









-
ADP-ribosylation is a class of NAD+-dependent post-translational modifications that can be rapidly installed and reversibly regulated, serving as a central hub in the DNA damage response, chromatin organization, transcriptional regulation, and cellular stress signaling[1−5]. Among these, poly(ADP-ribosyl)ation (PARylation) is catalyzed by a subset of PARP family enzymes, which synthesize poly(ADP-ribose) (PAR) chains on target proteins or on PARPs themselves, thereby coupling cellular metabolic state (NAD+ availability) to genome maintenance and cell-fate decisions[6−8]. Aberrant activation or dysregulation of PARylation has been repeatedly linked to tumorigenesis, cancer progression, and therapy resistance[9−13]. Importantly, PAR functions not only as a covalent modifier but also as a signaling scaffold that recruits and organizes a diverse set of PAR-binding reader proteins[12,14−16]. Locally produced PAR can rapidly assemble a dynamic and plastic interaction platform, promoting protein complex assembly and coordinating downstream responses[8,17−19]. Therefore, comprehensive profiling and identification of PAR-interacting proteins will not only substantially advance our understanding of PARylation's biological functions and cellular regulatory networks, but also expedite the development of novel therapeutic strategies for PAR-associated pathologies[20−23].
Recent advances have enabled the direct interrogation of PAR-interacting proteins through chemically or enzymatically synthesized PAR probes coupled with proteomic approaches[20,24,25]. One strategy employs chemically synthesized mono-, di-, and tri-ADP-ribose probes bearing a biotin tag to enrich interacting proteins from human cell lysates via streptavidin pulldown, followed by LC-MS/MS identification[26]. This structurally defined system enables reproducible and comparative profiling of chain length-dependent binding preferences. An alternative approach utilizes photoaffinity PAR probes of defined lengths (e.g., ~8-mer and ~40-mer) generated by site-specific incorporation of a benzophenone crosslinker into biotin-tagged PAR[27]. Following incubation with nuclear extracts and UV-induced crosslinking, streptavidin-based affinity isolation and LC-MS/MS allowed systematic comparison of length-dependent PAR interactomes and revealed proteins that selectively recognize long PAR polymers. A third methodology involves a bifunctional NAD+ analog bearing a C2-diazirine photo-crosslinker and a 3'-azido group, designed as an efficient substrate for PARP1 auto-PARylation to produce PAR chains equipped with both a photoactivatable moiety and a click-compatible handle[28]. This strategy preserves native PAR-protein interaction contexts and significantly enhances the recovery of direct, PARylation-dependent interactors. Together, these methodologies offer complementary capabilities in structural control, stabilization of weak or transient interactions, and systematic investigation of polymer-length effects. However, chemical modifications randomly introduced at internal ADP-ribose units of PAR may alter its native conformation, potentially compromising binding affinity and specificity toward certain readers and erasers, and thereby leading to reduced capture efficiency or biased outcomes[26,28,29]. When such PAR chains interact with PAR binding domains, the internal modifications may cause steric hindrance at the binding interface, leading to reduced binding affinity or false negative results. Therefore, further refinements are needed to better recapitulate the multivalent architecture of endogenous PAR, minimize heterogeneity introduced by probe synthesis and labeling, and improve the resolution and comparability of PAR-centered interaction networks. Based on these considerations, we envisioned that a natural PAR probe carrying a photo-crosslinker selectively positioned at the terminal ADP-ribose unit could serve as a more efficient and universally compatible tool for profiling PAR-interacting proteins under native interaction contexts.
Herein, we designed and synthesized a novel photo-clickable and chain-terminating NAD+ analogue (PCCT-NAD+) that enables the generation of PAR polymers bearing a terminal photo-clickable ADP-ribose unit (Fig. 1a). This design preserves the native multivalent architecture of PAR and minimizes structural heterogeneity by restricting functionalization to the terminal unit (Fig. 1b), thereby avoiding internal modifications that might interfere with the critical interactions between PAR and its binding proteins.
