









-
Traditional Chinese Medicine (TCM) has a history of several thousand years and has evolved into a distinct medical system through centuries of clinical practice and progressive theoretical refinement. Its formulations are mainly derived from natural sources, including plants, animals, and minerals[1]. The active ingredients in these natural materials are the basis of the clinical efficacy of TCM. From 1981 to 2019, over 60% of small-molecule drugs approved by the US Food and Drug Administration (FDA) were either directly or indirectly derived from natural products[2]. Accordingly, TCM represents an important natural compound source for innovative drug discovery and development[3].
In practice, TCM is commonly administered either as single herbs or as multi-herb formulas. Astragalus membranaceus is one representative medicinal herb commonly used for fatigue and related symptoms associated with qi deficiency[4]. Multi-herb formulas are more frequently used than single herbs in clinical practice[5]. For instance, Si-Jun-Zi-Tang is a representative TCM formula, which includes Panax ginseng, Atractylodes macrocephala, Poria cocos, as well as Glycyrrhiza uralensis[6]. It is used to treat spleen-stomach qi deficiency and related symptoms, including loss of appetite, abdominal distension, and indigestion[7]. Another representative formula is Niuhuang Jiedu Wan, which contains Bubali cornu, Coptis chinensis, Scutellaria baicalensis, Gardenia jasminoides, borneol, and several other ingredients, and is commonly used to clear heat, remove toxins, or treat conditions like acute upper respiratory tract infection[8]. Additional formulas and TCM preparations, including Yupingfeng Granule, Qingfei Paidu Decoction, Xuebijing Injection, and Qiliqiangxin Capsule, have also shown preventive or therapeutic potential across respiratory, infectious, inflammatory, and cardiovascular conditions[9−14].
Although TCM is widely used in clinical practice, its pharmacological mechanisms remain unclear, especially at the molecular level[1]. To achieve mechanistic understanding, ensure pharmacological standardization, and support clinical translation, systematic identification of TCM molecular targets has become essential[15]. However, this remains challenging[16]. Unlike many conventional drugs that act through relatively defined targets, TCM often works through multiple components, targets, and pathways. The isolation and characterization of bioactive constituents from TCM are often time-consuming, while their chemical composition and stability may differ from source materials and environmental conditions. Resource limitations may further constrain related TCM research in some settings[17].
Recent advances in analytical and experimental technologies have improved the precision and scalability of TCM target research. Cellular Thermal Shift Assay (CETSA), Drug Affinity Responsive Target Stability (DARTS), and other methods can directly verify the interactions between bioactive constituents and their targets. High-throughput screening, based on an automated platform, can rapidly evaluate a large number of compounds and accelerate the identification of promising compounds and potential targets[18]. Despite the continuous progress of modern analytical and experimental technology, TCM target-discovery research remains limited by chemical complexity, pharmacological diversity, and interindividual variability[16].
AI is an important branch of computer science that aims to develop intelligent systems with cognitive functions[19]. In 1956, John McCarthy and others first proposed the concept of 'artificial intelligence' at the Dartmouth Conference, marking the official birth of AI as an independent discipline[20]. In the past few decades, the field of AI has developed rapidly[21]. Recent advances in machine learning, deep learning, and generative AI have increased the ability to analyze complex biomedical and pharmacological data. These advances have created new opportunities for large-scale data mining, pattern recognition, and predictive modeling in drug discovery[22]. In data integration and analysis, deep learning (DL) can model complex feature representations, while machine learning (ML) remains effective for identifying informative patterns across multiple data sources[23]. With these technologies, researchers can extract valuable information from large and diverse datasets and improve decision-making in drug discovery and development[24].
In pharmacology, AI can integrate heterogeneous data through multimodal learning and knowledge mapping, enabling the analysis and interpretation of complex target discovery in TCM research[25]. AI algorithms can support virtual screening and docking-based prioritization of TCM-derived compounds against candidate protein targets, thereby facilitating lead identification and reducing time and cost in early-stage drug discovery[26]. The integration of graph neural networks with network pharmacology provides a framework to infer potential targets and system-level mechanisms of TCM. By integrating multi-omics data, including genomics, transcriptomics, proteomics, and metabolomics, with bioinformatics analysis and multimodal machine learning, researchers can identify potential targets of active TCM ingredients[27]. Current generative AI research is mainly centered on large language models (LLMs) based on Transformer architectures. Specialized generative AI models can efficiently predict potential targets related to TCM by utilizing multi-omics data and algorithms[28]. Although AI-based models still face challenges such as uneven data quality and insufficient algorithm interpretability, AI shows potential in TCM target discovery[29].
This review aims to summarize the application of AI in TCM target discovery and to help researchers in the field better understand its challenges and potential. It focuses on the roles of AI technologies in identifying targets and discusses experimental strategies for validating AI-based predictions. The importance of experimental confirmation for obtaining reliable and biologically relevant results is emphasized. Based on a systematic overview of current methods and research progress, the review highlights limitations of existing studies and proposes directions for future work to support the integration of AI and TCM and further exploration of TCM mechanisms. Figure 1 shows the schematic diagram of AI-assisted target discovery in traditional TCM.
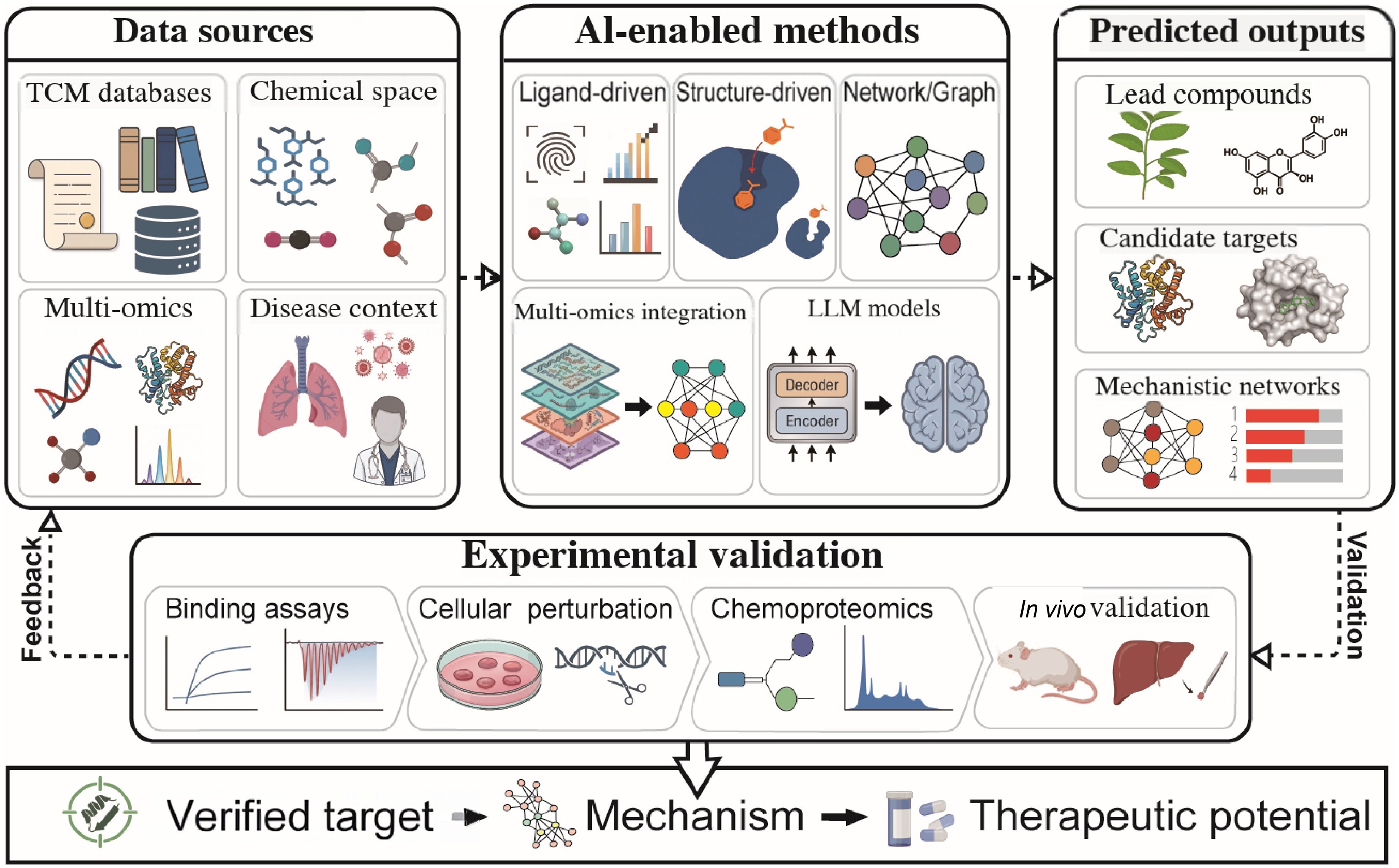
Figure 1.
Schematic diagram of AI-assisted target discovery in traditional Chinese medicine (TCM). The workflow integrates multiple data sources, including TCM databases, chemical space, multi-omics data, and clinical data, and infers potential TCM targets through ligand-driven, structure-driven, network-/graph-theory-based, multi-omics integration, large language model (LLM), and other AI-driven target prediction strategies. The predicted targets are then validated through biochemical binding experiments, cell-perturbation experiments, chemical proteomics, and in vivo pharmacological evaluation.
-
TCM databases are crucial data sources for target discovery research and are of great importance for AI-assisted target discovery[5]. With the rise of data-driven approaches in TCM research, a range of databases have been developed to collect data on TCM compounds, TCM formulas, chemical compounds, targets, and disease relationships[30]. These databases include ITCM[31], HIT 2.0[32], TCM Bank[33], TCMIO[34], ETCM v2.0[35], HERB[36], and SuperTCM[37], which contain data on TCM-derived compounds, predicted or verified potential targets, and pharmacological effects (Table 1).
Besides TCM-related sources, AI-assisted target discovery also benefits from a wide range of public biomedical resources that provide complementary chemical, pharmacological, genomic, and disease-related information[23]. These include chemical and bioactivity-related resources such as PubChem[38], ChEMBL[39], and BindingDB[40], which provide extensive information on chemical structures, bioactive constituents, and experimentally supported compound–target interactions. Similarly, drug-related resources such as DrugBank[33], DrugCentral[41], and SuperDrug2[42] offer a large amount of information regarding drug properties and mechanisms. On the other hand, target- and disease-related sources such as GeneCards[43], Therapeutic Target Database (TTD)[44], and DisGeNET[45] provide information on genes, targets, and diseases that support the understanding of targets and mechanisms.
Table 1. Summary of major TCM-related databases and biological resources for AI-driven target discovery.
