression of MDM2 in GBM, a ligand for MDM2 was selected for the design. This PROTAC showed a 95% MAGL degradation efficiency, was able to disrupt tumor lipid metabolism, and reduced the self-renewal capacity of cancer cells[77]. Indoleamine 2,3-dioxygenase 1 (IDO1) is another target; its high expression in GBM suppresses T-cell function and promotes immune evasion[79]. When designing a PROTAC targeting IDO1, ligands for both VHL and CRBN E3 ligases were tested. Since the CRBN ligand has a low molecular weight and few hydrogen-bond donors, it was expected to exhibit favorable pharmacokinetics and BBB permeability; consequently, the CRBN ligand was ultimately selected. During PROTAC design, linkers of varying lengths and chemical compositions were screened, and an 11-atom PEG linker with a rigid heterocyclic structure was selected for its superior degradation capacity. These rational design approaches enabled the PROTAC to reduce protein levels in the tumor microenvironment by 94% and eliminate immunosuppressive activity[80].
For proteins on the cell membrane and outside the cell, PROTACs are generally unable to achieve effective degradation. LYTACs represent a promising therapeutic approach. The introduction of an aryl sulfonyl fluoride (ASF) group into the PD-L1 ligand doubled its binding affinity. At the same time, the use of a D-amino acid peptide segment to target the transferrin receptor (TfR) improves proteolytic stability and eases penetration of the BBB. Following administration, LYTAC penetrates tumor tissue and simultaneously binds to both TfR and PD-L1. After entering the cell through the action of TfR, PD-L1 is directed to lysosomes, achieving a degradation efficiency of 91%[6]. Based on these findings, the TAC-based therapeutic strategy has shown excellent efficacy in preclinical GBM models.
TAC molecule design for the treatment of neurodegenerative diseases
-
The slow loss of neurons is a major problem in NDs, including AD, PD, frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS)[81]. Conventional therapies can only inhibit the activity of pathological targets and are unable to intervene in complex pathological cascades. TAC technology has the potential to serve as an alternative strategy for treating NDs (Fig. 5).
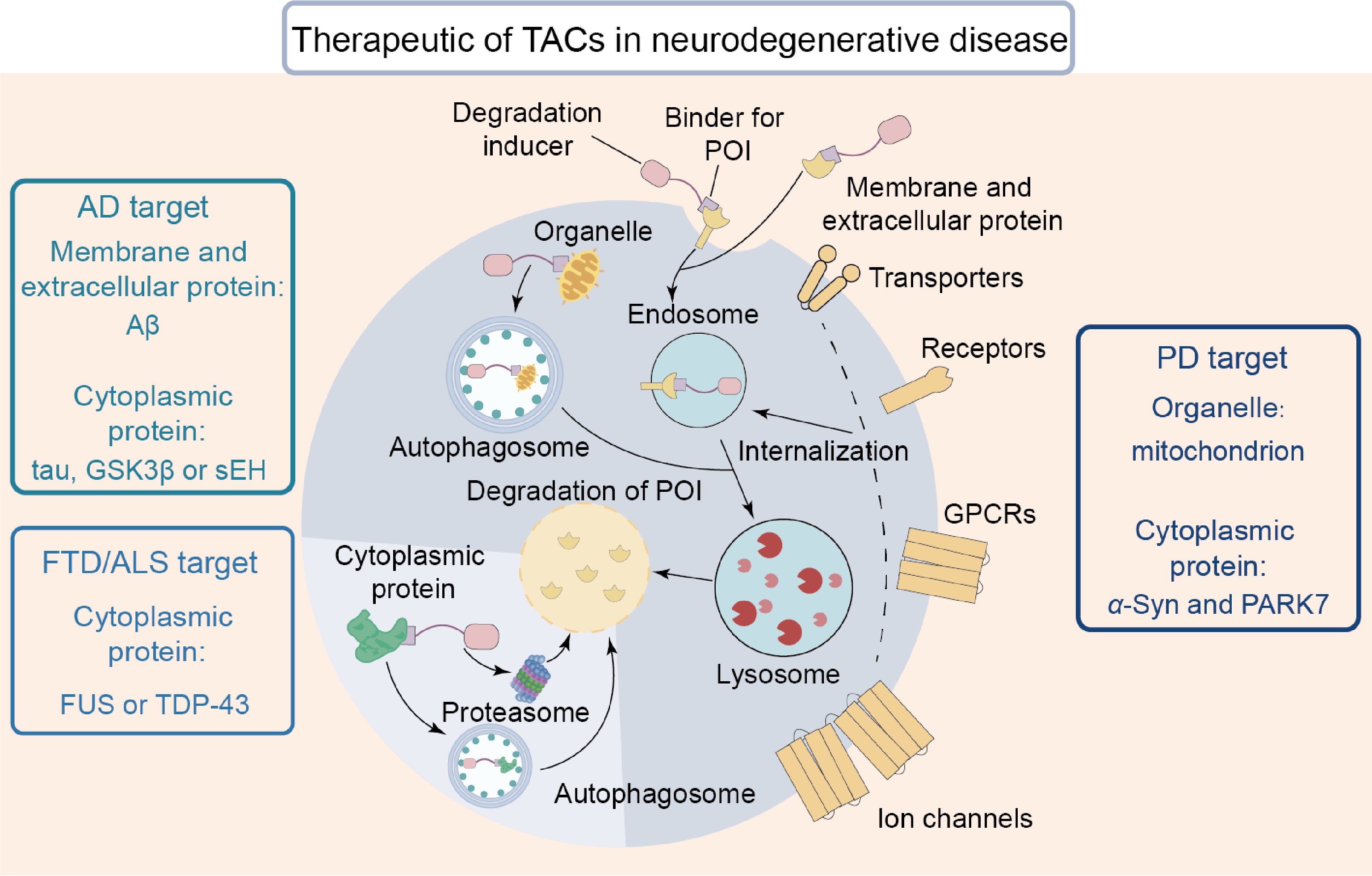
Figure 5.
By inducing the degradation of POIs, TAC serves as an effective approach for treating NDs. These POIs include membrane proteins, cytoplasmic aggregates, and damaged organelles.