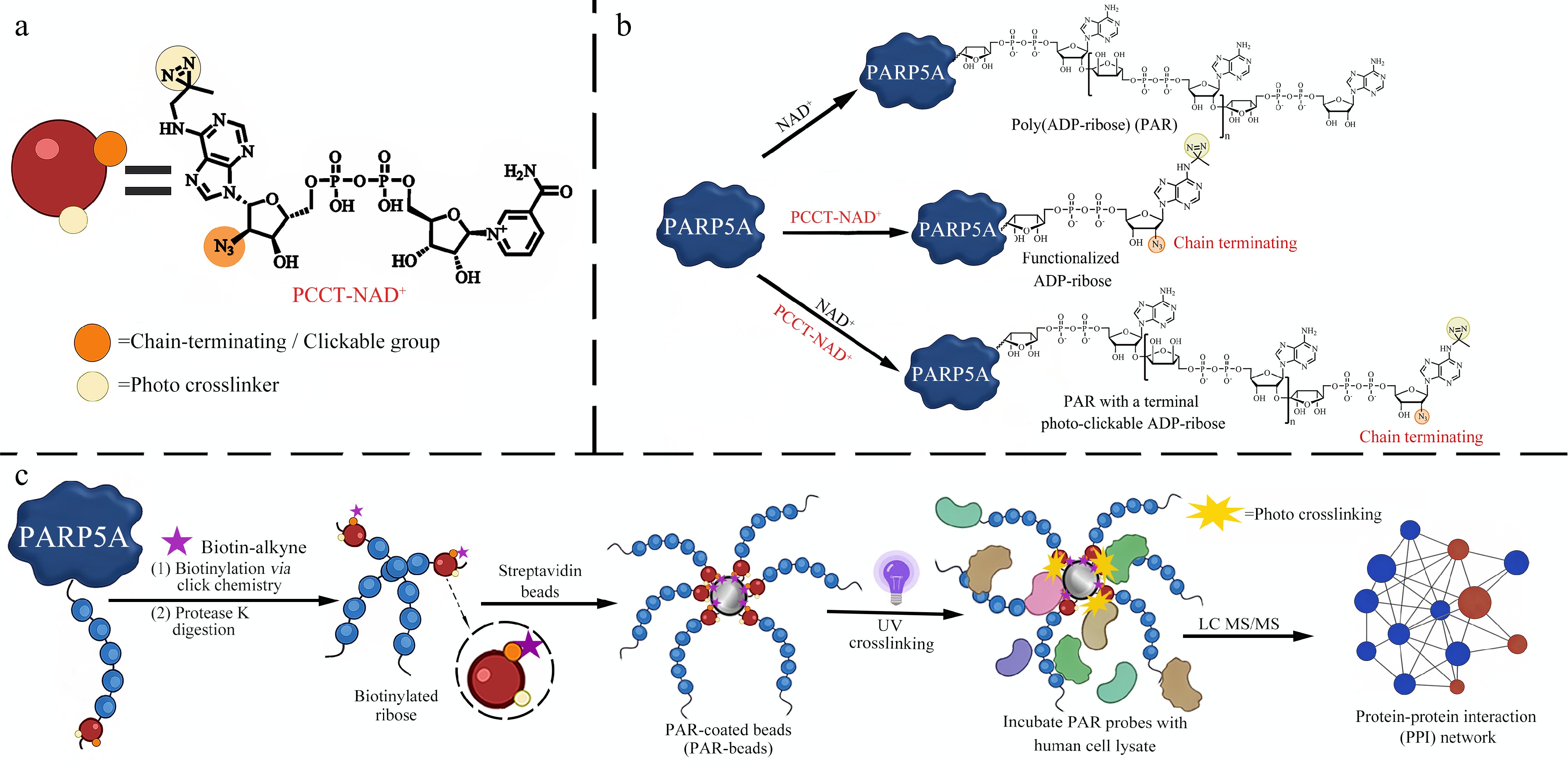
Figure 1.
Design and application of the PCCT-NAD+ probe for PAR interactome profiling. (a) Chemical structure of PCCT-NAD+. (b) ADP-ribosylation catalyzed by PARP5A using NAD+, PCCT-NAD+, or a mixture of NAD+ and PCCT-NAD+. (c) Workflow for PAR interactome profiling: functionalized PAR polymers are immobilized onto magnetic beads, followed by photo-crosslinking with cell lysates to enable efficient and unbiased capture of PAR-interacting proteins under native interaction contexts.
This design allows a more faithful representation of PAR-dependent interaction networks. Leveraging this probe, we developed a high-specificity, high-efficiency PAR interactome profiling strategy: following PARP5A-catalyzed synthesis using PCCT-NAD+, the resulting PAR polymers were immobilized on magnetic beads via click-chemistry-mediated biotinylation, subjected to photo-crosslinking with proteins in cell lysates, enriched, and identified by quantitative proteomics (Fig. 1c). We applied this platform to systematically profile PAR-interacting proteins under oxidative stress, revealing extensive remodeling of the PAR interactome and identifying translation-related pathways as major functional nodes. Our findings expand the functional paradigm of PARylation beyond DNA repair to encompass translational control and proteostasis regulation.
-
To characterize the reactivity of PCCT-NAD+ in PARP5A-catalyzed ADP-ribose chain extension, an in vitro PARP5A assay was established and analyzed via click chemistry labeling coupled with immunoblotting. Reactions were performed in a total volume of 100 μL in buffer containing Tris-HCl (100 mmol•L−1, pH 8.0), NaCl (200 mmol•L−1), MgCl2 (20 mmol•L−1), and dithiothreitol (DTT, 0.2 mmol•L−1). Unless stated otherwise, each reaction contained purified PARP5A catalytic domain (Supplementary Fig. S1a, Supplementary Table S1) (10 μmol•L−1, prepared in-house), NAD+ (1 mmol•L−1), and PCCT-NAD+ (0.2 mmol•L−1, synthesized in this study). To confirm NAD+ dependence and identify the incorporation stage of PCCT-NAD+ during chain extension, five conditions were tested: (1) PARP5A + NAD+ + PCCT-NAD+ (PCCT-NAD+ and NAD+ added simultaneously), (2) condition (1) supplemented with olaparib (100 μmol•L−1, Selleck Chemicals) as a catalytic inhibition control, (3) PARP5A + PCCT-NAD+ (without NAD+) to assess whether PCCT-NAD+ can be incorporated in the absence of chain extension, (4) PARP5A pre-incubated with NAD+ for 2 h to initiate elongation, followed by addition of PCCT-NAD+ and continued incubation to test incorporation during ongoing extension and its potential to terminate further elongation, and (5) PARP5A pre-incubated with PCCT-NAD+ for 2 h, followed by addition of NAD+ and continued incubation to evaluate whether PCCT-NAD+ pre-association affects subsequent chain extension and its mode of incorporation. Unless noted above, reactions were incubated at 30 °C for 2 h and were terminated by the addition of olaparib (final concentration, 100 μmol•L−1, Selleck Chemicals). All reactions were performed in triplicate (n = 3 independent experiments).