Database name Last update Data types Description Websites Target types Ref. ETCM v2.0 2023 48,442 prescriptions, 38,298 ingredients, 1,040 targets, 9,872 patent drugs Links TCM formulas and herbs to targets and diseases to reveal holistic mechanisms. www.tcmip.cn/ETCM2/front Predicted and experimentally validated [35] TCM bank 2023 9,192 herbs, 61,966 ingredients, 15,179 targets, 32,529 diseases Provides chemical structures and ADMET properties for screening
and target prediction.https://tcmbank.cn/ Predicted and experimentally validated [46] HERB (v2.0) 2024 7,263 herbs, 49,258 ingredients, 12,933 targets Integrates clinical trials with experimental data to validate herb–target interactions. http://herb.ac.cn/v2 Predicted and experimentally validated [47] ITCM 2023 25,857 prescriptions, 8,454 herbs, 43,430 ingredients, 1,488 sequencing profiles Focuses on the 'herb-ingredient-target' network to explain formula mechanisms. http://itcm.biotcm.net/browse.html Predicted and experimentally validated [31] Hit 2.0 2021 1,237 ingredients, 2,208 target sites, 10,031 active pairs Offers experimentally verified ingredient-target pairs with binding affinity data. www.badd-cao.net:2345 Experimentally validated [32] TM-MC 2.0 2024 635 herbs, 34,107 compounds, 13,992 targets, 27,997 diseases Connects herbs and ingredients to molecular targets and metabolic enzymes. https://tm-mc.kr/
Predicted[48] TCM-suite 2022 DNA marker sequences, Holmes/Watson module A cloud platform for drug property prediction and network pharmacology analysis. http://tcm-suite.aimicrobiome.cn/ Predicted [49] DCABM-TCM 2023 1,816 blood-entering components, 192 prescriptions, 194 herbs Bridges TCM syndromes and
medical cases with specific biological markers.http://bionet.ncpsb.org.cn/dcabm-tcm/#/Home Predicted and experimentally validated [50] TCMSID 2022 499 herbs, 20,015 ingredients, 3,270 targets Provides structural and botanical data for identifying TCM-specific active ingredients. https://bidd.group/TCMID/ Predicted [51] SuperTCM 2021 6,516 herbs, 55,000 ingredients, 254 pathways Supports target discovery through functional enrichment and network analysis tools. http://tcm.charite.de/supertcm Predicted and experimentally validated [37] SymMap (v2) 2019 1,717 symptoms, 499 herbs, 4,302 targets Maps TCM symptoms to modern diseases and targets via a large knowledge graph. www.symmap.org Predicted and experimentally validated [52] TCMID 2.0 2018 46,929 prescriptions, 43,413 ingredients, 17,521 targets,
MS spectraMaps TCM formulas and herbs to molecular targets and diseases to facilitate network pharmacology
and mechanism study.www.megabionet.org/tcmid Predicted and experimentally validated [53] YaTCM 2018 1,813 prescriptions, 6,220 herbs, 18,697 targets, 47,696 compounds Provides associations between TCM herbs, ingredients, and targets with a focus on ADMET properties and drug-likeness evaluation. http://cadd.pharmacy.nankai.edu.cn/yatcm/home Predicted and experimentally validated [54] TCMSP 2014 499 herbs, 29,384 ingredients, 3,311 targets, ADME properties Links herbs and ingredients to targets and diseases based on ADME properties to evaluate drug-likeness and screening potentials. https://old.tcmsp-e.com/tcmsp.php Predicted [55] ccTCM 2023 273 medicinal materials, 1,449 quantitative components Integrates chemical ingredients
and molecular targets with cancer-related pathways to facilitate TCM-based anti-tumor drug discovery.www.cctcm.org.cn Predicted and experimentally validated [56] PubChem 2026 >100 million compounds, bioassay data Provides extensive chemical structures, bioactivity data, and standardized identifiers to support virtual screening and target interaction studies. https://pubchem.ncbi.nlm.nih.gov/ Experimentally validated [57] ChEMBL 2026 2.8 million substance,
1.8 million bioassaysA large-scale bioactivity database linking drug-like molecules to therapeutic targets through experimentally validated binding and functional assays. www.ebi.ac.uk/chembl Experimentally validated [58] BindingDB 2026 3.2 million data points,
1.4 million compounds, 114,000 targetsFocuses on the quantitative binding affinities of drug-like molecules
with proteins to support molecular docking and QSAR model development.www.bindingdb.org/rwd/bind/index.jsp Experimentally validated [40] DrugBank 2026 13,575 substances, comprehensive target and pathway data Provides detailed drug-target information and pharmacological data to facilitate target identification and drug repurposing for TCM ingredients. https://go.drugbank.com/ Predicted and experimentally validated [33] DrugCentral 2026 Regulatory data, indications, side effects Integrates comprehensive information on approved drugs, including molecular targets, bioactivity, and clinical indications
to support target validation.https://drugcentral.org/ Experimentally validated [41] DRESIS (v2.0) 2025 > 20,000 drugs, metabolic reprogramming data Links TCM ingredients to gene expression profiles and disease-related targets to facilitate mechanism-based drug discovery and repositioning. http://dresis.idrblab.net/ Predicted and experimentally validated [59] DrugMap 2025 33,000 drugs, 50,180 interactions, tissue expression profiles Maps the relationships between drugs, targets, and diseases through integrated network pharmacology to support target identification and mechanism study. https://drugmap.idrblab.net/ Predicted and experimentally validated [60] SuperDrug2 2017 4,587 compositions, 2D/3D structure, PBPK simulation Provides extensive structural and pharmacological data for approved and experimental drugs to facilitate target prediction and repositioning for TCM ingredients. http://bioinf.charite.de/superdrug Predicted and experimentally validated [42] GeneCards 2025 Human genome disease target annotation A comprehensive human gene database providing detailed information on functions, pathways, and associated diseases to support target identification. www.genecards.org Predicted and experimentally validated [61] TTD 2025 932 Validation targets, QSAR model library Provides comprehensive information on clinical and experimental targets, including their associated drugs and clinical trial statuses. https://ttd.idrblab.cn/ Predicted and experimentally validated [44] DisGeNET 2024 GWAS data, clinical variant association Integrates human gene-disease associations from curated databases and literature to support disease-target discovery and mechanism analysis. https://disgenet.com/ Predicted and experimentally validated [45] BATMAN-TCM (v2.0) 2023 Predictive interactions, GO/KEGG analysis An online bioinformatics tool for predicting TCM molecular targets and functional mechanisms through network-based association analysis. http://bionet.ncpsb.org.cn/batman-tcm/index.php Predicted and experimentally validated [62] NPASS 2017 35,032 NPs, 25,041 species, quantitative activity values Provides curated bioactivity and structural data for natural products and their targets to support drug discovery and mechanism studies. http://bidd2.nus.edu.sg/NPASS Experimentally validated [63] Despite differences among these databases in scope, curation strategies, update frequency, and data quality, they collectively provide a comprehensive data resource for AI-driven studies in TCM target discovery. The integration of heterogeneous data from these resources enables more comprehensive analyses of compounds, targets, and diseases, thereby facilitating the identification and validation of potential targets in TCM research[64].
-
Ligand-driven models use known active ingredients as references to infer biological activities or potential target associations without requiring specific structural information about the target. These models are primarily based on the similarity principle, which states that compounds with similar structures tend to exhibit similar biological activities[65]. This consideration is especially relevant in TCM research, as many compounds remain incompletely characterized and may lack corresponding structural information for their targets. Therefore, ligand-driven methods are particularly valuable for hypothesis generation, multi-target strategies, and virtual screening of TCM compounds.
Chemical similarity remains one of the most commonly used approaches in ligand-driven target discovery. It is typically quantified using molecular fingerprints, such as extended connectivity fingerprints (ECFP), combined with similarity metrics like the Tanimoto coefficient[66]. Graph neural networks (GNNs) have demonstrated strong potential for learning molecular representations and can identify compounds with similar biological activities, thereby enhancing chemical similarity-based approaches[67,68].
Ligand-based virtual screening (LBVS) represents another major branch of ligand-driven target discovery (Fig. 2a). This approach ranks candidate compounds according to their similarity to known bioactive constituents, generally using similarity search methods or machine learning classifiers. LBVS is particularly useful when target structural information is unavailable, which is a common situation in TCM-related research. Compared with conventional screening methods, AI-assisted LBVS improves the enrichment of bioactive constituents by capturing nonlinear relationships between molecular features and target-binding activity[69]. Active learning strategies have also been integrated with LBVS to systematically explore TCM compound libraries. This combination facilitates the identification of bioactive constituents while maintaining cost-efficiency[70].
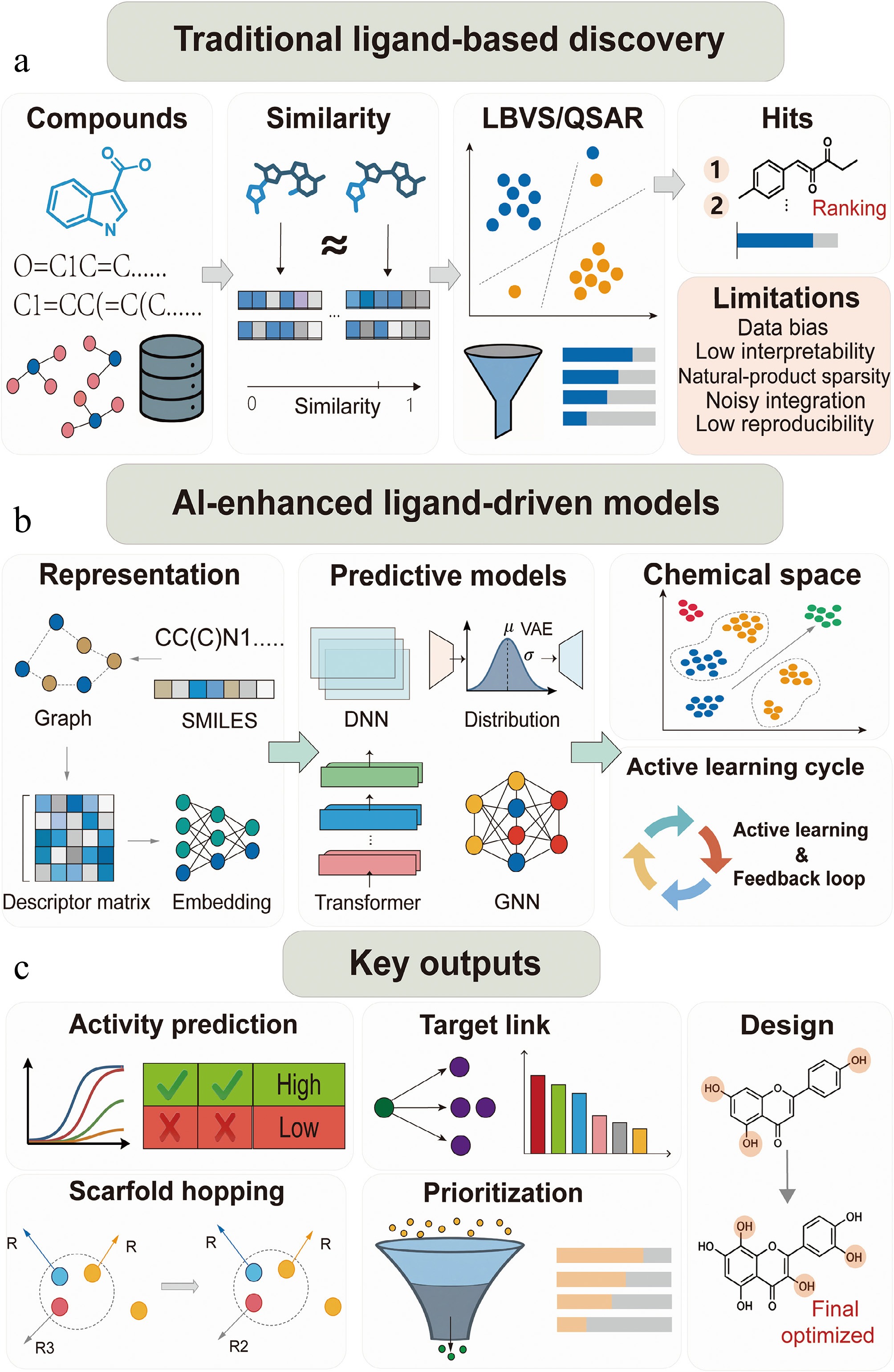
Figure 2.