The pathogenesis of AD is complex and includes multiple driving factors such as tau, glycogen synthase kinase 3β (GSK3β), soluble epoxide hydrolase (sEH), and so on. Conventional treatments often face questions in terms of efficacy or toxicity[82]. TAC technology has emerged as a valuable strategy for addressing AD. Tau aggregation disrupts synaptic function and accelerates neuronal death[83]. In transgenic mouse models of AD, a PROTAC degrader using a tau-binding small molecule as the POI ligand reduced tau levels by approximately 50% after 8 d of treatment; it also restored synaptic integrity and reversed cognitive deficits[84]. In the design of a PROTAC targeting GSK3β, E3 ligase ligands and PEG linker lengths were systematically screened, and the PROTAC composed of the CRBN ligand pomalidomide and a five-unit PEG linker was found to be optimal. At a concentration of 1 μmol·L−1, the PROTAC induced 94.3% degradation of GSK3β, reduced phosphorylated tau levels by 40%–60%, and provided neuroprotective benefits[85]. In designing an sEH-targeting PROTAC, a small-molecule inhibitor was utilized as the POI ligand, which possesses both short- and long-branch exit binding sites. PROTACs with long-branch exits did not exhibit degradation activity, whereas those with short-branch exits would successfully degrade the POI. Subsequently, an ether bond linked the POI ligand to the CRBN ligand; the degradation efficiency of this PROTAC reached 75%, producing an effective anti-inflammatory candidate for treating AD[86]. These findings indicated that TACs are able to restore synaptic function and reduce cognitive decline by degrading POIs in AD treatment.
α-Syn aggregates and human Parkinson's disease protein 7 (PARK7) are two critical pathogenic factors in PD progression. Current treatment procedures focus only on easing symptoms but do not stop disease progression[87]. TACs potentiate the halting of the progression of PD. To address the death of dopaminergic neurons resulting from abnormal intracellular α-syn aggregation[88], a PROTAC targeting α-syn was combined; this was achieved by linking an E3 ligase ligand to the meso site of the POI ligand via a click chemistry reaction. The resultant PROTAC exhibited low toxicity and reduced α-syn aggregation by 44%[89]. Pathogenic oxidation of PARK7 also impaired its neuroprotective function and accelerated disease progression[90]. In order to address these conditions, a PARK7-targeting PROTAC was combined. It was constructed by linking a PARK7 inhibitor to a CRBN ligand via a two-unit-length PEG linker; the resultant molecule facilitated approximately 80% degradation of PARK7[91]. AUTOTAC is another therapeutic option capable of degrading pathological targets in PD that are resistant to PROTACs. During synthesis of an AUTOTAC molecule targeting α-syn, researchers evaluated four α-syn-binding compounds and finally selected anle138b. The resultant AUTOTAC induced p62 aggregation, promoted autophagy, and cleansed harmful α-syn aggregates into the autophagy-lysosomal pathway. Ultimately, the extracellular α-syn level was reduced by 50%, and its intercellular spread was halted[87]. All the above findings highlight the potential of TAC technology as a viable therapeutic option for treating PD.
FTD and ALS share numerous similarities in symptoms, neuropathology, and genetic causes[92]; one of the key pathological features is their abnormal aggregation of TAR DNA-binding protein 43 (TDP-43) or fused in sarcoma (FUS). Even though there is a comprehensive understanding of the progression of these two diseases, effective treatment options remain elusive. TAC-based therapies may offer a viable approach for addressing FTD and ALS. When the FUS gene is mutated, the protein becomes mislocalized in the cytoplasm and forms harmful aggregates[93,94]. In order to address this pathogenic mechanism, an RNA-based PROTAC targeting FUS was devised, in which the RNA sequence with the highest binding affinity was selected from five candidates, and then the CRBN ligand was linked. This PROTAC promoted the ubiquitination of FUS and achieved 80% degradation efficiency following a single administration[92]. Abnormal accumulation of TDP-43 C-terminal fragments is also a major pathological factor in ALS[95]. To target this pathway, a PROTAC was designed to specifically recognize the β-sheet structures abundant in TDP-43 aggregates. During the design process, researchers systematically evaluated the linker length ranging from two to five units and found that a PROTAC with a three-unit linker was the most effective candidate, which reduced TDP-43 C-terminal fragment aggregation by 62% and increased cell viability by 30%[96]. TAC-based candidates have offered viable therapeutic options for both FTD and ALS; more specifically, they are capable of removing TDP-43 aggregates and mislocalized FUS.
TAC molecules have shown promising potential in treating ND. However, it is important to note that the animal models used in these studies may differ from humans in terms of POI expression levels, and functional proteins such as E3 ligases may also vary in their cellular distribution and expression levels. Consequently, the optimization strategies for TACs and the data obtained from preclinical studies may not be fully applicable to the clinical phase. Further experiments using human brain organoids or non-human primate models are needed to confirm the therapeutic efficacy of TACs. Future research may expand the scope of POIs to target extracellular and transmembrane proteins. These protein targets are critical in influencing disease progression, which include amyloid-β (Aβ) peptides[97], transporters[98], receptors, G protein-coupled receptors (GPCRs)[99], ion channels[100], and damaged mitochondria[101]. Recent research aims at designing TACs to target those theoretically difficult-to-degrade proteins and represents a transformative and significant field of study. Furthermore, in preclinical studies of ND, safety evaluations of TAC molecules are often of short duration, which may lead to numerous adverse consequences. For example, TACs may induce the degradation of unintended new substrates, resulting in severe off-target toxicity. Short-term TAC studies struggle to capture such cumulative effects. The POIs degraded by TACs sometimes possess important physiological functions, and long-term POI degradation can easily lead to serious consequences, whereas short-term POI clearance differs fundamentally from the results observed under long-term conditions. This poses a major obstacle to the translation of TAC-related research findings into clinical studies. Therefore, it is necessary to conduct further long-term safety evaluations of TACs. For example, long-term treatment of neuronal cell lines with TAC molecules based on specific functional proteins, followed by whole-cell proteomic analysis, can map the TAC degradation profile, identify unintended degradation targets interacting with the TAC, and enable early screening and optimization. For chronic diseases such as ND, the dosing intervals should be further extended, and changes in POI levels and the activity of relevant signaling pathways should be closely monitored to establish a multi-tiered, time-extended toxicology research system. These measures will facilitate the translation of TACs into clinical applications. By combining TAC designs with advanced delivery systems, researchers have created opportunities to improve therapeutic efficacy in the complex CNS microenvironment. For example, by incorporating TACs into mesenchymal stem cell carriers[102], it is possible to take advantage of stem cells' natural tendency to penetrate the BBB and migrate toward areas of brain injury, ultimately enabling repair and targeted therapy of the damaged regions. Co-administering TACs with bioactive natural compounds[103] may also create a supportive environment through their inherent neuroprotective capabilities, hence aiding TAC degradation of POIs.