After termination, reactions were subjected to click-chemistry labeling at 37 °C for 2 h in a total volume of 45 μL containing 30 μL of the reaction mixture, tris(3-hydroxypropyltriazolylmethyl)amine (THPTA, 2 mmol•L−1, Sigma-Aldrich), CuSO4 (1 mmol•L−1, Sigma-Aldrich), alkyne-biotin (100 μmol•L−1, Click Chemistry Tools), and sodium ascorbate (10 mmol•L−1, Sigma-Aldrich) prepared in phosphate-buffered saline (PBS, HyClone). Labeled samples were resolved by SDS-PAGE (12% gels) and transferred onto PVDF membranes (Millipore). Incorporation of PCCT-NAD+ was detected using a streptavidin-HRP conjugate (1:10,000, SA00001-0, Proteintech), and total PARP5A was detected with a mouse anti-His tag monoclonal antibody (1:5,000, AE003, ABclonal Technology) as a loading control, followed by an HRP-conjugated goat anti-mouse IgG (H + L) secondary antibody (1:5,000, AS003, ABclonal Technology). Chemiluminescence was developed using BeyoECL Star (P0018AM, Beyotime).
Synthesis, purification, and bead-based immobilization of photo-crosslinkable PAR for capture of interacting proteins
-
PAR was synthesized under the same reaction conditions as described for condition (1) above, with the total reaction volume scaled up to 2 mL. To prevent residual PARP5A in the reaction mixture from interfering with downstream pull-down experiments, PARP5A was digested with proteinase K (100 μmol•L−1, P6157, Macklin) by incubating the reaction at 60 °C for 30 min. The mixture was then transferred to a 10 kDa molecular-weight-cutoff centrifugal filter unit, and buffer exchange was performed with 10 mL phosphate-buffered saline (PBS) to remove proteinase K, free biotin, and other low-molecular-weight contaminants. The sample was concentrated by centrifugation at 4,500 × g, and the final volume was adjusted to 2 mL for subsequent pull-down assays. To characterize PAR chains' length, an aliquot of the PAR complex was analyzed by native 20% polyacrylamide gel electrophoresis (native PAGE [20% gel, 1 × TBE]) using xylene cyanol, orange G, and bromophenol blue as migration markers, followed by silver staining. Chain lengths were determined via orange G (corresponding to an ADP-ribose monomer), bromophenol blue (corresponding to an ADP-ribose 8-mer), and xylene cyanol (corresponding to an ADP-ribose 20-mer). PAR was indirectly quantified prior to PARP5A removal and then immobilized onto magnetic beads at a ratio of 200 μL beads per 400 μg PAR to generate PAR-conjugated beads. To remove unbound/excess PAR, beads were separated on a magnetic rack and subjected to sequential high-stringency washes at 4 °C with gentle end-over-end rotation: two washes with PBS containing 4 mol•L−1 urea (10 min each), three washes with PBS containing 1% NP-40 (P0013F, Beyotime, 10 min each), two washes with 50 mmol•L−1 ammonium bicarbonate (10 min each), two washes with PBS (10 min each), and finally two washes with 20% acetonitrile (10 min each). The resulting beads were referred to as PAR-beads and used for subsequent capture assays.
To validate photo-crosslinking-based capture, 50 μL PAR-beads were incubated with 50 μg of the established PAR-binding protein GST-WWE (Supplementary Fig. S1b; Supplementary Table S1). Bead-protein mixtures were preincubated for 10 min to facilitate complex formation and then irradiated with 365 nm UV light for 15 min (lamp-to-sample distance, 2 cm). After irradiation, samples were returned to 4 °C and incubated overnight with gentle end-over-end rotation. Non-irradiated controls were processed in parallel under identical conditions without UV exposure. Beads lacking immobilized PAR were included as negative controls and were similarly divided into UV-irradiated and non-irradiated groups to assess the contribution of the PCCT-NAD+-encoded photo-crosslinking moiety to protein capture. After incubation, beads were washed extensively, and bound proteins were eluted for immunoblotting; captured GST-WWE was detected using an anti-GST-HRP conjugate (K000478P, Solarbio). Parallel pull-down assays using GST-Ub (Supplementary Fig. S1c; Supplementary Table S1) under identical conditions were performed as a non-specific binding control.
Cell culture and oxidative stress treatment
-
HeLa cells (QuiCell-H860, QuiCell) were maintained in DMEM supplemented with 10% fetal bovine serum at 37 °C in a humidified incubator with 5% CO2. Cells were seeded in 10-cm culture dishes and grown to approximately 70%–80% confluency before treatment. To induce oxidative stress, cells were treated with H2O2 (final concentration, 500 μmol•L−1) for 2 h, while DMSO vehicle control cells received an equal volume of DMSO as the vehicle control. After treatment, cells were rapidly washed with ice-cold PBS and collected for preparation of cell lysates for subsequent enrichment of PAR-interacting proteins. Each condition was prepared in triplicate (n = 3 independent biological replicates).