Integrated ligand-driven target discovery workflow for TCM compounds. (a) Traditional ligand-based discovery: compounds from TCM libraries are screened via chemical similarity and ligand-based virtual screening (LBVS)/quantitative structure–activity relationship (QSAR) models to rank potential bioactive molecules. (b) AI-enhanced ligand-driven models: graph- or simplified molecular input line entry system (SMILES)-based embeddings feed deep neural network (DNN), graph neural network (GNN), or transformer models to predict activity, explore chemical space, and enable scaffold hopping. Active learning cycles iteratively refine predictions with experimental feedback. (c) Key outputs: generates activity predictions, target links, scaffold hopping opportunities, prioritized leads, and informs rational compound design for experimental validation.
For instance, quantitative structure–activity relationship (QSAR) models describe quantitative relationships between molecular properties and bioactivities and have been a fundamental component in ligand-driven discovery for a long time. Traditional QSAR methods rely on statistical models built from predefined molecular properties, whereas recent AI-based QSAR models heavily utilize deep learning neural networks that learn directly from chemical structures[71,72]. GNN-based models have been shown to more accurately describe the nonlinear relationship between TCM molecule structures and target-binding affinities by treating TCM molecules as molecular graphs[73]. Therefore, it is reasonable to believe that AI-based QSAR models have enhanced the robustness and scope of application for ligand-driven target discovery from various types of compounds in TCM (Fig. 2a, c). The Ligand-Transformer model proposed by Zhang et al. does not require compound structures and instead uses protein sequences and small molecule topologies for virtual screening of compounds. It has shown a hit rate greater than 50% and inhibitors with low nanomolar potency[74].
Chemical-space analysis allows for more effective visualization and understanding of the diversity and bioactivities present in a large set of compounds. Dimensionality reduction techniques such as principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and uniform manifold approximation and projection (UMAP) are often used for this purpose, and AI-based embeddings have also been proposed as a tool for better organization and understanding of chemical space and identification of biologically active regions within it[75]. More recent developments in generative models have extended this concept to allow for the projection of TCM bioactive constituents into continuous spaces. Variational autoencoders and other such models have been proposed as a tool for better visualization and understanding of activity cliffs and structurally informative regions and for target-based exploration of compounds (Fig. 2b, c). Thus, it has extended the scope of chemical-space analysis from purely descriptive visualization to more predictive and target-based understanding.
Significantly, AI has greatly enhanced ligand-driven target discovery by improving learning and predictability in molecular representations. Latest reports have shown that DL techniques, especially GNN and transformer models, have the potential to extract various molecular representations from chemical structures and SMILES strings (Fig. 2b), thereby facilitating better ligand similarity assessment and activity prediction compared to other models that use descriptor-based techniques[76,77]. Moreover, AI-driven analysis in chemical spaces has enhanced scaffold-hopping and diversity discovery, especially in the case of natural products and TCM-related compound libraries[78].
Despite these advancements, there are a few challenges. The public bioactivity datasets may be biased toward synthetic drug-like compounds, while the diversity of TCM is not well represented, making it difficult to generalize the results to TCM-derived compounds[79]. In addition, the interpretability of DL algorithms, particularly in QSAR and similarity-based analysis, is not well understood, which may affect the understanding and translation of the results[80]. The most challenging issue is the data integration of different data types, including ligand properties, phenotypic response, and pathway data, into a unified predictive framework, particularly in the presence of inconsistent annotations and noisy data[81].
Future development of ligand-driven AI methods is likely to move toward multimodal or hybrid methods, which integrate chemistry, biology, omics technology, and network data, so as to build richer background information for target inference. This may be particularly useful for understanding the complex formulation characteristics commonly found in TCM-derived compounds. Deep learning methods presented in the form of conditional chemical language models may also contribute to target-directed molecular design because they are able to generate candidate compounds based on specific protein families or disease states. In addition, the integration of ligand similarity analysis, QSAR prediction, structure-based methods, and closed-loop methods for experimental target validation also helps to improve the reliability of computational target prediction. Another important aspect is adaptive or active learning methods, which help reduce false positives and improve the translational relevance of AI methods.
Structure-based target discovery in TCM
-
Structure-driven target discovery employs structural data related to biomolecular targets to predict ligand binding, as well as to clarify the binding process using atomic detail. Unlike ligand-driven methods, where activity is predicted using similarity between compounds, structure-driven methods offer more direct mechanistic insights using physical models of compound interaction within binding sites, as well as changes in conformation that affect ligand recognition[82]. The data integration of AI with conventional physics-based methods has enhanced predictions of protein flexibility, interaction energetics, and even large-scale compound virtual screening[76]. Thus, structure-driven methods have become critical tools for the discovery of binding sites, refining target hypotheses, and even exploring allosteric or cryptic sites, especially in TCM pharmacology[82].
Molecular docking aims to predict how ligands are embedded in protein binding pockets and score potential binding modes according to geometric and energy criteria[83]. Traditional docking algorithms sample the ligand conformational space and rank the resulting poses using scoring functions (Fig. 2a)[84]. Although classical docking tools (e.g., AutoDock, Glide) are still widely used, recent frameworks based on deep learning have improved the accuracy of conformation prediction and scoring. AI models trained based on protein–ligand complexes are able to infer binding interactions that are more closely related to experimental affinity, thereby reducing reliance on approximate scoring functions[82]. The LABind model, which explicitly integrates ligand coding into binding site prediction, was developed by Zhang et al. By combining a graph converter with a cross-attention mechanism, the model can learn local protein–ligand interaction modes and maintain good generalization ability to unseen ligands[85].
New AI-native docking approaches, like transformer-based approaches and unified scoring approaches, show promising efficiency in ligand virtual screening and binding pose prediction for a wide range of targets[86]. With the help of Geometric deep learning and Equivariant Neural Networks, AI-aided docking approaches have unraveled the multi-target regulatory mechanism of Ginsenosides targeting the ACE2 receptor and Mpro protease (Fig. 2b, c)[87].
In structure-based virtual screening (SBVS), ligands are prioritized based on the predicted interactions with a specified target structure. A standard process for SBVS is composed of target preparation, pocket characterization, docking, and ranking (Fig. 3a)[84]. AI improves SBVS by using scoring functions and pose filters that eliminate the problems of heuristic docking score predictions. DL approaches that combine protein–ligand interaction patterns are able to differentiate between binders and false positives (Fig. 3b, c). SBVS is useful for assessing the interactions of various TCM compounds with complex targets and identifying novel associations in complex TCM formulas[83].
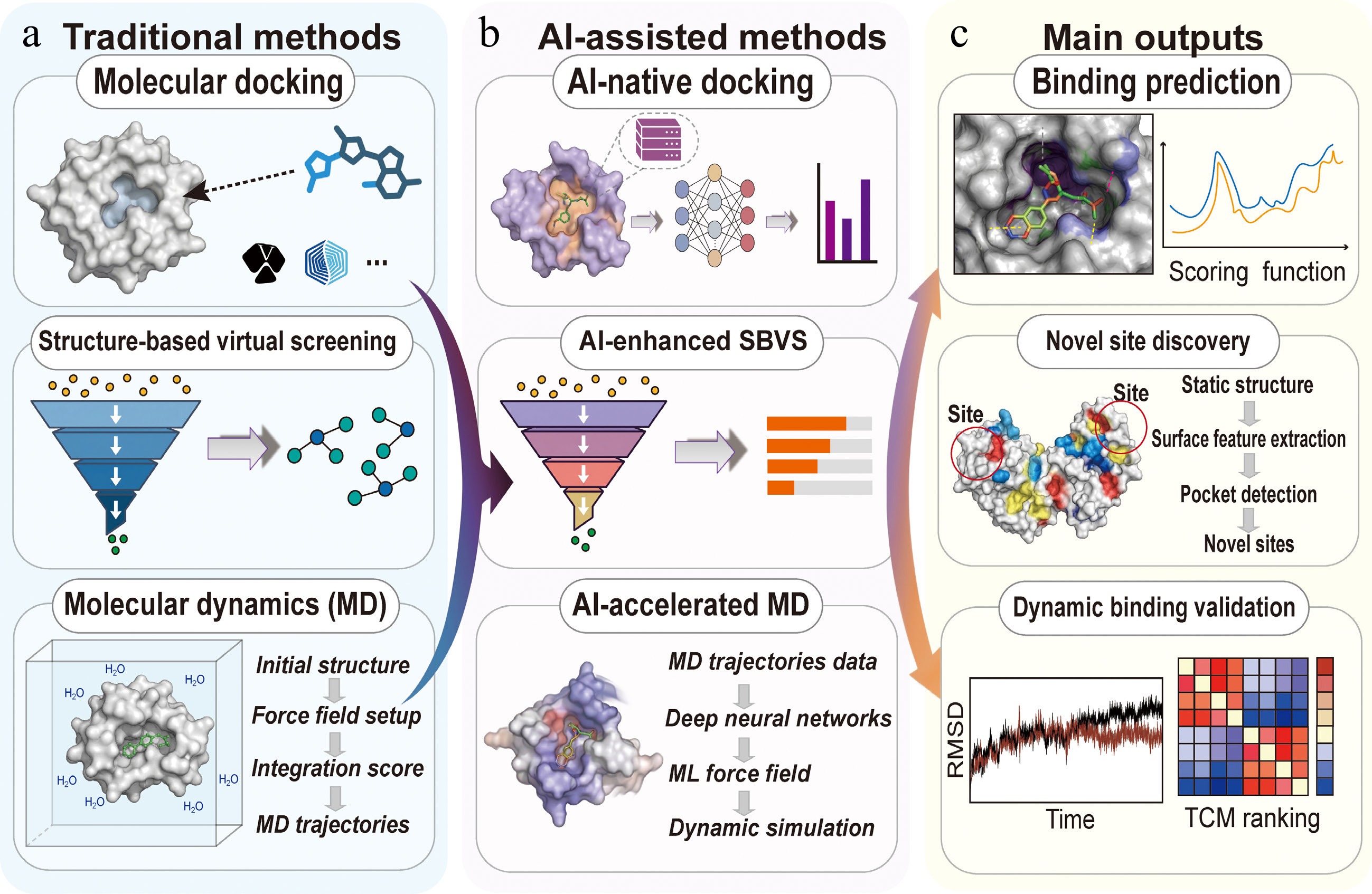
Figure 3.
Integrated structure-driven target discovery workflow. (a) Traditional methods: combines molecular docking, structure-based virtual screening (SBVS), and molecular dynamics (MD) to evaluate ligand-protein binding, rank candidate compounds, and assess interaction stability over time. (b) AI-assisted methods: integrates AI-native docking, AI-enhanced SBVS, and AI-accelerated MD to predict binding affinities, efficiently prioritize compounds, and model dynamic interactions with deep learning refinement. (c) Output applications: highlights core outcomes of target discovery, including binding prediction, binding-site discovery, and dynamic validation, supporting experimental prioritization of TCM compounds while reducing visual complexity for journal readability.
Molecular dynamics (MD) simulation studies are based on the dynamics of the protein-ligand systems, including the time dependence of the systems, induced fit effects, and the stability of the binding process, in a dynamic manner[88]. AI helps MD simulation studies achieve quantum accuracy in TCM natural products while maintaining the speed necessary for protein-ligand systems (Fig. 3b, c). ML helps in the acceleration of MD studies, the identification of important interaction patterns, and the development of enhanced sampling methods, which reduce the computational cost while maintaining the dynamics[89]. In a recent structure-based drug discovery (SBDD) study, MD and SBVS were used on phytocompounds targeting a receptor involved in cancer, showing stable binding with dynamic properties, as observed in vitro studies, highlighting the role of MD in validating target prediction[90].
In structure-based modeling, the accuracy of pose prediction is enhanced by the use of AI for the prediction of conformational patterns, scoring and ranking with deep interaction models that include physicochemical and data-driven features, and the efficiency of library virtual screening with the use of transformer and GNN-based scoring approaches[82].