-
Recent advances in TAC-based treatments have created immense potential for treating CNS disorders. TAC molecules utilize an event-driven mechanism to promote the formation of a ternary complex between POI and degradation-related proteins, which would subsequently accelerate POI degradation and thereby produce a desired therapeutic effect[104]. There are still deficiencies in clinical applications of TACs, insufficient BBB penetration, instability, and off-target toxicity risk, just to name a few. TACs of large molecular size and high polarity will make it difficult to cross the BBB[10]; the complex structure of TACs is also readily recognized and degraded by enzymatic and immune systems[105]. In addition, functional proteins that are ubiquitous in healthy tissues may cause off-target toxicity[10]. Current research has shifted from improving TAC molecular structures to developing advanced delivery systems, with goals of improving targeting accuracy, enhancing pharmacokinetic properties, and ultimately supporting clinical applications of TAC-based treatments (Fig. 6).
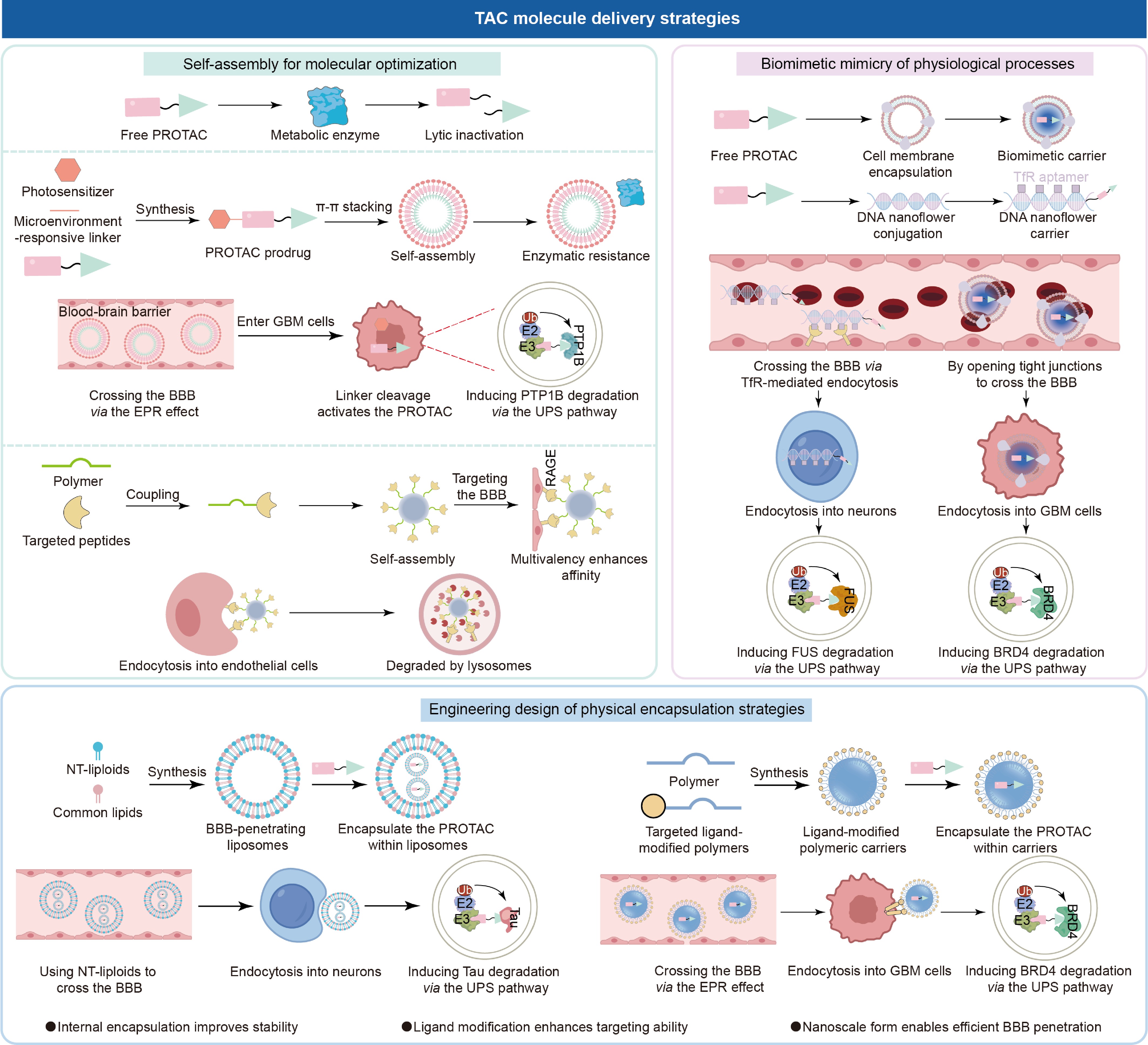
Figure 6.
Nano-delivery carriers offer a solution to the questions of TAC therapy. These carriers use self-assembly methods to improve molecular properties, employ biomimetic materials to mimic physiological processes, and improve therapeutic efficacy through engineered design.
Carrier-free self-assembled TAC delivery for molecular improvement
-
Chemical modification of TAC molecules can endow them with self-assembly characteristics. Programmed π–π stacking or hydrophobic interactions would enable TAC molecules to autonomously form supramolecular nanoparticles[106], which results in a significant transition from passive drug encapsulation to active engineering of nanostructures. This carrier-free preparation could exhibit multiple advantages; for example, close packing of TAC molecules conceals enzyme-susceptible cleavage sites, preventing enzymatic recognition and degradation in vivo, and removal of inactive excipients leads to higher drug delivery efficiency[107]; nanoscale dimensions enhance permeability and retention (EPR)[108], consequently increasing BBB permeability under pathological conditions. These strategies have made productive progress in advancing the design and development of TACs intended for efficient therapy of CNS diseases.
Protein tyrosine phosphatase 1B (PTP1B) has been investigated as a valuable therapeutic target because an elevated expression level of PTP1B is observed in both GBM cells and infiltrating T lymphocytes, where it eases tumor advancement and contributes to T cell exhaustion[109,110]. A prodrug was engineered by covalent conjugation of a PTP1B-targeting PROTAC to a photosensitizer via a peptide linker, which could supposedly enable tumor microenvironment-specific activation. This particular construct undergoes spontaneous self-assembly into nanoparticles owing to intermolecular π–π stacking, resulting in a substantial drug loading capacity of 88.6% and exhibiting considerable stability in a murine GBM model. In GBM, tumor cells activate immune cells in brain tissue by secreting pro-inflammatory cytokines such as interleukin-6 (IL-6), thereby inducing an inflammatory response. These inflammatory cytokines, in turn, activate the Signal Transducer and Activator of Transcription 3 (STAT3) signaling pathway in BBB cells, leading to the downregulation of transporters and tight junction proteins on the BBB. This disrupts the integrity of the BBB, resulting in increased permeability[111]. Consequently, the resultant nanoparticles exploited the EPR effect, improved BBB crossing efficiency by a factor of 2.16, and increased accumulation within GBM tissue. Upon exposure to light, the system also triggered significant ROS production while concurrently facilitating PTP1B degradation through PROTAC-intervened proteolysis, leading to an 80% reduction in cell viability and complete tumor regression. Furthermore, it decreased critical T cell exhaustion markers by 27% and 35%, respectively, thereby improving cytotoxic T cell function[108]. The self-assembly principle has also been adapted to multivalent targeting, including targeting membrane proteins associated with AD pathology. In AD, the receptor for advanced glycation end products (RAGE), which is highly expressed on the BBB, guides Aβ entry into the brain; a low level of LRP1 would hinder Aβ clearance[5,112−114], thereby driving the progression of AD pathology. Multiple RAGE-binding peptides were conjugated to a polymer backbone through rational engineering, and functional nanoparticles self-assembled loaded with simvastatin (Fig. 7a). This multivalent design not only improved interaction with receptors on the BBB (by increasing local retention time 1.2 times, Fig. 7b) but also reduced RAGE expression (by 1.25 times), decreased Aβ influx (by 22%), and upregulated LRP1 expression (by increasing Aβ efflux 2.81 times, Fig. 7c and d)[5]. It is apparent that successful chemical modification will endow TAC molecules with self-assembly capability and enable them to effectively penetrate the CNS; this approach can offer new avenues for the application of TACs in treating CNS diseases.