Microscale thermophoresis (MST) assay
-
Auto PARylation reactions of purified PARP5a at 10 µmol•L−1 were carried out overnight at 30 °C in 100 µL of assay buffer containing 100 mmol•L−1 Tris-HCl at pH 8.0, 50 mmol•L−1 NaCl, 20 mmol•L−1 MgCl2, 0.2 mmol•L−1 DTT, and a total NAD+ (native NAD+ or a mixture of NAD+ probe and native NAD+) concentration of 5 mmol•L−1. Two different NAD+ analogue-to-native NAD+ mixtures were prepared: the control mixture was PC-NAD+ mixed with native NAD+ at a ratio of 1 to 1; the test mixture was PCCT-NAD+ mixed with native NAD+ at a ratio of 1 to 5. A blank control containing 100 µmol•L−1 BSA in the same assay buffer was processed in parallel. Purified GST-AF1521 Macro-EGFP, GST-WWE-EGFP, or RPLP-EGFP proteins (Supplementary Fig. S1b and S1d, Supplementary Table S1) were dialyzed into PBS before use. Each auto-PARylation reaction mixture was serially diluted twofold in PBS to generate 16 concentration points. For binding assays, 10 µL of each dilution was combined with 10 µL of the respective purified protein at a final concentration of 0.1 µmol•L−1 and incubated at room temperature for 15 min. MST data were then acquired at 20% infrared laser power using a Monolith NT.115 instrument. Data analysis was performed with MO. Affinity Analysis Software v2.3 to determine the dissociation constant Kd. The binding affinities of the PAR polymers generated from the two mixtures to both the AF1521 Macro and the WWE domain were determined in parallel.
Enrichment and photo-crosslinking capture of PAR-interacting proteins using PAR-beads
-
Following validation of PAR immobilization and UV-induced photo-crosslinking capture, PAR-beads were used to enrich putative PAR-interacting proteins from HeLa cell lysates. Cell lysates were prepared using RIPA lysis buffer (P0038, Beyotime) supplemented with 1 mmol•L−1 PMSF, incubated on ice for 30 min, and centrifuged at 12,000 × g for 15 min at 4 °C to remove insoluble debris. Lysates were prepared from an H2O2-treated group and a DMSO-treated control group, and each condition was prepared in triplicate. For each enrichment experiment, 100 μL of PAR-immobilized beads were incubated with 400 μg of total cellular protein lysate at 4 °C with gentle end-over-end rotation overnight to allow formation of PAR-protein complexes. To stabilize PAR-protein interactions, lysate-beads mixtures were preincubated for 10 min and then irradiated with 365 nm UV light for 15 min at an approximate distance of 2 cm, after which samples were returned to 4 °C and incubated with rotation for the remaining time.
After photo-crosslinking, beads were separated using a magnetic rack and subjected to sequential high-stringency washes to remove nonspecifically bound proteins. Washing steps included two washes with PBS containing 4 mol•L−1 urea (10 min each, rotating at 4 °C), three washes with PBS containing 1% NP-40 (P0013F, Beyotime, 10 min each, rotating at 4 °C), two washes with 50 mmol•L−1 ammonium bicarbonate (10 min each, rotating at 4 °C), two washes with PBS (10 min each, rotating at 4 °C), and two washes with 20% acetonitrile (10 min each, rotating at 4 °C). After the final wash, beads were split into two portions: one portion was boiled in 2 × SDS sample buffer (62.5 mmol•L−1 Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol) at 95 °C for 10 min and analyzed by SDS-PAGE followed by silver staining using a silver stain kit (P0017S, Beyotime) to qualitatively assess protein enrichment, and the remaining portion was processed for subsequent LC-MS/MS analysis. Parallel experiments using beads without immobilized PAR were performed as negative controls to assess nonspecific binding.
On-bead digestion and peptide desalting
-
Separation of the magnetic bead was carried out on a magnetic rack for 2 min. The bead was resuspended in reduction and alkylation reagent (20 µL, 100 mmol•L−1 ammonium bicarbonate, 5 mmol•L−1 tris[2-carboxyethyl]phosphine [TCEP], 10 mmol•L−1 chloroacetamide [CAA], 0.1% sodium deoxycholate [SDC]) and incubated at 95 °C/1,300 r•min−1 for 5 min. After heating, the sample was cooled to room temperature. A 3 µL amount of Trypsin/Lys-C mix (Thermo Scientific [Rockford, USA]) was added and mixed well, then the sample was digested at 37 °C/1,300 r•min−1 for 2 h. A 2.3 µL amount of 25% trifluoroacetic acid (TFA) was added and mixed to terminate the digestion reaction. The sample was centrifuged at 12,000 r•min−1 for 15 min, and the supernatant was collected for subsequent desalting. 100 μL of Wash Buffer (0.2% TFA) was added to the desalting column, followed by centrifugation at 700 × g for 1 min; this step was repeated once. A 50 μL amount of elution buffer (90% ACN, 0.2% TFA) was added to the desalting column, followed by centrifugation at 700 × g for 1 min; elution was performed twice, and the final sample volume was 100 μL. The sample was concentrated using a vacuum freeze centrifuge concentrator, reconstituted with Mobile Phase A, and subjected to LC-MS analysis.