The major challenge for structure-based AI is target flexibility. Many existing AI models are based on a rather static description of proteins, but the interaction of TCM-derived compounds with their molecular targets is often highly dynamic. Furthermore, the 'Solvation Effect' of the complex physiological environment of TCM decoctions is hard for existing AI approaches to simulate[91]. End-to-end generative models, such as AlphaFold 3, might provide new opportunities for a joint description of protein structure and ligand binding[92]. Further progress might depend on closer data integration with machine learning force fields (MLFFs) and simulation approaches[93].
Network pharmacology-based target discovery in TCM
-
Network pharmacology (NP), a term first proposed by Hopkins in 2007, is grounded in systems biology and network science concepts[94]. It treats drugs, targets, pathways, and diseases as interconnected networks, aligning with the multi-component and multi-target characteristics of TCM[24]. A key feature of NP is the integration of biomolecular interaction data, which enables the combined analysis of multiple compounds and their potential targets, helping to identify major bioactive constituents and their mechanisms of action[15]. In addition, NP provides a new perspective for visualizing complex interactions between TCM-derived compounds and molecular targets[95]. By integrating the concepts of systems biology, multi-omics approaches, and high-throughput analysis, NP has become an important tool for investigating the holistic mechanisms of TCM[96]. However, conventional NP has limitations, including noise accumulation, high dimensionality, and difficulty in integrating dynamic processes[97]. AI provides new opportunities to enhance target discovery (Fig. 4c). The advance is achieved through the integration of ML, DL, and GNNs for the analysis of complex interactions among compounds, targets, pathways, and diseases[27].
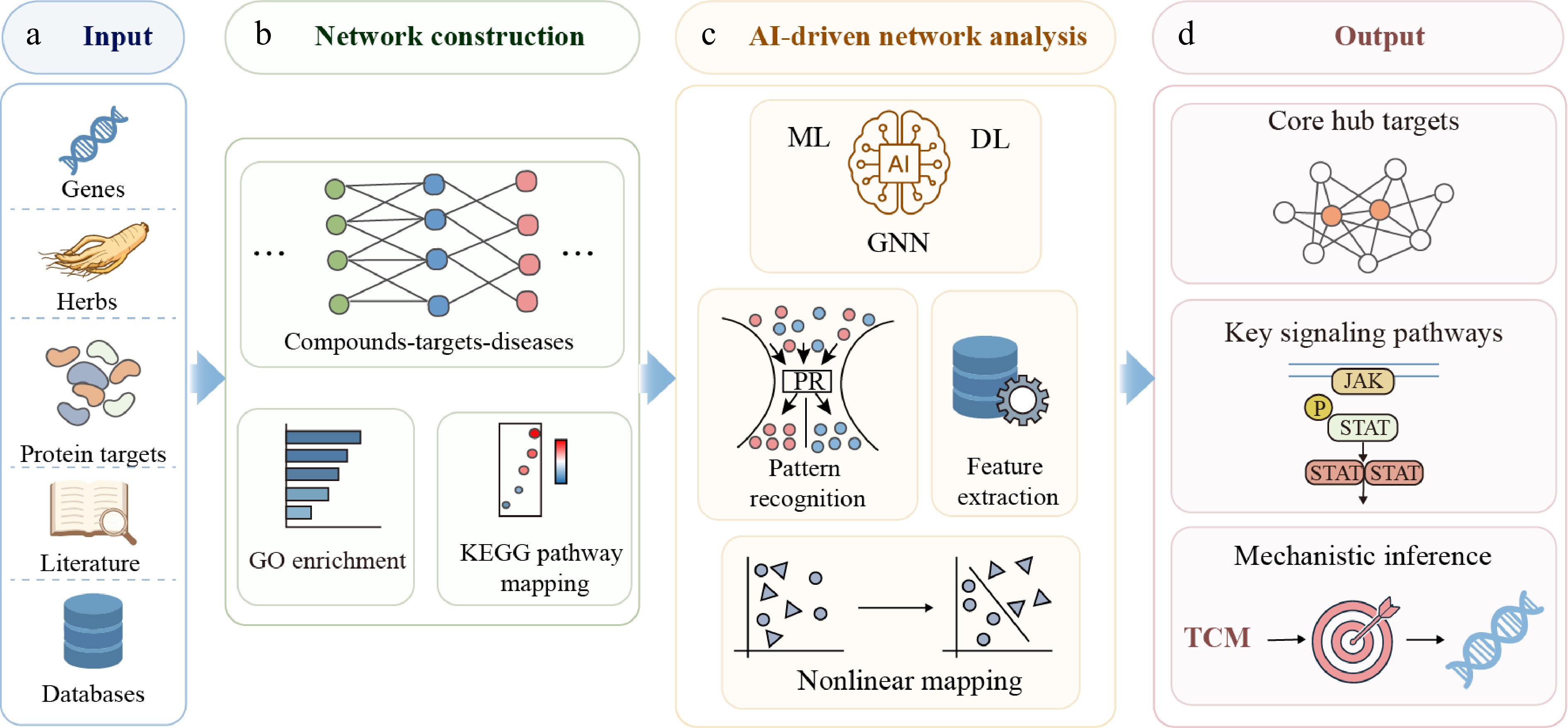
Figure 4.
Schematic workflow of AI-assisted network pharmacology for target discovery in traditional Chinese medicine (TCM). (a) Multi-source input data include genes, herbs, protein targets, literature, and databases. (b) Network construction integrates compound-target-disease associations with Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway mapping. (c) AI-driven network analysis incorporates machine learning (ML), deep learning (DL), and graph neural networks (GNNs) for pattern recognition, feature extraction, and nonlinear mapping. (d) The resulting outputs include core hub targets, key signaling pathways, and mechanistic inference, thereby supporting target identification and biological interpretation in TCM.
ML has become increasingly important in NP for target prioritization, association inference, and network-based prediction (Fig. 4c, d). These methods have shown promise in providing deeper insights into the mechanisms of TCM by identifying potential interactions between compounds, targets, and pathways based on patterns extracted from high-dimensional omics datasets and complex biomolecular networks[98]. For example, Ying et al. analyzed TSGJ-related bioactive components and breast cancer-related targets and identified 160 common targets. Subsequently, they identified five key genes using four ML algorithms, including support vector machines (SVM) and random forests (RF)[99]. A study by Ding et al. identified feature genes and key therapeutic pathways for idiopathic pulmonary fibrosis. They used a combination of LASSO logistic regression and SVM-recursive feature elimination (SVM-RFE) for this analysis[100]. In another study by Zhao et al., potential targets for treating colorectal cancer were identified by combining database-derived targets with differential gene expression analysis and weighted gene co-expression network analysis[101].
DL has the potential to extract and learn complex nonlinear relationships (Fig. 4c)[102]. For instance, Chen et al. proposed a functional representation method for gene features based on DL. The authors showed that applying this method to predict compound–target interactions using the Broad Institute's L1000 dataset was more efficient than using models based solely on gene identity. This DL-based approach led to a substantial increase in the number of predicted compound–target interactions. DL models have also been applied to evaluate the effectiveness of TCM formulas in treating liver cancer by integrating multidimensional data from sources such as The Cancer Genome Atlas (TCGA) and the Traditional Chinese Medicine Systems Pharmacology Database (TCMSP), which may facilitate the identification of potential targets for compound combinations[103]. Qu et al. proposed a DL model based on convolutional neural networks for the data integration of heterogeneous network data and the accurate prediction of drug-target interactions[104]. Zhang et al. proposed a DL-based hybrid model using graph convolutional networks (GCN) and RNN for the accurate target prediction of the meridian associations of bioactive constituent metabolites[105].
Among various DL techniques, GNNs have shown promise in network pharmacology due to their ability to handle graph-structured biological data and identify key hub nodes within complex biological networks (Fig. 4c, d). Graph theory-based GNN models can learn feature representations by propagating information across graph-structured data according to inter-node connections[106]. In this regard, Guo et al. have shown that a GNN-based DL model could be applied for target prediction and virtual screening of compounds with anti-Helicobacter pylori activity using molecular graphs and extended connectivity fingerprints (ECFP)[107]. Similarly, Duan et al. proposed a GNN-based target prediction framework consisting of three modules[108].
However, some challenges still exist in AI-assisted NP analysis. Most AI models are highly dependent on the quality and quantity of input data[109]. Issues such as data imbalance, inconsistent annotation, high collinearity among input features, and substantial noise can compromise the robustness and introduce bias in ML model predictions[23]. Other challenges involve the strong reliance of models such as LASSO regression, SVM-RFE, decision trees, and ensemble methods on structured and high-quality input data[110]. Similarly, clustering approaches may perform poorly in high-dimensional or noisy data environments[24]. In deep learning models, additional limitations include restricted interpretability, dependence on structured experimental datasets, and a high risk of overfitting due to limited training samples. RNN models may be less effective in capturing long-range dependencies in data, and GNN models may be computationally expensive in high-dimensional heterogeneous networks[22].
Recent studies have increasingly emphasized the integration of multiple database resources, cross-modality data fusion, and model-based target validation across various computational approaches[5]. For example, Huang et al.[111] explored the molecular targets and mechanisms of isoliquiritigenin (ISO) in chronic obstructive pulmonary disease (COPD) by using a combination of weighted gene co-expression network analysis, differential expression analysis, and target discovery from TCMSP, Comparative Toxicogenomics Database (CTD), PubChem, GeneCards, and Molecular Signatures Database (MSigDB). In addition, multi-modal fusion frameworks have been proposed. These frameworks integrate genomics, protein structure data, chemical information, pathway networks, and biomarkers into comprehensive input representations. For instance, Lu et al.[112] proposed a multimodal fusion deep learning model that combined SMILES encoding vectors, ECFP fingerprints, and molecular graph information using a transformer encoder together with a bidirectional gated recurrent unit (BiGRU) and graph convolutional networks (GCNs). Their results suggest that integrating information from multiple modalities can improve predictive performance[113].
Multi-omics-based target discovery in TCM
-
Although AI-driven network pharmacology has built a systematic framework for analyzing the multi-component and multi-target properties of TCM, most existing methods still rely mainly on relevance-based inference, which makes it difficult to distinguish between true treatment drivers and spurious associations caused by confounding factors (Fig. 5). This methodological bottleneck makes multi-omics integration and causal inference the next key step to promote the discovery of TCM targets from correlation prediction to causal validation.