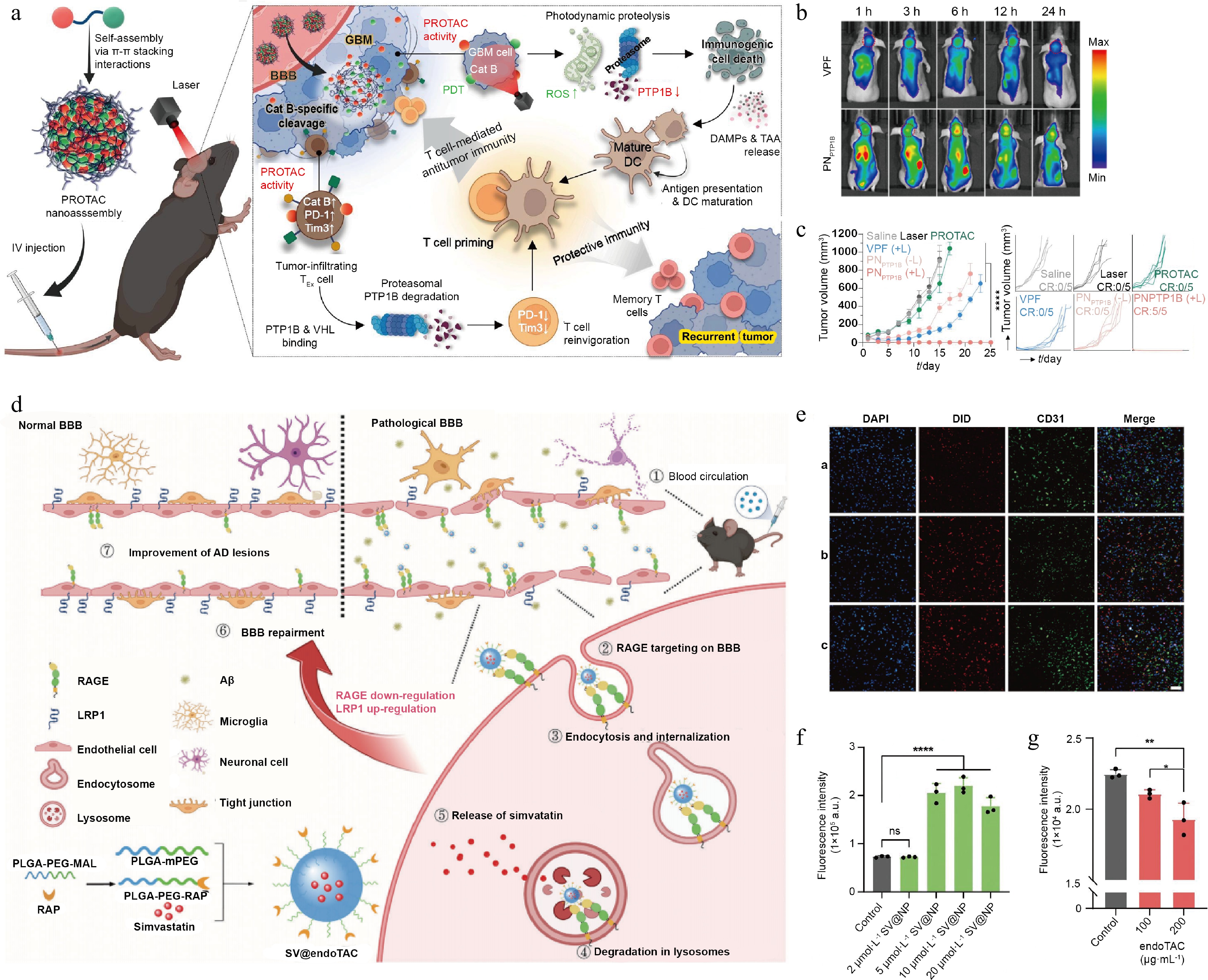
Figure 7.
Characterization of TAC-based self-assembling nanoplatforms and their performance. (a) Design strategy of endoTAC and its mechanistic role in AD therapy. (b) Intracellular distribution of nanoparticles in brain vascular endothelial cells. (c) Flow cytometric analysis of FITC-Aβ uptake in bEnd.3 cells following 24-h co-incubation with SV@NP. (d) Flow cytometry was used to analyze FITC-Aβ uptake in RAGE-overexpressing bEnd.3 cells after 24 h of co-culture with endoTAC. Reprinted with permission from Ref.[5]. Copyright 2024 Wiley.
In addition to the strategies mentioned above, research on ischemic stroke has shown that antisense oligonucleotides (ASOs) targeting caspase-3 can self-assemble into spherical nanostructures for drug delivery via ring-forming amplification technology. After being modified with TfR-targeting aptamers on their surface, these nanostructures can effectively cross the BBB and enter neurons, silencing the caspase-3 gene and inhibiting neuronal apoptosis, thereby addressing both the challenges of brain delivery and gene therapy[115]. Furthermore, in studies on intracerebral hemorrhage, rolling circle amplification technology enables the combination of signal regulatory protein α (SIRPα) DNAzyme with Mg2+, leading to the self-assembly of a nanomodulator with Mg2+ at its core and SIRPα DNAzyme as its scaffold. Upon entering microglia, this nanomodulator dissociates into Mg2+ and SIRPα DNAzyme. Mg2+ can inhibit inflammatory pathways in microglia, promoting their transition to an anti-inflammatory phenotype. It also activates the cleavage activity of the SIRPα DNAzyme, silences the SIRPα gene, and promotes the phagocytosis of red blood cells by microglia, leading to the efficient clearance of hematomas[116]. Based on this design concept, it is promising to attempt the self-assembly of aptamer-based TAC molecules, thereby significantly increasing the drug-loading capacity of aptamer-based TACs and providing a new approach for their delivery.