LC-MS/MS analysis and mass spectrometry data research
-
For each sample, 500 ng of total peptides were separated and analyzed with a nano-UPLC (Vanquish neo) coupled to an Astral zoom instrument (Thermo Scientific) with a nano-electrospray ion source. Separation was performed using a reversed-phase column (EASY-Spray™ HPLC [150 μm × 15 cm], Thermo Scientific, USA). Mobile phases were H2O with 0.1% FA (phase A) and 80% ACN with 0.1% FA (phase B). Data independent acquisition (DIA) was performed in profile and positive mode with an Orbitrap analyzer at a resolution of 240 K and a m/z range of 380−980 for MS1, and an m/z range of 150−2,000 for MS2; absolute AGC value: 5E4; maximum injection time: 3 ms. The MS/MS spectra were acquired using HCD with a normalized collision energy (NCE) of 25% and an isolation window of 2 m/z.
For DIA data processing, a species-specific spectral library was constructed using Data-Dependent Acquisition (DDA) from samples matching our experimental conditions. DIA data analysis was performed using DIA-NN software v1.8.1. False discovery rate (FDR) filtering criteria were set at < 0.01 at both the precursor and protein levels. In this study, the corresponding proteins were obtained after applying the relevant FDR threshold controls. Missing values were then filled with half of the minimum value. Additionally, a normalization method was employed in the data analysis (optional). The final dataset, containing protein numbers, sample names, and normalized protein quantification information, was imported into the SIMCA18.0.1 software package (Sartorius Stedim Data Analytics AB, Umea, Sweden) for multivariate analysis. Data was scaled and logarithmically transformed to minimize the impact of both noise and high variance of the variables. After these transformations, PCA (principal component analysis), an unsupervised analysis that reduces the dimension of the data, was carried out to visualize the distribution and the grouping of the samples. A 95% percent confidence interval in the PCA score plot was used as the threshold to identify potential outliers in the dataset.
In order to visualize group separation and find significantly differentially expressed proteins, Student's t-test (When there are biological replicates) was applied. Thresholds of P-value < 0.05 and (FOLD CHANGE ≤ 0.83 or FOLD CHANGE ≥ 1.2) will be applied to screen for differentially expressed proteins. Considering the errors brought by multiple hypothesis testing, the R package 'fdrtool' will be used to correct the P-values. In addition, databases including KEGG (
www.genome.jp/kegg ) were used for pathway enrichment analysis. Public databases GO (www.geneontology.org ), COG (www.ncbi.nlm.nih.gov/research/cog ), and STRING (https://cn.string-db.org/ ) will be used for functional analysis of the identified proteins.Data analysis and validation of candidate PAR-interacting proteins
-
Differential enrichment between the H2O2-treated and control groups was assessed based on three independent biological replicates. Volcano plot analysis was performed using thresholds of FC ≥ 1.2 or FC ≤ 0.83 and P < 0.05, with P values subjected to multiple-testing correction, yielding 1,576 differentially enriched proteins. To select representative candidates for validation, proteins were prioritized by integrating enrichment magnitude and direction, physicochemical properties relevant to PAR binding (including net charge characteristics), subcellular localization (nuclear/cytoplasmic distribution), and functional annotations. RPLP1 and FCF1 were chosen as proteins showing pronounced downregulation and upregulation, respectively, whereas PRIM2 and EIF2S1 were included as additional candidates displaying comparatively modest changes.
To validate PAR binding by the candidate proteins, recombinant proteins were expressed and purified in vitro. RPLP1 was cloned into the pET-28 vector to generate a His-tagged construct (plasmid synthesized by Jiutian Gene), whereas FCF1, PRIM2, and EIF2S1 were cloned into pGEX-6P-1 to express GST-tagged proteins (Supplementary Fig. S1e−S1h, Supplementary Table S1). Recombinant proteins were purified using Ni-NTA or GST affinity chromatography (resins were purchased from Tiandirenhe). PAR-containing substrates for membrane overlay were generated in vitro using a PARP5A- and NAD+-containing reaction system, followed by removal of PARP5A as described above. Reaction products were resolved by SDS-PAGE and transferred onto PVDF membranes. Membranes were blocked with 5% BSA and incubated with purified RPLP1, FCF1, PRIM2, or EIF2S1 diluted to a final concentration of 20 μmol•L−1 in PBST containing 0.05% Tween-20 at room temperature for 3 h. As a negative control, recombinant ubiquitin (UB) was incubated with a separate membrane under identical conditions. After washing, membranes were incubated with an anti-GST tag antibody-HRP conjugate (1:5,000, K000478P, Solarbio) at room temperature for 1 h. An anti-His antibody was used to detect the substrate protein PARP5A as a loading control (1:5,000, AE003, ABclonal Technology). Signals were developed using a chemiluminescent substrate, and band intensities were quantified using ImageJ software.