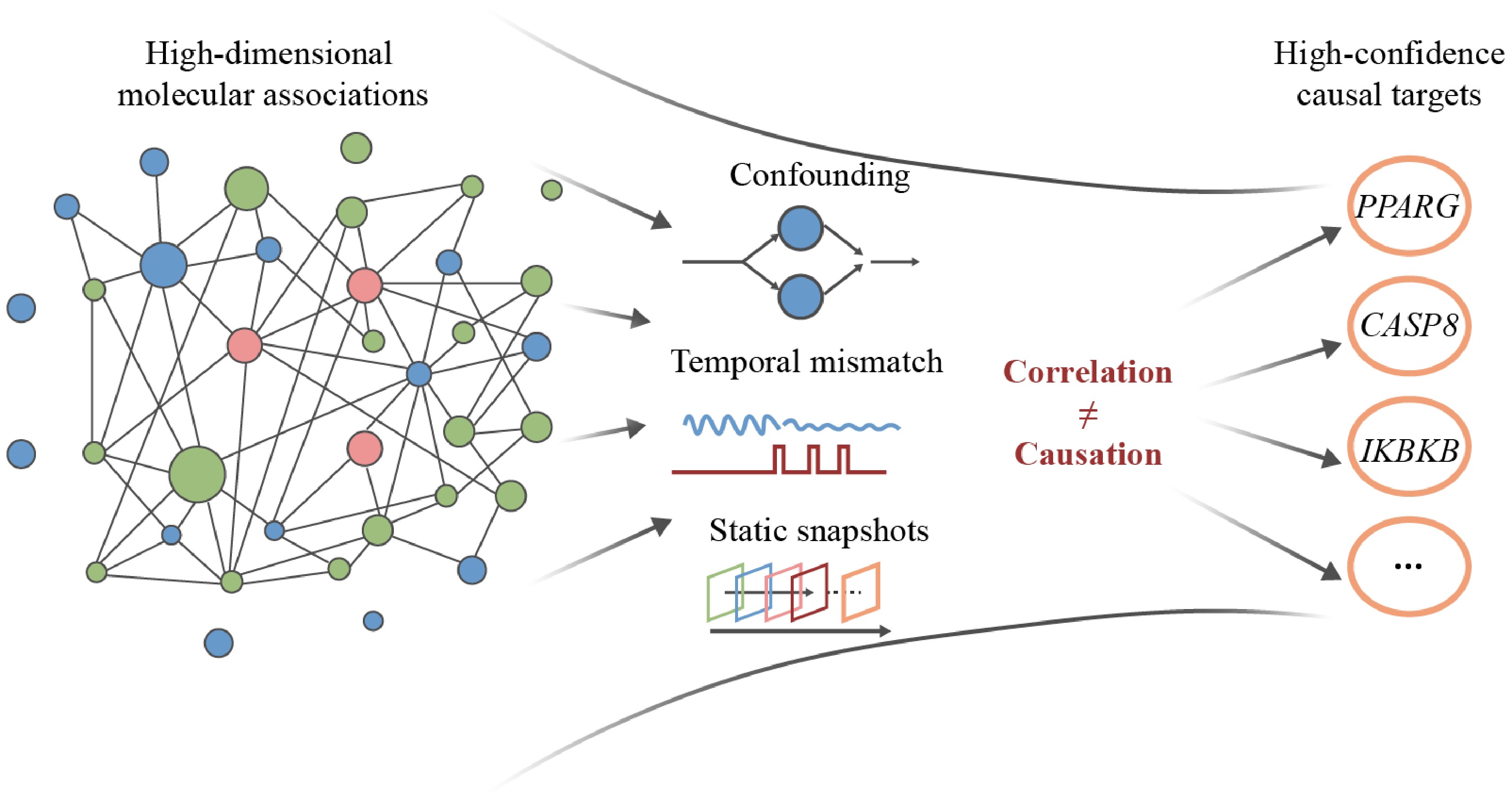
Figure 5.
Schematic overview of methodological challenges in multi-omics-based target discovery in traditional Chinese medicine (TCM). High-dimensional molecular associations are subject to confounding, temporal mismatch, and static omics snapshots, which complicate the distinction between correlation and causation and hinder the identification of high-confidence causal targets.
However, applying multi-omics technology and causal inference to TCM research presents significant challenges. TCM interventions are inherently multi-component, time-dependent, and dynamically adjustable, and are often tailored to patient responses[114], while the vast majority of omics assays can only reflect static molecular snapshots. In addition, Mendelian randomization and other methods are based on lifelong genetic exposure models, which are difficult to directly match with the mode of short-term therapeutic intervention of TCM. If not handled properly, issues including high-dimensional noise, confounding factors, and time-scale mismatch will not only fail to reveal the true causal relationship but also generate spurious associations[114].
Despite these inherent methodological challenges, the integration of multi-omics and AI technology provides an indispensable high-dimensional database for the mechanism analysis of TCM. In recent years, a comprehensive multidimensional analysis framework has been systematically established, which integrates genomics, transcriptomics, proteomics, and metabolomics, providing solid methodological support for decoding the multi-target characteristics of TCM[115]. Under this multidimensional approach, the combination of multi-omics and network pharmacology successfully mapped the intervention network of the TCM system. For example, the comprehensive omics approach revealed that Fuyuan Xingnao Decoction and Huangqi Longdan granules play a role in the treatment of cerebral ischemia by regulating the gut microbial-metabolite axis and systemic signaling pathways, including the Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) pathway and T helper 17/interleukin 17 (Th17/IL-17) axis[116]. In addition, AI algorithms have pushed this convergence upstream to decipher the complex material basis of TCM. Machine learning models have been combined with multi-omics data to decode the highly complex metabolic network of bioactive compounds in Ganoderma lucidum, providing an intelligent prediction model for identifying the effective material basis of TCM[117].
However, although these AI-driven multi-omics studies have greatly enriched the pharmacological network of TCM, they mainly construct high-resolution correlation maps. Therefore, causal inference is needed as a key bridge from complex molecular associations to biologically meaningful target prioritization.
In order to bridge the key gap from statistical correlation to biological causality, researchers began to adapt the causal reasoning framework, especially Mendelian randomization (MR)[118,119], to the unique characteristics of TCM pharmacology. It is worth noting that the high-dimensional multi-omics foundation may support important causal inference analysis: multi-layer omics data may help prioritize candidate genes, improve biological interpretation, and support complementary analyses, such as colocalization and triangulation[120,121], thereby improving the robustness of causal target identification (Fig. 5). However, the core limitation of traditional MR is that it relies on a lifelong genetic exposure model, which cannot directly simulate the short-term therapeutic intervention mode of TCM[122]. In order to address this time-scale mismatch, a practical research paradigm has emerged. MR is first used to identify causal targets associated with disease at the population genetic level and is then used to evaluate the potential of the multi-component regulatory network of TCM formulas to regulate these genetically supported nodes. Recently, this two-step strategy has been applied to multi-omics driven target research in TCM[123].
For example, one study identified seven putative causal targets of rheumatoid arthritis through MR analysis, including CASP8, PPARG, and IKBKB. Subsequent network-based analysis showed that the regulatory network of Guizhi Shaoyao Zhimu Decoction was significantly enriched in these genetically supported targets and may regulate some of these key nodes. Molecular docking further supported the potential interaction between the main active compounds in the formula (such as quercetin and kaempferol) and the identified targets[124]. Although the evidence itself cannot establish direct target involvement or therapeutic causality, it provides a feasible framework for linking genetically supported disease targets with the multi-component action characteristics of TCM formulas. This reverse matching strategy helps to solve the key methodological bottleneck of applying MR to TCM research and provides a feasible framework for linking omics associations with genetically supported therapeutic hypotheses.
However, an ongoing limitation of current TCM omics research is the excessive reliance on cross-sectional sampling[125], which fails to capture the dynamic, time-dependent nature of TCM treatment responses[126]. In order to fully tap the potential of AI-driven causal inference in TCM target discovery, future research efforts must turn to longitudinal and perturbation research designs, combining time-series molecular monitoring and controlled therapeutic intervention[127]. Only by inputting time-resolved multi-omics data into an AI causal reasoning framework[128] can dynamic biological responses be described, rather than static correlations, thus providing a solid and operable foundation for causal target identification and mechanism exploration of TCM.
Generative AI-based target discovery in TCM
-
Whereas multi-omics and causal inference strengthen biologically grounded target prioritization, generative AI offers a complementary route by transforming large-scale unstructured TCM knowledge into computable representations for hypothesis generation and mechanistic reasoning. In recent years, great progress has been made in the application of large language models (LLMs) in TCM research[129]. In addition to their powerful natural language processing capabilities, LLMs can also transform unstructured TCM literature and clinical knowledge into computable structured data representations, thereby providing a basic framework for TCM target inference and pharmacological mechanism analysis. In addition, when combined with the curated structured database and domain knowledge base, LLMs can promote the construction and expansion of TCM knowledge graphs (Fig. 6a, b).
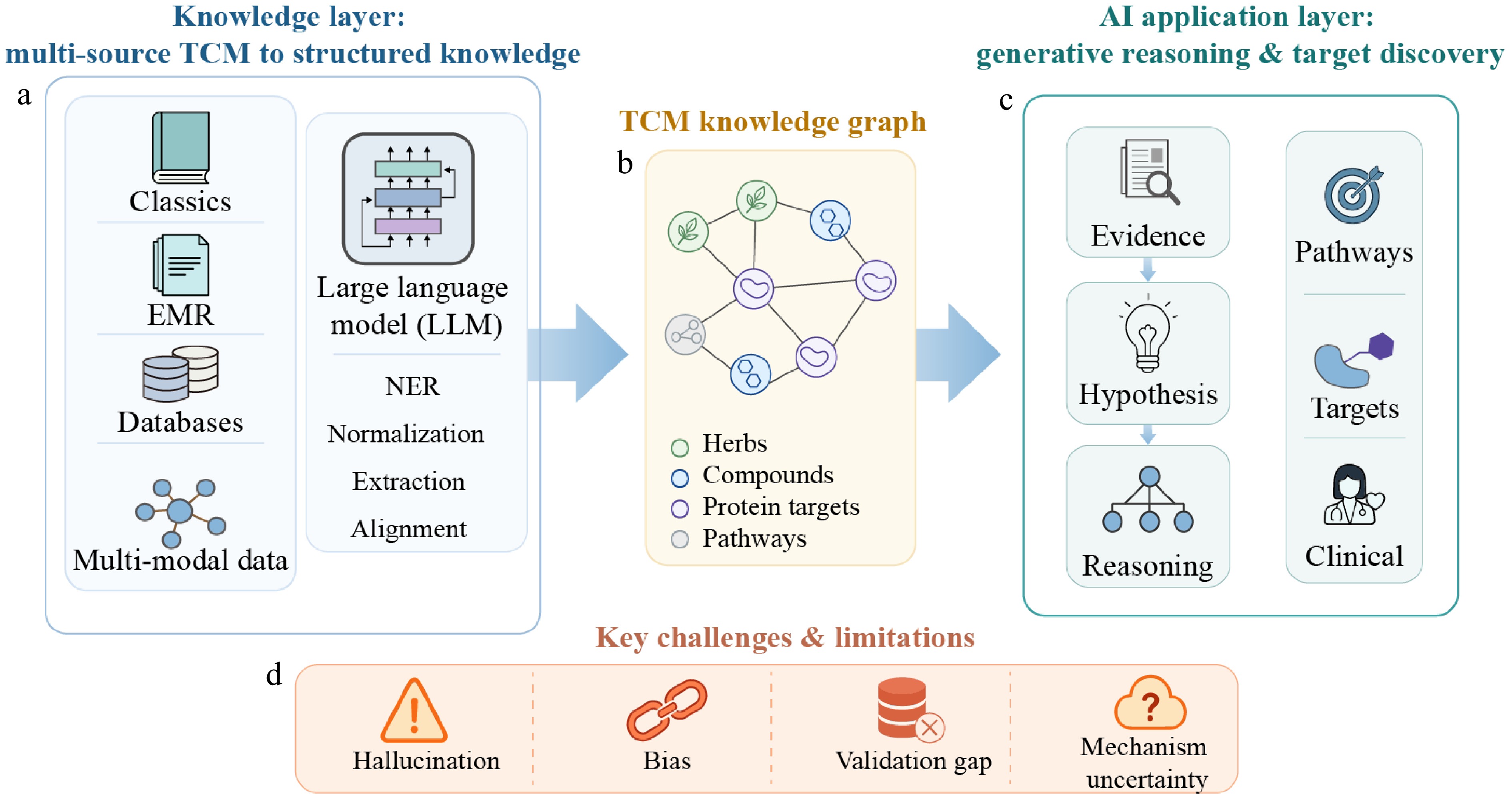
Figure 6.
Schematic framework of generative AI and knowledge graph-based modeling for traditional Chinese medicine (TCM) target discovery. (a) Knowledge layer for transforming multi-source TCM information, including classical texts, electronic medical records (EMRs), databases, and multimodal data, into structured representations with the assistance of large language models (LLMs). (b) Construction of a TCM knowledge graph linking herbs, compounds, protein targets, and pathways. (c) AI application layer for evidence integration, hypothesis generation, reasoning, and downstream applications in pathway analysis, target discovery, and clinical decision support. (d) Key challenges and limitations, including hallucination, bias, validation gaps, and mechanistic uncertainty.