Physical encapsulation in an engineering carrier for TAC delivery
-
Drug delivery systems made of exogenous biomaterials may offer many benefits; these delivery carriers can be functionally engineered to solubilize poorly soluble TAC molecules and prolong their circulation times in the body. In addition, modification of carrier surface with targeting ligands would shift drug delivery from passive accumulation dependent on the EPR effect to active targeting capability intended for sites of pathology, further enhancing TACs' in vivo use, safety, and efficacy[10].
A liposomal delivery system is regarded as an effective solution to solve the problem of peptide-based PROTACs crossing the BBB and to minimize their susceptibility to degradation in the bloodstream. Incorporating neurotransmitter-derived lipidoids (NT lipidoids) into liposomal formulation improved brain delivery efficiency and promoted the accumulation of various therapeutic drugs[117]. Furthermore, the pathological progression of AD is often accompanied by neuroinflammation. These abundant inflammatory mediators not only cause pericytes to contract and die but also lead to the loss of polarized distribution of the AQP4 aquaporin in astrocytes, resulting in depolarization of astrocyte dendrites. These changes collectively increase BBB permeability, facilitating the passage of liposomal carriers through the BBB into the brain parenchyma[118]. Based on these mechanisms, researchers encapsulated peptide-based PROTAC inside liposomes through electrostatic interaction with calf thymus DNA and obtained a stable nanocomposite preparation (Fig. 8a). The resultant liposomal delivery platform protected the peptide-based PROTAC from enzymatic degradation and prolonged the drug's half-life to 17–18 h. In an animal model, the liposomes produced stable drug release, maintained the desired concentration range of PROTAC, eased the formation of ternary complexes, and reduced the 'hook effect'. Moreover, this liposomal delivery strategy also enhanced BBB permeability by converting systemic administration into brain-targeted accumulation and increased cellular uptake efficiency by 2.13-fold (Fig. 8b). After 12 h of treatment with the liposomal formulation, intracellular tau level decreased by approximately 75%; in comparison, treatment with free PROTAC reduced tau level by only around 50%. The findings conclude that liposomal carriers can improve PROTAC-intervened degradation efficiency (Fig. 8c and d)[119].
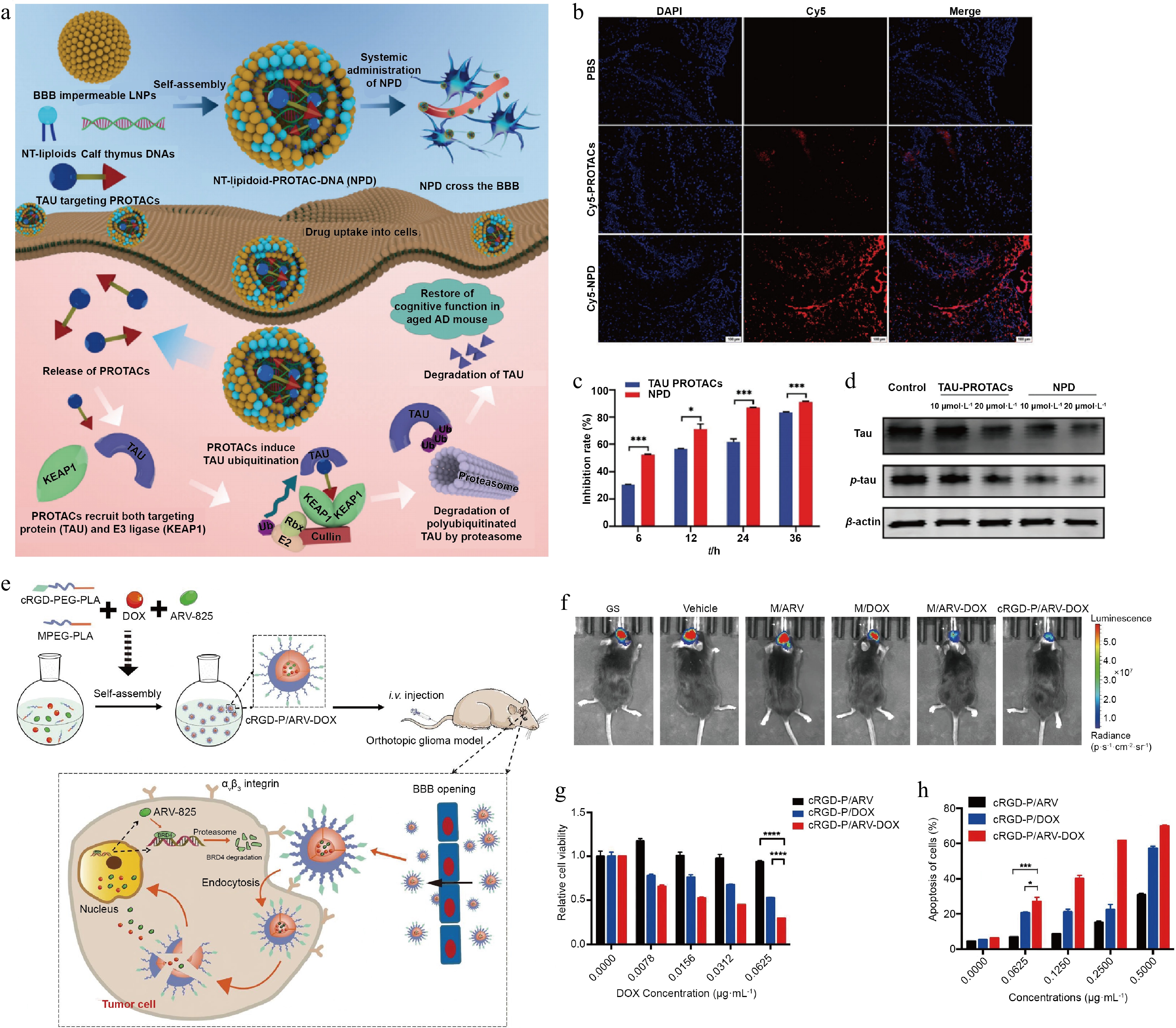
Figure 8.