To validate interactions at the endogenous level, pull-down assays were performed using PAR-conjugated beads. HeLa cells were treated with or without 1 mmol•L−1 H2O2 for 1 h, lysed in RIPA buffer with protease inhibitors. PAR-conjugated beads (50 μL) or unconjugated control beads were incubated with 500 μg lysate (BCA-normalized) in binding buffer (PBS-0.1% NP-40) overnight at 4 °C. After washing (PBS-0.1% NP-40, then PBS), bound proteins were eluted by boiling in 2 × SDS loading buffer, separated by SDS-PAGE, and immunoblotted with antibodies against RPLP1, FCF1, PRIM2, and EIF2S1 (1:1,000 each), followed by HRP-conjugated secondary antibody. Signals were detected by ECL and quantified. To quantify total protein levels of the candidates, whole cell lysates (20 μg per lane) were immunoblotted with the same antibodies and GAPDH as loading control; relative levels were normalized to GAPDH.
Analysis of abundance alteration of cellular RPLP1, FCF1, PRIM2, and EIF2S1 under oxidative stress
-
Cells treated with or without H2O2 were lysed in RIPA lysis buffer supplemented with 1 mmol•L−1 PMSF on ice for 30 min, and lysates were centrifuged to collect supernatants. Protein concentrations were determined using a BCA protein assay kit purchased from Beyotime. Equal amounts of protein were separated on SDS-PAGE gels and transferred onto PVDF membranes. Membranes were blocked with 5% BSA in TBST (0.1% Tween-20) and incubated overnight at 4 °C with primary antibodies against RPLP1, FCF1, PRIM2, and EIF2S1 purchased from ABclonal Technology. After washing, membranes were incubated with HRP-conjugated secondary antibodies (goat anti-rabbit IgG or goat anti-mouse IgG, ABclonal Technology) for 1 h at room temperature. Immunoreactive bands were visualized using enhanced chemiluminescence reagent purchased from Beyotime and quantified using GraphPad Prism 10.
Multi-level validation of RPLP1 expression under oxidative stress
-
To evaluate changes in cellular RPLP1 expression under oxidative stress, RPLP1 levels were examined in HeLa cells at the cellular, protein, and transcript levels. HeLa cells (QuiCell-H860, QuiCell) were maintained in DMEM supplemented with 10% fetal bovine serum at 37 °C in a humidified incubator with 5% CO2. Cells were seeded in 10 cm dishes and grown to 70%−80% confluency before treatment. To induce oxidative stress, cells were treated with hydrogen peroxide (H2O2, final concentration, 500 μmol•L−1) for 2 h, while DMSO vehicle control cells received an equal volume of DMSO as the vehicle control.
For confocal immunofluorescence microscopy, cells were fixed with 4% paraformaldehyde (PFA, G1101, Servicebio) for 30 min after H2O2 treatment, permeabilized with 0.2% Triton X-100 for 10 min, and blocked with 3% bovine serum albumin (BSA) in PBS for 60 min at room temperature. Cells were then incubated with a rabbit anti-RPLP1 primary antibody (1:200, A6725, ABclonal Technology) at 4 °C for 12 h. The primary antibody was diluted in TBST containing 5% BSA (Tween-20, 0.1%). After washing three times with PBST (Tween-20, 0.1%, 10 min each), cells were incubated with a Cy3-conjugated goat anti-rabbit IgG (H + L) secondary antibody (1:200, SAB43732, Bioswamp) for 1 h at room temperature. Nuclei were counterstained with DAPI (1 μg•mL−1) for 20 min at room temperature. Coverslips (diameter, 1.5 cm) were mounted, and images were acquired using a Leica DMi8 confocal laser scanning microscope (Leica Microsystems) equipped with a 63×/1.4 NA PL APO oil-immersion objective. Fluorescence signals were collected in the DAPI (375−435 nm) and Cy3 (541−551 nm) channels, and image acquisition and processing were performed using LAS X software.
For western blot analysis, cells were lysed in RIPA lysis buffer (P0038, Beyotime) purchased from Beyotime supplemented with 1 mmol•L−1 PMSF on ice, and lysates were centrifuged at 12,000 × g for 15 min at 4 °C to remove insoluble debris. Protein concentrations were determined using a BCA protein assay kit (P0010S, Beyotime) purchased from Beyotime. Equal amounts of protein were separated on 12% SDS-PAGE gels and transferred onto PVDF membranes. Membranes were blocked with 5% BSA in TBST (0.1% Tween-20) and incubated overnight at 4 °C with a rabbit anti-RPLP1 antibody (1:2,000, A6725, ABclonal Technology) and a mouse anti-β-tubulin antibody (1:5,000, AC021), which were purchased from ABclonal Technology. After washing, membranes were incubated with HRP-conjugated secondary antibodies (1:10,000, goat anti-rabbit IgG or goat anti-mouse IgG, ABclonal Technology), which were purchased from ABclonal Technology, for 1 h at room temperature. Immunoreactive bands were visualized using an enhanced chemiluminescence reagent (P0018AM, BeyoECL Star, ultra-sensitive ECL chemiluminescence kit) purchased from Beyotime and quantified using GraphPad Prism 10.