BATMAN-TCM 2.0 is a TCM knowledge resource that focuses on compound–target interactions. By integrating manually curated and computationally predicted data, the platform systematically organized more than 2.3 million high-confidence records of TCM ingredient–target protein interactions[62]. This structured resource provides important support for knowledge graph construction, high-precision knowledge retrieval, and relational reasoning in TCM target discovery and pharmacological mechanism research. The knowledge graph supported by LLM and structured databases may help to represent the complex associations among formulas, herbs, ingredients, targets, and pathways, thus supporting multi-target discovery and mechanism interpretation in TCM research[130].
Although large-scale language models and knowledge graph technologies have made rapid progress, TCM is still one of the most challenging knowledge engineering environments faced by AI systems. The characteristics of TCM knowledge include semantic ambiguity, inconsistent terminology, historical language changes, and a lack of standardized ontology. These characteristics make entity resolution, relationship extraction, and knowledge construction much more difficult than in the modern biomedical field[131]. Therefore, building a reliable TCM knowledge graph is not a simple digitization process but a complex interpretive task that requires careful normalization, expert involvement, and validation. In order to systematically address these challenges and bridge the gap between historical literature and computable insights, a specialized modeling framework is needed (Fig. 6b).
However, the complexity of TCM theory, the profundity of syndrome differentiation, and ancient and abstruse medical classics have brought great challenges to the general LLMs applied in clinical diagnosis and treatment scenarios. In some cases, these problems may even lead to misinformation and model hallucinations, resulting in unsafe clinical recommendations[129, 132,133]. Therefore, in order to solve these key problems, researchers have designed and developed a series of generative AI models specifically used in the field of TCM (Fig. 6c). For example, TCMChat has been developed, which is a large generative model specially tailored for the field of TCM[134].
In order to further improve the organization and reasoning ability of the AI models for complex TCM knowledge, researchers have developed a full-dimensional large TCM model named Tianyi, based on a massive amount of ancient Chinese medicine literature and real-world electronic medical record data. Benchmark evaluations suggest that the model performs well in complex tasks such as syndrome prediction, clinical decision support, and TCM formula generation[135]. The above research results show that the model can help organize and clarify the complex correlation network among syndrome types, formulas, TCM, ingredients, and targets, which may be valuable for mechanistic interpretation and candidate target discovery in TCM research.
In addition, to improve AI-driven medical consultation, researchers have proposed BianQue, a health-oriented model centered on the logic of continuous inquiry. By simulating the iterative questioning process between clinicians and patients, BianQue may help overcome the limitation of single-step recommendations commonly seen in conventional large models. Preliminary studies suggest that, in health consultation scenarios, this strategy may improve the safety of computer-aided diagnosis and the relevance of personalized recommendations[136].
From a research perspective, such interactive models may also facilitate more structured and traceable acquisition of clinically relevant TCM information. However, hallucinations in LLMs remain a major bottleneck for both TCM target discovery and clinical application (Fig. 6d). False compound–target interactions, unsupported mechanistic claims, and plausible but incorrect medical recommendations may all arise if model outputs are generated without retrieval augmentation, graph-based reasoning, traceability, and expert review[130,132]. Therefore, although LLMs and TCM knowledge graphs provide unprecedented opportunities for organizing large-scale unstructured knowledge and exploring complex multi-component relationships, they should currently be primarily positioned as hypothesis generation and knowledge aids, rather than autonomous decision-making systems. Any new targets or mechanistic hypotheses generated by these models must still be validated through a rigorous experimental framework.
In summary, AI-based analytical methods can generate a large number of candidate targets and hypotheses regarding their potential mechanisms of action within a relatively short period[24,137]. However, prediction results often differ in evidence strength, model confidence, cross-method consistency, and biological interpretability[25]. In this context, the transition from computational prediction to experimental investigation should not aim to validate all candidate targets in parallel[137,138]. Rather, it should rely on a stepwise prioritization framework that integrates evidence strength, model confidence, cross-method consistency, biological relevance, druggability, and experimental feasibility[24,25,138]. Within this framework, experimental research serves not merely as a passive confirmation of computational outputs (Supplementary Table S1) but as an active process for target filtering, prioritization, mechanistic refinement, and causal validation[18,138].
-
Following computational prediction and target prioritization, experimental validation is essential to determine whether predicted targets are genuinely engaged and functionally relevant in TCM research. However, computational predictions are often prone to false-positive results because of algorithmic limitations, data bias, and the multicomponent, multitarget nature of TCM systems[25,139]. Therefore, a critical task in AI-driven TCM research is to convert in silico predicted targets into experimentally supported targets through a hierarchical evidence framework. This process requires a progressive, multilevel validation strategy that advances logically from in vitro biochemical binding assays to intracellular target engagement profiling, cellular functional validation, and ultimately, system-level in vivo pharmacological evaluation.
A range of experimental strategies are available to support the hierarchical validation of AI-predicted targets in TCM research, including in vitro biochemical and biophysical assays, intracellular target engagement approaches, cellular perturbation methods, and in vivo pharmacological evaluation (Table 2)[140].
Table 2. Experimental validation strategies for AI-predicted targets in TCM research.
Stage Method Measured readout Strengths and TCM-specific pitfalls Recommended application scenario Ref. In vitro binding SPR Direct binding kinetics (KD, kon, koff) real-time quantitative binding; prone to nonspecific adsorption or aggregation with some polyphenols, quinones, and flavonoids Orthogonal validation of AI-predicted binders/Single compound ★★★/Formula $\chi $ [141] ITC Binding thermodynamics (KD, ΔH, stoichiometry) Gold-standard thermodynamic readout; limited sensitivity for weak-affinity ligands; high concentrations may cause precipitation Secondary confirmation of AI-predicted binders/Single compound ★★★/Formula $\chi $ [140] Intracellular engagement (probe-based) ABPP Active-site occupancy High enzyme specificity; limited applicability to non-covalent compounds and probe-incompatible TCM ingredients Enzyme-target validation/Single compound ★/Formula $\chi $ [146,147] PAL Reversible noncovalent interactions Captures reversible binding; probe derivatization may reduce bioactivity or permeability Target fishing when chemical modification is feasible/Single compound ★/Formula $\chi $ [146,147, 149] Intracellular engagement (label-free) CETSA Cellular thermal stabilization (ΔTm) Label-free in situ engagement; low intracellular exposure may yield false negatives Cellular target engagement validation/Single compound ★★★/Formula ★ [151,152] DARTS Protease resistance shift Simple and scalable; sensitive to high-concentration artifacts and nonspecific protein protection Rapid preliminary target screening/Single compound ★★/Formula ★ [18, 147, 153] TPP Proteome-wide thermal shift profiling Unbiased proteome-wide target discovery; indirect responders or stress-related proteins may appear as hits Identification of indirect or missed targets/Single compound ★★★/Formula ★★★ [151,152] Functional necessity RNAi Knockdown-associated phenotypic change Operationally accessible; incomplete knockdown and off-target effects may compromise causal interpretation Preliminary functional validation of AI-predicted targets/
Single compound ★★/Formula★[150, 154] CRISPR knockout Knockout-dependent loss of drug response Strong causal evidence; partial attenuation may still be informative in multi-target settings Core causal validation of AI-predicted targets/Single compound ★★★/Formula ★ [150, 156, 161] CRISPR screening Functional network hub identification Supports systems-level interpretation; herbal synergy complicates hit annotation Pathway and module validation/Single compound ★★★/Formula ★★★ [156,157, 169] In vivo validation Target-dependent in vivo efficacy Target-linked pharmacological efficacy High translational relevance; multi-component exposure obscures single-ingredient contributions Final in vivo validation of AI-derived hypotheses/Single compound ★★★/Formula ★★★ [150, 161, 174] PK-PD modeling Exposure-response relationship Clarifies formula-level synergy; herb-herb PK interactions complicate modeling Validation of formula compatibility mechanisms/Single compound ★/Formula ★★★ [158, 162] Applicability was rated as follows: ★★★, highly suitable; ★★, moderately suitable; ★, limited suitability; $\chi $, generally not recommended. Ratings reflect practical feasibility, interpretability, and compatibility with AI-guided target validation in TCM research. Methods are organized as a progressive workflow from direct binding to intracellular engagement, functional necessity, and in vivo confirmation. TCM, traditional Chinese medicine; AI, artificial intelligence; SPR, surface plasmon resonance; ITC, isothermal titration calorimetry; ABPP, activity-based protein profiling; PAL, photoaffinity labeling; CETSA, cellular thermal shift assay; DARTS, drug affinity responsive target stability; TPP, thermal proteome profiling; RNAi, RNA interference; PK-PD, pharmacokinetic-pharmacodynamic; PAINS, pan-assay interference compounds. Biochemical and biophysical validation of binding activity
-
It is essential to confirm the in vitro interaction of compounds with their purified target proteins during the early stages of validation, as this provides direct molecular support for predicted targets (Fig. 7a). SPR is a real-time, label-free technique for the quantitative analysis of biomolecular interactions and binding kinetics[141]. SPR has been widely adopted for the validation of target molecules for bioactive components in TCM. While the binding kinetics are primarily characterized by SPR, the thermodynamic analysis is carried out by ITC. ITC is used to measure the changes in heat during binding to determine simultaneously important parameters such as affinity (KD), binding stoichiometry (n), and enthalpy change (ΔH)[142].
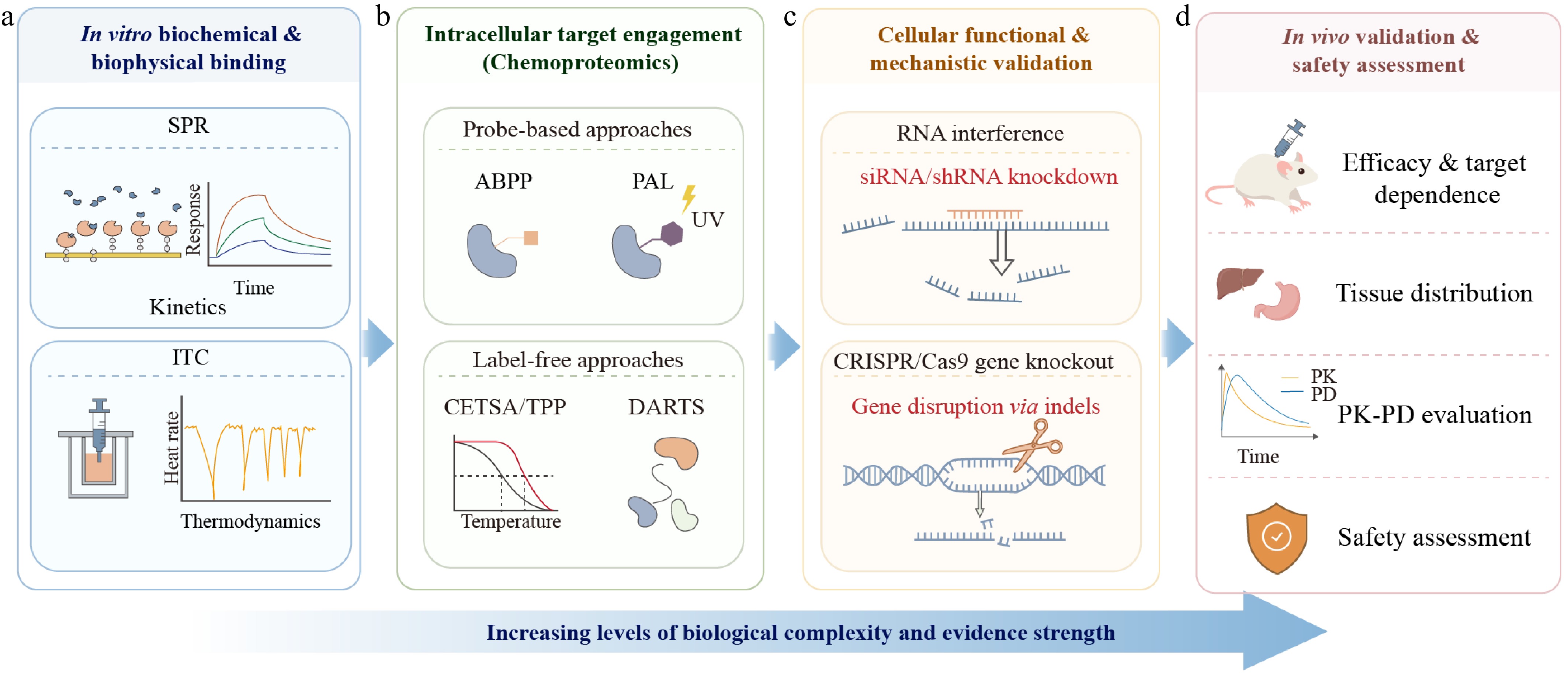
Figure 7.