Design and experimental confirmation of TAC delivery via liposomes and polymer carriers. (a) Design of the liposome-based delivery platform and its mechanism of action in AD treatment. (b) Representative fluorescence images of coronal brain sections from C57 mice, showing Cy5-labeled PROTAC (red) and nuclear staining (blue). (c) Quantitative flow cytometric analysis of fluorescence intensity in TAU-EGFP-overexpressing N2a cells following PROTAC treatment at specified time points. (d) Western blot analysis was performed to assess the extent of tau protein degradation. Reprinted with permission from Ref.[119]. Copyright 2024 Wiley. (e) Design of the polymer delivery carrier and its mechanism of action. (f) Representative in vivo bioluminescence imaging of tumor-bearing mice after treatment. (g) Cytotoxicity of cRGD-P/ARV, cRGD-P/DOX, and cRGD-P/ARV-DOX in GL261 cells after 24-h incubation was assessed using the MTT assay at concentrations ranging from 0 to 0.0625 μg·mL−1. (h) Flow cytometric analysis of apoptosis in GL261 cells using Annexin V/PI double staining. Reprinted with permission from Ref.[120]. Copyright 2022 Elsevier.
Carriers constructed from natural and synthetic polymers also offer a functionally tunable delivery option capable of delivering hydrophobic drugs, including TAC molecules[10]. In GBM, bromodomain-containing protein 4 (BRD4) is highly expressed in tumor cells and localized to multiple oncogene promoters, where it promotes tumor proliferation[121]. PROTAC targeting BRD4 degrades this protein and potentiates it as a key therapeutic option for GBM. However, PROTAC may demonstrate low solubility, instability, and inability to effectively target tumor tissue, which will complicate their clinical applications. Polymeric carriers have been developed to address these technical issues[122]. The carriers are hence fabricated from biomaterials such as poly(lactic-co-glycolic acid) (PLGA) and poly(ε-caprolactone) (PCL) and are further surface-modified with PEG to improve their hydrophilicity and prolong their circulation time in the body. This resultant preparation encapsulated PROTAC, improving its solubility and stability; the delivery utilized the EPR effect to penetrate the BBB and passively targeted the GBM, where drug concentration within the tumor was increased, BRD4 protein level decreased by 59%, and approximately 65% cell cycle arrest efficiency was achieved[123]. In addition to passive targeting of the GBM, functionalizing the carrier surface with peptide ligands also facilitates active tumor targeting by PROTAC. Researchers constructed a polymeric carrier by combining PEG and poly(d,l-lactic acid) (PDLLA) and conjugated it with a peptide targeting the neurokinin-1 receptor (NK-1R). In GBM, this receptor is overexpressed on both vascular endothelial and GBM cells[124]. This delivery carrier improved the solubility and stability of PROTAC through physical encapsulation, reducing the drug release rate by 33%. Through NK-1R-intervened endocytosis, the carrier successfully crossed the BBB and facilitated selective accumulation of the PROTAC in tumor tissues while also minimizing exposure to normal tissues. In a GBM model, this delivery system exhibited 75% inhibition of cell proliferation, 77.5% reduction in tumor volume, and 75% reduction in tumor weight[76]. Similarly, modification with a cycle (Arg-Gly-Asp-d-Phe-Lys) (cRGDfk) peptide enabled the polymer carrier to actively target αvβ3 integrin, a receptor overexpressed in GBM (Fig. 8e). This design enhanced tumor-specific delivery of PROTAC and decreased drug distribution in healthy tissues (Fig. 8f); the delivery achieved 75% inhibition of tumor cell viability and 55% apoptosis (Fig. 8g and h)[120].
Delivery carriers made of natural and synthetic materials may also improve the solubility and stability of TAC. By modifying the delivery system with targeting ligands, it is possible for TAC to permeate the BBB and be delivered to the site of the lesion; this will enhance localized accumulation of TAC at pathological sites and reduce off-target effects. Hydrogels composed of polymers can also be used for nucleic acid delivery. Cross-linking agents prepared using aptamers that specifically target hemoglobin can bind to DNA chains grafted onto polyacrylamide, thereby enabling the preparation of hydrogels for the loading of therapeutic drugs. In the event of postoperative rebleeding following a cerebral hemorrhage, the aptamers respond to elevated hemoglobin levels, causing the DNA chains to dissociate and release the therapeutic drugs[125]. Based on this concept, it is possible to design hydrogels composed of aptamer-based TACs that respond to specific elements in the microenvironment of CNS diseases, thereby achieving efficient loading of TACs and controlled release within the disease environment. The successful development and application of mRNA vaccines also represents a major advancement in liposomal drug delivery systems, which has reignited interest in using nanocarriers for TAC molecule delivery. Such delivery systems allow for flexible administration by multiple routes, including intravenous injection, oral ingestion, and intranasal administration. As such, delivery platforms could be carefully designed to meet the specific needs of a given disease, further expanding the potential for treating CNS disorders.
Biomimetic TAC delivery for simulating physiological process
-
Bio-inspired TAC delivery carriers combine biological components with synthetic materials to demonstrate technical advantages, such as a simple manufacturing process, high biocompatibility, excellent targeting capability, and low immunogenicity. These preparation characteristics help TACs evade immune detection and improve their stability and safety in vivo[126].
DNA nanoflowers, a class of DNA-based hybrid nanomaterial, are designed through co-crystallization of DNA and pyrophosphate to form a petal-like hierarchical structure[127]. Their programmability at both the sequence and functional level, combined together with substantial drug-loading capacity, low toxicity, and superior biocompatibility, has made DNA nanoflowers a versatile nucleotide-based drug delivery platform[128]. To improve the stability of aptamer-based PROTAC, multiple TfR aptamers were incorporated into the DNA nanoflowers, and an aptamer-PROTAC targeting FUS was attached by base-pairing. The resultant delivery preparation increased the stability of the PROTAC threefold and nearly doubled its circulation half-life. By exploiting the high expression of TfR on brain endothelial cells, the construct also permeated the BBB via TfR-mediated endocytosis and enhanced PROTAC accumulation in the brain (Fig. 9a and b). A single dose of the DNA nanoflower carrier maintained a sustained therapeutic effect for up to 2 weeks, decreased the proportion of FUS aggregate-positive cells to 10%, and reached 70% FUS degradation efficiency (Fig. 9c and d)[92].