For transcript-level analysis, total RNA was extracted using TRIzol RNZol Universal RNA Reagent (lot: B0121A01) purchased from Yeasen Biotechnology according to the manufacturer's instructions. RNA concentration and purity were assessed using an automatic microplate reader (UMR-9600) purchased from Youmi Instruments. Reverse transcription with genomic DNA removal was performed using Hifair® AdvanceFast One-step RT-gDNA Digestion SuperMix for qPCR purchased from Yeasen Biotechnology. Briefly, 10 ng total RNA was used for reverse transcription in a 20 μL reaction containing RNase-free water, 4× Hifai® AdvanceFast One-Step RT SuperMix (5 μL), and gDNA Remover Mix (1 μL). Quantitative PCR was performed using Hieff UNICON® Advanced qPCR SYBR Master Mix purchased from Yeasen Biotechnology on a qPCR instrument, with a total reaction volume of 20 μL. The thermocycling conditions were as follows: initial denaturation at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 10 s and annealing/extension at 60 °C for 30 s. A melting curve analysis was performed using the instrument's default settings to confirm amplification specificity. The following primers were used: RPLP1 forward, 5'-TGGCCTGGCTTGTTTGC-3' and reverse, 5'-CTC GGATTCTTCTTTCTTTGCTT-3', β-actin forward, 5'-GCGTGACATTAAGGAGAAG-3' and reverse, 5'-GAAGGAAGGCTGGAAGAG-3'. Reactions containing RNase-free water instead of cDNA served as no-template controls. Relative mRNA expression levels were calculated using the 2−ΔΔCᴛ method, with β-actin as the internal reference gene, and β-actin and β-tubulin were confirmed to remain stable under oxidative stress conditions. All experiments were performed with three biological replicates. Statistical analyses were conducted using an independent samples t-test, as appropriate.
-
Previous studies have shown that PARP family enzymes exhibit a relatively high tolerance to chemical modification on the adenine moiety of NAD+[30−38]. Meanwhile, PAR chain elongation mechanistically relies on the 2'-hydroxyl group of the terminal ADP-ribose unit as a nucleophile to form the characteristic 2'-1" ribose-ribose glycosidic linkage. Therefore, masking or replacing the 2'-OH is expected to markedly impair chain extension and confer a chain-terminating effect. Consistent with this notion, prior studies have reported that NAD+ analogues lacking a critical hydroxyl group can be processed by ARTs/PARPs to yield chain-terminated PAR products, thereby reducing PAR heterogeneity and enabling subsequent installation of fluorophores or affinity tags via click chemistry[39]. On this basis, we designed and synthesized the novel NAD+ analogue, PCCT-NAD+ (Fig.1a; Supplementary Scheme 1, Supplementary File 1), to enable the generation of PAR polymers that are selectively photo-clickable only at the terminal ribose moiety, thereby closely preserving the structural and functional features of native PAR.
The substrate activity of PCCT-NAD+ was first evaluated through PARP5A auto-PARylation assays (Fig. 2a). Immunoblot analysis demonstrated that incubation of PARP5A with a mixture of PCCT-NAD+ and native NAD+ led to robust PAR polymer formation and confirmed the incorporation of the analogue into the PAR chains (Fig. 2a, lane 1). Treatment with olaparib, a PARP inhibitor, resulted in suppression of PARP5A auto-PARylation by the mixture (Fig. 2a, lane 2). In the absence of native NAD+, PAR chain elongation was completely blocked, with only a single band corresponding to mono-ADP-ribosylated PARP5A detectable upon PCCT-NAD+ incubation (Fig. 2a, lane 3). To delineate the incorporation mechanism of PCCT-NAD+, we systematically varied the substrate addition sequence (Fig. 2a, lanes 4−5). When PCCT-NAD+ was added following a 2-h pre-incubation of PARP5A with native NAD+ at 35 °C, subsequent biotinylation via click chemistry and detection revealed clear PAR signals, demonstrating that the analogue can be incorporated into growing PAR chains during the elongation phase (Fig. 2a, lane 4). Conversely, pre-incubation of PARP5A with PCCT-NAD+ prior to native NAD+ addition completely abolished further chain extension (Fig. 2a, lane 5). Collectively, these results establish PCCT-NAD+ as a photo-clickable, chain-terminating substrate that is incorporated as the terminal unit into growing PAR chains, thereby preventing subsequent ADP-ribose addition.