Hierarchical experimental framework for validation of AI-predicted targets from traditional Chinese medicine (TCM)-derived compounds. (a) Biochemical and biophysical binding validation via surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) using purified target proteins. (b) Intracellular target engagement profiling via chemoproteomics, including probe-based (activity-based protein profiling, ABPP; photoaffinity labeling, PAL) and label-free (thermal proteome profiling, TPP/CETSA; drug affinity responsive target stability, DARTS) approaches, for intracellular target profiling under near-physiological conditions. (c) Cellular functional and mechanistic validation via gene perturbation (RNA interference, siRNA/shRNA; CRISPR/Cas9-mediated gene knockout) to confirm target necessity for the observed pharmacological effects. (d) In vivo validation and safety assessment in animal models, evaluating target-dependent efficacy, pharmacokinetic-pharmacodynamic (PK-PD) profiles, tissue distribution, and safety. From left to right, the framework indicates increasing levels of biological complexity and evidence strength.
However, these methods have inherent limitations when applied to TCM compounds. In particular, polyphenols and quinones may exhibit Pan-Assay Interference Compounds (PAINS)-like behavior or assay-interference risks under certain experimental conditions[143]. They may undergo non-specific colloidal aggregation or nonspecific binding to SPR sensor chips, resulting in deceptive false-positive binding signals[144]. Therefore, it is strongly recommended to add detergents (e.g., Tween-20) to the running buffer or use dynamic light scattering (DLS) to exclude aggregation[145]. In addition, binding to purified proteins does not guarantee binding to targets in living cells, so subsequent intracellular validation is crucial.
Chemoproteomics-based evidence for target engagement
-
Although in vitro biophysical assays, such as SPR and ITC, can confirm the direct interaction between compounds and purified proteins, they cannot guarantee that such binding events actually occur in a highly complex intracellular environment. Chemical proteomics can capture the interaction between drugs and proteins under near-physiological cellular conditions, thus converting in silico predictions into verifiable intracellular molecular evidence and providing direct experimental verification for bridging the gap between in vitro binding and cellular function (Fig. 7b)[146].
Probe-based chemical proteomics approaches
-
In competitive activity-based protein profiling (ABPP), it is possible to determine whether the test compound interacts with the active site of a specific enzyme by analyzing the fluorescence intensity difference of the fluorescently labeled probe compound before and after drug treatment. The traditional ABPP method is most effective when used with ligands that can covalently bind to their target proteins. However, many bioactive compounds derived from TCM bind to proteins through reversible, noncovalent interactions[147]. To extend the utility of ABPP to noncovalent ligands, photoaffinity labeling (PAL) was adapted. PAL involves the use of photoaffinity probes. These probes form covalent bonds with the target amino acids under UV irradiation, thereby enabling the capture of non-covalent interactions that would otherwise be lost[148].
However, the application of probe-based methods to the research of TCM faces a major practical bottleneck. When TCM molecules with complex structures are modified with bulky photosensitive groups and reporter tags (e.g., biotin or alkyne), they often suffer from significant steric hindrance. This modification frequently alters their intrinsic biological activity, target binding profile, or membrane permeability[149,150]. Therefore, for many bioactive ingredients of TCM, the successful synthesis of probes is still challenging, which requires an alternative strategy without modification.
Label-free target engagement approaches
-
Label-free target engagement approaches bypass the need for ligand modification, and can avoid potential interference with endogenous binding interactions. For example, in Cellular Thermal Shift Assay (CETSA), the thermal stability of proteins is usually improved when ligands are bound to proteins. This principle can be used to identify direct interactions between candidate compounds and their predicted targets in whole cells or cell extracts[151]. Thermal proteomics, also known as thermal proteome analysis (TPP), further expands its application by combining this principle with quantitative mass spectrometry. This method can simultaneously measure the thermal stability changes of thousands of proteins in one experiment, thus comprehensively and unbiasedly revealing the interactions between drugs and targets in the proteome[147,152]. Drug Affinity Responsive Target Stability (DARTS) is another major label-free method for target identification. When the ligand binds, the target protein will change its structure, making it resistant to proteolytic degradation. This technique is often used for target validation to demonstrate the direct interaction between small molecules and their target proteins without thermal denaturation[153].
However, in TCM research, it is critical to interpret the results of label-free assays[150,151]. In order to observe significant changes in thermal stability or resistance to proteolysis, methods such as CETSA and DARTS usually require incubation of cells with relatively high concentrations of TCM compounds (e.g., 10−100 μmol·L−1). These experimental doses may significantly exceed their true physiological steady-state concentrations in vivo. Therefore, confirming that these interactions are functionally essential for the pharmacological phenotype is the next step.
Cellular perturbation and mechanistic validation
-
Although chemical proteomics and label-free methods can confirm the physical binding of compounds to intracellular targets, they cannot verify whether such binding events are functionally necessary to drive the pharmacological actions of compounds. To establish a clear causal relationship between target binding and phenotypic outcomes, cellular perturbation techniques at the transcriptional or genomic level must be utilized (Fig. 7c).
RNA interference (RNAi) technologies
-
RNA interference reagents, such as small interfering RNA (siRNA) and short hairpin RNA (shRNA), are widely used tools for post-transcriptional gene silencing, which enable researchers to rapidly test computational predictions at an early stage. For example, studies have shown that siRNA knockdown of AMPK can significantly inhibit berberine-induced apoptosis and autophagy in hepatocellular carcinoma cells. This suggests that the AMPK pathway may mediate an important part of the pharmacological response to berberine[154]. However, RNAi reagents are not always effective enough to knock down target genes and may also exhibit sequence-dependent off-target effects. This problem is particularly prominent in TCM because TCM has the characteristics of being multi-component and multi-target, which may lead to compound off-target effects[155]. Therefore, the data obtained from RNAi-based experiments should generally be regarded as preliminary evidence and need further rigorous verification.
CRISPR/Cas9-mediated gene knockout and screening
-
Unlike RNAi technology, CRISPR/Cas9-mediated gene editing can precisely, irreversibly, and stably disrupt gene function at the genomic level, providing a more reliable method for functional verification. Cell lines with complete gene knockout can be generated to evaluate the effect of gene deletion on the biological activity of compounds, so as to directly determine whether the target gene is necessary for drug activity[155,156]. Genome-wide CRISPR screening has been used to systematically identify important regulatory nodes from a pool of targets identified by computational prediction or omics analysis[157]. Because most bioactive compounds in TCM have the characteristics of a synergistic multi-target network, knockout of a single predicted target gene may only partially attenuate the pharmacological phenotype, but cannot eliminate it[94]. Therefore, researchers should comprehensively interpret CRISPR results and recognize that partial loss of efficacy still constitutes effective evidence for target validation of TCM, rather than rigidly adopting the typical 'all-or-nothing' validation criteria for single-target small-molecule drugs.
In vivo validation and safety assessment
-
Although in vitro and cellular experiments provide strong evidence for target binding and mechanistic necessity, active compounds often fail to translate into in vivo efficacy due to off-target effects, complex tissue distribution, and poor pharmacokinetic (PK) properties. This makes in vivo research the final and indispensable checkpoint in the hierarchical validation framework. Animal models enable researchers to simultaneously assess pharmacokinetics, pharmacodynamics (PD), pharmacokinetic–pharmacodynamic (PK-PD) relationships, and disease progression, thus bridging the critical translational gap between computational predictions and true pharmacological value[158,159].
For bioactive single compounds of TCM, the core goal of in vivo validation is to confirm the target dependence of drug efficacy (Fig. 7d). The validation of GLI1, the core target of berberine against colorectal cancer, is a typical case. Following AI-based target prediction and subsequent validation of target binding and cellular engagement, researchers used tumor-bearing mouse models to confirm that berberine exerts significant antitumor effects by inhibiting the GLI1/STAT3-ferroptosis negative regulator (FNR) pathway, inducing ferroptosis, and interfering with energy metabolism[160]. The key is that the in vivo inhibition of this specific pathway verifies the target dependence of its efficacy[161]. However, for complex TCM formulas, a single rigid target validation paradigm is not enough to meet the research needs. In vivo validation should focus on the compatibility and synergy between the overall target network predicted by AI and TCM formulas. For example, a multi-dimensional PK-PD model was used to verify the compatibility mechanism of Lianhua Qingwen (LHQW). The study provides in vivo system-level evidence for the AI-predicted multi-target network and the principle of TCM compatibility[162].
In addition to validating the efficacy, in vivo studies are equally crucial for evaluating the safety and druggability of AI-predicted targets. Because most AI-predicted targets participate in a variety of physiological processes, long-term pharmacological regulation may produce unexpected biological effects. AI toxicology models can predict potential organ toxicity and off-target risk in advance, but these predictions still need to be systematically evaluated in animal models to comprehensively evaluate their tissue specificity and metabolic stability[115]. This final in vivo verification link closes the translational loop from computational hypothesis to preclinical confirmation and serves as the critical pathway to translate AI-driven TCM target discovery into clinical application.
-
Although the hierarchical experimental validation framework discussed provides a robust way to validate computational predictions, the fundamental reliability of AI-driven target discovery in TCM depends not only on downstream experimental validation but also on upstream parameters, including data quality, computational robustness, modeling bias, and generalization ability, as well as interpretability, traceability, and reproducibility[163,164]. Therefore, these considerations are not only technical barriers but also the basic requirements for the scientific credibility of AI-driven target research in TCM.
Data quality, heterogeneity, scarcity, and concept drift
-
The reliability and upper bound of prediction performance of any AI model is inherently constrained by the quality of its underlying training data[139]. Inconsistent terminology, missing structural annotations, and serious data noise are common problems in TCM datasets[138]. A major downstream consequence of the lack of a universal gold standard for target validation of TCM is that false-positive component–target interaction records continue to accumulate, thereby systematically biasing model training. In addition, there is a serious lack of validated negative interaction data in the research of TCM. Therefore, most AI models have to treat the unobserved interactions as negative samples, thus introducing significant systematic bias. The third persistent challenge is concept drift: the dynamic distribution of newly acquired real-world clinical and experimental data often deviates from the static distribution of the original training dataset, resulting in a gradual decline in the performance of the model in practical applications[165].