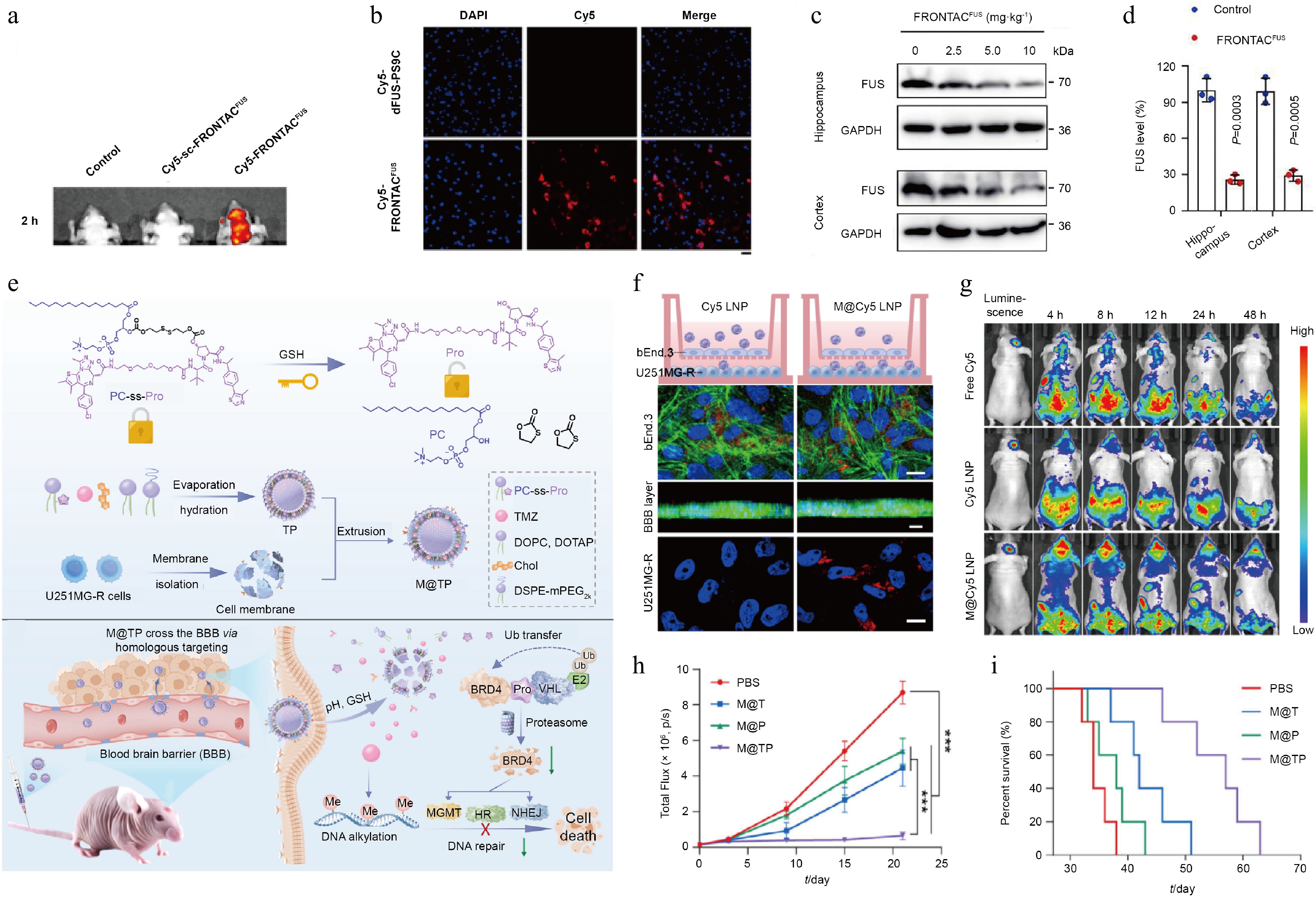
Figure 9.
Characterization of TAC-based biomimetic carrier delivery platforms and their experimental confirmation. (a) In vivo and ex vivo fluorescence imaging of mice following tail vein injection of PBS, sc-FRONTACFUS, and FRONTACFUS. (b) Fluorescence imaging of brain tissue sections from mice administered Cy5-labeled dFUS-PS9C or Cy5-labeled FRONTACFUS via tail vein. (c) Immunoblot analysis of FUS level in hippocampal and cortical tissues from mice administered escalating doses of FRONTACFUS via tail vein injection. (d) Quantitative comparison of Western blot data analyzing differences in FUS degradation in hippocampal and cortical tissues following tail vein injection of NF control vs 10 mg·kg−1 FRONTACFUS. Reprinted with permission from Ref.[92]. Copyright 2025 Springer Nature. (e) Mechanism of action of the TAC biomimetic cell membrane delivery platform. (f) In vitro evaluation of the BBB penetration capacity of Cy5 LNP and M@Cy5 LNP using the Transwell assay. (g) In vivo distribution of free Cy5, Cy5 LNP, and M@Cy5 LNP measured at specified time points following tail vein injection in mice with orthotopic U251MG glioma. (h) Quantitative analysis of tumor bioluminescence signals in each treatment group. (i) Kaplan–Meier survival analysis for mice in each treatment group. Reprinted with permission from Ref.[129]. Copyright 2025 Wiley.
Biomimetic cell membrane carriers also provide a means of delivering therapeutic drugs, using the natural properties of cell membranes to cross the BBB and target specific lesions. To solve the questions associated with TAC therapy in GBM, a biomimetic hybrid nanovesicle called M@TP has been developed. The construction of this platform involves two key steps, namely, the conjugation of a BRD4-targeting PROTAC to lipid (1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine) to generate an amphiphilic prodrug capable of liposomal combination, and co-extrusion-intervened fusion of drug-resistant GBM cell membranes with the prodrug-loaded liposomes to produce the final biomimetic system (Fig. 9e). This biomimetic membrane coating allows M@TP to avoid being cleared by the immune system, which extends the PROTAC's half-life by 6.5 times and significantly improves its stability in the body. Moreover, by leveraging the homogeneous targeting of GBM by native tumor cells and their ability to downregulate tight junction proteins on brain endothelial cells, M@TP enhances the permeability of the BBB, effectively crosses the BBB, and selectively builds up in GBM tissue, increasing the uptake of the drug by tumor cells by 2.3 times (Fig. 9f and g). This targeted delivery leads to marked suppression of tumor growth and a significant extension of median survival in mouse models to 59 d (Fig. 9h and i)[129].
The biomimetic delivery system uses naturally occurring biological elements to functionalize TAC nanoparticles. This type of delivery creates a stealth coating that evades immune detection and prolongs circulation time inside the body. The drug carrier is thus capable of crossing the BBB and preferentially accumulating at pathological sites by using its intrinsic homing capability or functionalized targeting ligands. A combined high biocompatibility and active targetability can not only improve therapeutic precision but also lead to developing safer and more effective TAC therapies for CNS diseases as well. Researchers may take advantage of other biological carriers, in particular engineered probiotics and functional cells, as TAC delivery platforms in the future. For example, naturally occurring exosomes have shown therapeutic values in applications; these vesicles can deliver PROTAC in treating triple-negative breast cancer[130] or deliver small-molecule drugs across the BBB to treat brain diseases[131]. These findings suggest that exosomes may serve as an effective delivery system for TAC targeting CNS diseases. These biomimetic delivery systems are able to maintain activity in vivo for extended time periods and use their natural capability to autonomously migrate to the site of lesion and release drugs in a controlled manner, thereby resulting in new possibilities for TAC therapy in CNS diseases.