In order to solve these data-related bottlenecks, a number of targeted strategies have been proposed. First, the establishment of a standardized TCM terminology ontology and a multi-center data-sharing alliance may help alleviate data heterogeneity, inconsistent labeling, and the lack of universal validation standards. Second, zero-shot learning and few-shot learning frameworks based on transfer learning (e.g., Therapy-related Graph Neural Network [TxGNN]) have successfully improved model generalization in new tasks with limited data[166]. Third, matched negative sampling strategies based on TCM compound structural similarity and target functional classification have been developed to mitigate systematic bias caused by the lack of verified negative samples. Finally, to address concept drift, a continuous quality control and performance evaluation system should be established, with regular model retraining or updating and Bayesian uncertainty quantification to reduce the risks of dynamic data distribution shifts[167].
Computational demands and the need for shared resources
-
Deep learning models for TCM target discovery often require substantial computational resources, relying heavily on graphics processing units (GPUs) and tensor processing units (TPUs), with considerable hardware and operating costs. The interdisciplinary nature of TCM requires substantial computational power: the multi-component and multi-target characteristics of TCM lead to an exponential growth of the potential component-target interaction space[168], while target prediction requires the integration of cross-domain heterogeneous data, including chemical structures, protein targets, gene expression profiles, and TCM syndrome (Zheng) information. At present, there is a lack of open-source pre-trained molecular base models for TCM. This means that most research teams must either spend additional computational resources to build the model architecture from scratch or perform extensive fine-tuning on general chemical models to adapt to the unique multidimensional characteristics of TCM.
There are several practical strategies to address these computational bottlenecks. First, the establishment of an open, shared computing platform and cloud-based TCM-specific pre-trained models can significantly lower the hardware barrier for researchers and avoid redundant model training and waste of computing resources. Second, the development of lightweight AI models for TCM through model pruning and knowledge distillation can reduce the computational requirements while maintaining the prediction performance. Third, the open-source collaborative framework for TCM target prediction can standardize the model training workflow, promote the sharing of pre-trained weights and validation datasets, and further reduce the redundant computational overhead.
Modeling bias, overfitting, and generalization ability
-
Robustness and generalization ability are the core methodological criteria of AI-driven target prediction. Due to data-related factors and model-related factors, there are often significant deviations between in silico predictions and experimental verification results. Overfitting and poor generalization ability are closely related to data bias and data leakage: if the molecular distribution of the training dataset overlaps with the test set too much, the performance of the model will be spuriously inflated[25].
Beyond these general challenges, the unique challenge of TCM lies in its syndrome differentiation system: the same disease may correspond to different TCM syndrome types, and different syndrome types may reflect partially different molecular network states[169]. If the model cannot correctly stratify and identify syndrome types, the efficacy of TCM formulas may be incorrectly attributed to unrelated target pathways, and the model bias may also be manifested in the so-called 'Clever Hans' phenomenon; that is, the model relies on spurious associations to obtain high prediction performance, but does not capture the real biological mechanism[170]. To address these challenges, several targeted strategies have been proposed. First, entropy-based methods and Monte Carlo (MC) dropout have been widely used to estimate prediction uncertainty and improve the reliability of AI models[171]. Second, in order to reduce data leakage and overfitting problems, structure-based dataset splitting strategies have been developed, which exclude training samples with high structural similarity to the test set to ensure the unbiased evaluation of the model[25]. Finally, syndrome-stratified model training and multi-center external validation should be included in the model development workflow to ensure that the prediction results are generalizable across different TCM syndrome types and clinical scenarios.
Interpretability, traceability, and reproducibility
-
The development of interpretable, traceable, and reproducible AI models is the basis for bridging the gap between in silico hypotheses and scientific validity. Interpretability mainly aims at the black-box nature of deep learning: the opaque decision-making process of AI models hinders the biological interpretation of predicted targets and mechanisms[172]. At present, there are two main methods to improve model interpretability: one is to design an inherently interpretable model architecture under the guidance of knowledge in the fields of TCM and biomedical knowledge; the other uses post hoc methods to generate visual and mechanistic interpretations. However, accurately linking abstract AI-derived mathematical features to specific physiological processes in TCM remains a major bottleneck[137].
Traceability refers to the ability to trace the prediction results back to the original training data, processing steps, and model versions. For complex natural products in TCM, a traceable computational workflow can ensure that every step of target prediction is auditable, which is a prerequisite for subsequent experimental verification[173]. To this end, Findable, Accessible, Interoperable, and Reusable (FAIR) principles must be strictly applied to all training data, model codes, and processing protocols to ensure reliable data sharing and result verification[137].
Reproducibility, the cornerstone of scientific inquiry, is facing a severe crisis in the AI era. To address this, AI-driven standardized reporting guidelines for TCM target prediction should be developed, requiring researchers to disclose complete model codes, training datasets, and detailed experimental protocols. At the same time, publishing large-scale, standardized and annotated TCM datasets can effectively reduce batch effects, ensure the consistency of model input, and support robust external validation of AI models[137].
Ultimately, solving the challenges of interpretability, traceability and reproducibility in TCM is not only a technical necessity; it is a prerequisite for legitimately integrating AI-identified targets into both modern pharmacological research and TCM theoretical frameworks. This represents a key way for AI to bridge the gap between high-dimensional modern omics data and the holistic and multi-target therapeutic principles of TCM.
Validation gaps, experimental inconsistencies, and translational challenges
-
Although the above hierarchical experimental evidence framework provides a systematic method to reduce the high false-positive rate in AI-driven target prediction, it still has significant limitations in practical application[174]. First, the current validation technology is largely derived from the research and development of the paradigm of conventional single-target drugs, and its applicability is still limited in fully analyzing the inherent multi-target synergy effect of complex TCM formulas[94]. Second, for the whole chain validation analysis from in vitro biophysical binding to in vivo target-dependent confirmation, there is still a lack of standardized and reproducible experimental protocols between laboratories. This lack of standardization directly leads to poor reproducibility and inconsistent conclusions when AI-driven computational predictions are translated into reliable preclinical and clinical outcomes[94].
To overcome these bottlenecks, future optimization of this framework should integrate cutting-edge biological models. For example, patient-derived organoids (PDOs) can bridge the translational gap between 2D cell cultures and whole-animal models[175], provide a physiologically relevant human-based platform to simultaneously verify the multi-target synergistic effect of TCM formulas and standardize validation workflows[176]. This will not only systematically reduce false positives in AI-driven TCM target prediction but also accelerate the modernization and clinical transformation of TCM.
-
From the perspective of methodological integration and transformation of experimental validation, this review systematically summarizes the application pathways, advantages, and limitations of AI in TCM target discovery, as well as the research framework of synergetic mechanism analysis and target confirmation with experimental validation. Meanwhile, biochemistry, cell biology, chemical proteomics, and in vivo validation strategies are still indispensable for translating computational predictions into targets with biological significance and therapeutic value.
Despite these advances, there are still some major challenges in AI-assisted target discovery of TCM. There are still problems of heterogeneity and uneven quality in the underlying data ecosystem. There are differences in annotation standards, update frequency, experimental evidence level, and the representation of natural products in different databases. Furthermore, extracting true causal targets from high-dimensional multi-omics associations is frequently hindered by confounding variables, temporal mismatches between static omics snapshots and dynamic physiological responses, and the fundamental difficulty of distinguishing correlation from causation. Many existing models still have problems such as overfitting, bias, limited generalization ability, and insufficient representation of chemical diversity and pharmacological complexity of CHM-derived compounds and prescriptions. Black box prediction is still a key bottleneck because its poor interpretability, lack of traceability, and reproducibility make it difficult to translate the computational predictions into experimentally verifiable and clinically meaningful hypotheses. In addition, the dynamics of protein conformation, the complexity of biological background, and the multi-target characteristics of TCM also continue to challenge the existing structure-based and network-based AI models.
Rigorous experimental validation therefore remains indispensable for confirming AI-derived predictions. Current experimental strategies can assess the binding reliability, biological activity, and physiological relevance of candidate targets at multiple levels, thereby providing critical support for computational results and reducing false-positive discoveries. Nevertheless, existing validation technologies still possess methodological limitations, highlighting the need for more robust, scalable, and standardized experimental frameworks. AI prediction and experimental validation can be more closely integrated through iterative feedback mechanisms that continuously improve model training and optimization. Such collaborative workflows may improve the reliability and translational potential of AI-assisted target discovery.
Future progress may depend on several converging directions. A high-quality TCM database is one of the most fundamental requirements, and it is important to build a higher-quality, standardized, and interoperable special dataset for TCM. At the same time, generative AI and foundation models can further broaden the scope of target discovery by improving knowledge extraction, hypothesis generation, candidate drug prioritization, and even targeted molecular design. However, before these models can be reliably applied to research or clinical practice, they need to control their possible hallucinations more strictly, enhance their domain adaptability, and conduct more rigorous benchmarking.
In addition, experimental validation may become more tightly integrated with AI by incorporating iterative feedback to improve model training and establishing a collaborative optimization strategy between AI and experiments. Ultimately, combining AI predictions with rigorous experimental validation and a robust, standardized data infrastructure will accelerate the translational impact of TCM. This integration will bridge the gap between historical empirical practice and modern clinical applications, providing clear, actionable insights for personalized and precision medicine.
-
Not applicable.
-
The authors confirm contributions to the work as follows: conception and design: Zhang W, Tian S; draft manuscript preparation: Guo C, Li Q, analysis and interpretation: Liang W, Lu J, Yue W. All authors reviewed and approved the final version of the manuscript.
-
Data sharing is not applicable to this review as no datasets were generated or analyzed.
-
This research was funded by the Innovative Drug Research and Development-National Science and Technology Major Project (2025ZD1800300), the National Natural Science Foundation of China (82574713, 82430119), the Computational Biology Program (25JS2830400) of Science and Technology Commission of Shanghai Municipality (STCSM), the Shanghai Municipal Science and Technology Major Project (ZD2021CY001), the Chenguang Program of Shanghai Education Development Foundation, and Shanghai Municipal Education Commission (23CGA45, Saisai Tian), the Natural Science Foundation of Shanghai (25ZR1402574), the ability establishment of sustainable use for valuable Chinese medicine resources (2060302), and the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (2023-I2M-3-009).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/targetome-0026-0027.
-
#Authors contributed equally: Chengyang Guo, Qingmeng Li
- Supplementary Table S1 Comparison of AI-enabled target discovery methods in TCM research.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Guo C, Li Q, Liang W, Lu J, Yue W, et al. 2026. AI-enabled target discovery in traditional Chinese medicine: from computational prediction to experimental validation. Targetome 2(3): e029 doi: 10.48130/targetome-0026-0027 

AI-enabled target discovery in traditional Chinese medicine: from computational prediction to experimental validation
- Received: 26 April 2026
- Revised: 29 May 2026
- Accepted: 03 June 2026
- Published online: 30 June 2026
Abstract: Traditional Chinese Medicine (TCM), with a history of thousands of years, has been increasingly supported by modern clinical studies for its effectiveness in disease prevention, treatment, and rehabilitation. However, the complex, multi-component, and multi-target characteristics of TCM make it challenging to identify bioactive compounds and understand the molecular mechanisms underlying their effects. These features also complicate the standardization of clinical applications. However, investigation of TCM targets remains limited. Systems biology and network pharmacology have become valuable approaches for analyzing the complex multi-component and multi-target interactions of TCM. Omics-based approaches, including transcriptomics and single-cell sequencing, have further improved the ability to characterize gene expression changes and cell heterogeneity. Recently, artificial intelligence (AI) has emerged as a powerful tool for identifying potential targets within TCM. The continuous improvement of professional TCM databases supports the integration of information, including prescriptions, bioactive constituents, target interactions, disease associations, and pharmacological annotations, gradually building a more comprehensive data system for TCM research. These databases serve as key resources, allowing AI-based approaches to identify targets in TCM. This review summarizes recent progress in AI-assisted TCM target identification, discusses experimental strategies for validating computational predictions, and outlines the main methodological challenges and future directions.