-
This review has demonstrated that TAC can effectively eliminate POIs via the UPS or lysosomal pathways; progress and success of TAC candidates in clinical trials highlight their therapeutic potential in treating CNS diseases. Rational and systematic improvement to TAC ligands and linkers would ease the formation of ternary complexes, and effective utilization of various delivery methods can also enhance TAC biocompatibility and targetability. Strategic planning of TAC designs and delivery platforms will lead to key pathways creating innovative and intelligent therapeutics for CNS disorders. Compared to peripheral diseases, the BBB is the primary barrier that molecules must overcome to exert their therapeutic effects in TAC design for CNS diseases; therefore, brain penetration of TAC molecules is a prerequisite. However, the physiological state of the BBB varies across different types of CNS diseases. For example, in GBM, the BBB's physiological function is impaired, increasing its permeability, whereas in the early stages of some neurodegenerative diseases, the BBB structure may remain relatively intact, requiring receptor-mediated active transport to deliver molecules into the brain. Consequently, TAC administration strategies must be tailored to the pathological state of each specific disease. Furthermore, in CNS diseases, POIs within the brain often exist as protein aggregates. Conventional PROTACs that rely on the UPS pathway may be unable to address these high-molecular-weight protein aggregates, necessitating a shift toward molecules such as AUTOTACs that depend on the autophagy-lysosomal pathway. In this context, the ligand must be capable of precisely recognizing the structural features unique to protein aggregates to effectively clear the pathogenic targets. Furthermore, TAC-induced sustained protein degradation resulted from event-driven molecular functions and may inadvertently cause excessive clearance of POIs and trigger toxicity. A reversible termination of the process could be predesigned such that once the POI level has met the desired therapeutic requirement, free TAC will be captured and inactivated, thereby achieving controlled termination of the degradation and creating a precise approach for safer use of TAC in clinical applications[132].
In addition to achieving therapeutic goals through direct degradation of proteins, TAC also plays a role in enzymatic cleavage of POIs, which consequently generates a large number of antigenic peptides, improves antigen cross-presentation, and stimulates a robust cellular immune response. These immunological processes lead to opportunities for TAC technology to be used in vaccine development and therapeutic protein production, thus opening avenues for innovative biopharmaceutical manufacturing. It may also allow POIs associated with CNS diseases to be redirected into antigen-presentation pathways, enabling the establishment of long-lasting immune protection and advancing therapeutic applications for CNS diseases.
Furthermore, in current preclinical research on CNS diseases, the evaluation of TAC molecule efficacy and the assessment of various delivery strategies to improve TAC drugability are often conducted in rodent models. These animal models exhibit significant structural differences from humans in the CNS; for example, the expression levels of certain drug efflux transporters in the human BBB differ by nearly threefold compared to those in rats, while the expression levels of transporters responsible for delivering substances into the brain can differ by as much as fivefold. This necessitates caution when interpreting data obtained from preclinical rodent models and limits the translation of preclinical drug candidates into human applications[133,134]. Microfluidic chips and related technologies should be employed to closely mimic the human physiological environment, or primate models, which exhibit a protein expression profile in the BBB more closely aligned with that of humans, should be utilized to conduct in-depth safety and efficacy evaluations of TAC candidate molecules, thereby accelerating the transition of TACs to the clinical stage.
Future TAC research should continue to focus on enhancing its suitability for the CNS. Attempts could be made to introduce small rings or fused ring structures into the molecular architecture to constrain molecular conformations, and these conformational constraints could be validated through molecular dynamics simulations to further reduce the polar surface area of TACs and improve their BBB permeability. Through in-depth analysis of transcriptomic or proteomic data from the BBB and target brain cells using bioinformatics tools, endocytic receptors with significantly elevated expression under disease conditions can be identified. This enables the design of corresponding targeted ligands for integration into delivery carriers, thereby advancing the development of stable, precise, and efficient brain delivery technologies. Concurrently, efforts are focused on integrating TAC with anti-inflammatory strategies, such as inhibiting excessive microglial activation, and antioxidant strategies, such as using nanozymes to mimic antioxidant enzymes, in CNS disease treatment. This approach aims to improve the microenvironment of CNS diseases while clearing pathogenic proteins, thereby achieving synergistic therapeutic effects. It is anticipated that emerging innovative preparations will surely provide more and better treatment options for the management and treatment of CNS diseases in the near future.
-
Not applicable.
-
The authors confirm their contributions to the work as follows: study conception: Ding Y, Han G, Gu X; writing original draft: Liu S, Xu W, Wang M; review and editing: Ding Y, Han G, Gu X. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
-
This study was supported by the National Natural Science Foundation of China (82372113), the National Ten Thousand Talents Program for Young Top-notch Talents, Lingang Laboratory (Grant No. LGL-2614-09), the Natural Science Foundation for Distinguished Young Scholars of Jiangsu Province (BK20240096), and the Joint Fund of Zhejiang Provincial Natural Science Foundation of China (LKLY25H180014). In addition, we thank the Jiangsu 333 High-level Talent Training Project ([2022] 3-16-190), Fundamental Research Funds for the Central Universities (2632025PY02, 2632026TD05), China Postdoctoral Science Foundation (Grant Nos 2025T180968 and 2025M783610).
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Shuo Liu, Wei Xu, Mengxi Wang
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Liu S, Xu W, Wang M, Mao Z, Zhou J, et al. 2026. Design and delivery of targeting chimeras therapeutics in managing central nervous system disorders. Targetome 2(3): e028 doi: 10.48130/targetome-0026-0028 

Design and delivery of targeting chimeras therapeutics in managing central nervous system disorders
- Received: 10 March 2026
- Revised: 11 May 2026
- Accepted: 20 May 2026
- Published online: 27 June 2026
Abstract: Site-specific degradation by targeting chimeras (TACs) has become a desirable alternative to occupancy-driven inhibition for central nervous system (CNS) diseases due to the possibility of completely deactivating pathogenic proteins, rather than complete blocking. The catalytic mechanism of TACs is dynamic, meaning that they can provide sustained therapeutic effects at low doses in clearing key pathogenic proteins associated with CNS diseases, while resisting the development of resistance mechanisms. However, the physicochemical properties of TACs pose a high delivery question for the brain, limiting brain bioavailability and impairing clinical progress. To address this issue, this review presents the structure-function relations of TAC modules, focusing on their therapeutic potential in various CNS diseases such as glioblastoma, Alzheimer's disease, and Parkinson's disease, as well as the role of novel nanocarriers and delivery platforms in overcoming biological barriers. The review is intended to inform future brain-penetrant TAC therapeutics.