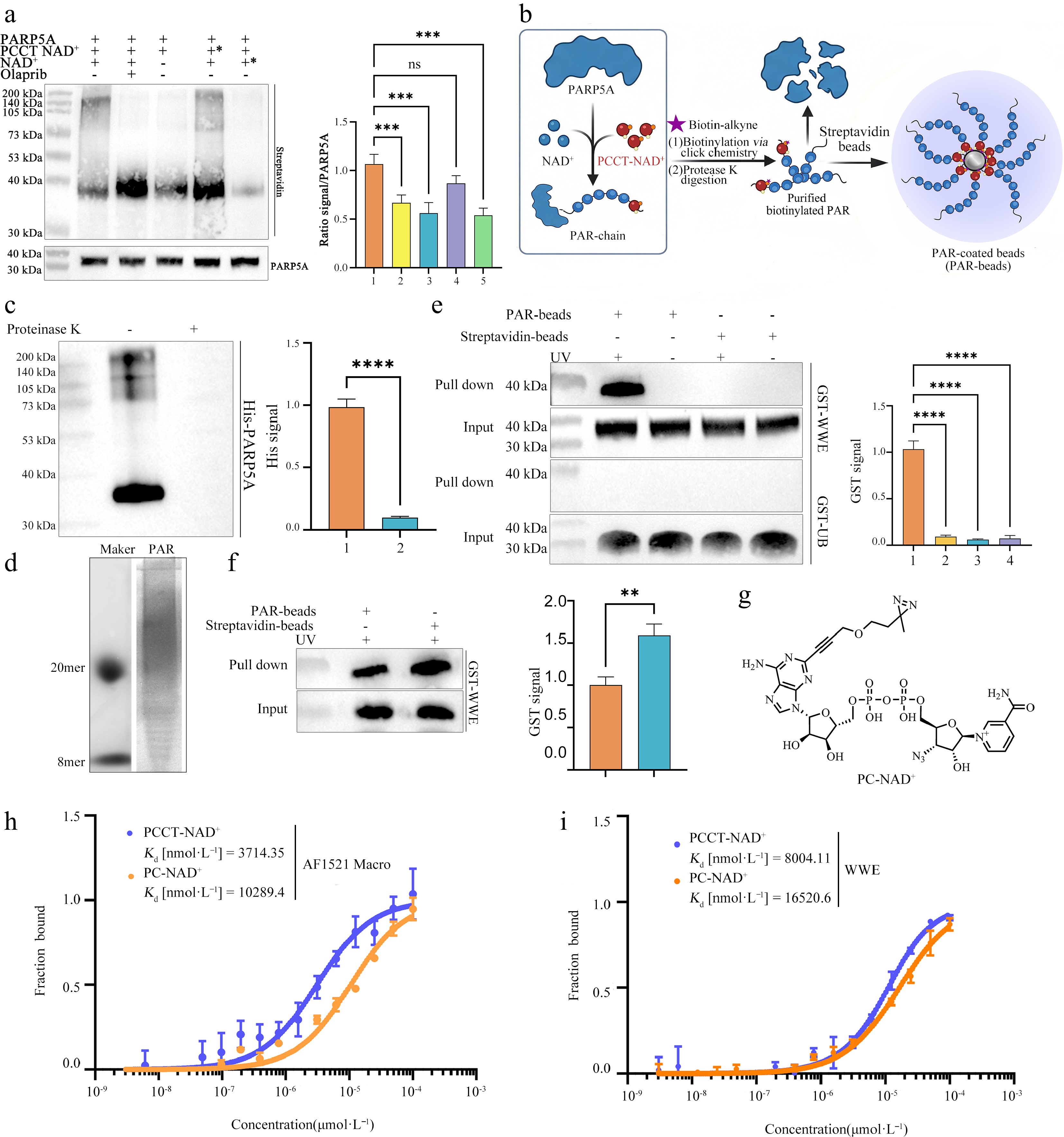
Figure 2.
Construction and functional validation of a bead-immobilized, photo-crosslinkable PAR platform derived from PCCT-NAD+ for specific capture of PAR-binding proteins. (a) In vitro PAR synthesis reactions were performed using recombinant PARP5A (10 μmol•L−1) incubated with NAD+ (1 mmol•L−1) or/and PCCT-NAD+ (0.2 mmol•L−1) at 30 °C for 2 h. Control conditions were included to evaluate NAD+ dependence, inhibition by the PARP inhibitor olaparib (100 μmol•L−1), and the effect of delayed substrate addition. Conditions marked with an asterisk (*) indicate that the indicated component (PCCT-NAD+ or NAD+) was added after an initial 2-h incubation. Following the reactions, samples were subjected to click chemistry at 37 °C for 2 h. Reaction products were resolved by SDS-PAGE and analyzed by streptavidin blotting to detect PCCT-NAD+-labeled PAR polymers. PARP5A was detected as a loading control (lower panel). Right panel shows densitometric quantification of streptavidin signals in the left panel normalized to PARP5A input. (b) Schematic workflow: PAR polymers were synthesized by PARP5A using NAD+ and PCCT-NAD+, followed by proteinase K digestion to remove PARP5A and purify protein-free PAR. The resulting PAR was then covalently immobilized on magnetic streptavidin beads for subsequent interactor capture. (c) Immunoblot analysis of samples before and after proteinase K treatment (100 μg·mL−1, 60 °C, 30 min) using an anti-His antibody to monitor removal of His-tagged PARP5A (left). Right, densitometric quantification of His signal (**** P < 0.0001). (d) Native PAGE analysis showing the chain length distribution of purified PAR polymers. (e) Photo-cross-linking of PAR-beads with model interacting proteins. A 50 μL sample of PAR-beads was incubated with 50 μg GST-WWE for 10 min, followed by 365 nm UV irradiation for 15 min. After overnight incubation at 4 °C, samples were analyzed by immunoblotting with anti-GST-HRP under different PAR/UV conditions. Top, GST-WWE as a PAR reader; bottom, GST-Ub as a non-binder control; right, densitometric quantification (**** P < 0.0001). (f) Assessment of binding efficiency enhancement by bead immobilization. Residual GST-WWE remaining in the supernatant after incubation with PAR-beads or free PAR was detected by immunoblotting with an anti-GST-HRP conjugate (left). Densitometric quantification of residual supernatant GST signal (right). (** P < 0.01). (g) Chemical structure of the previously reported photo-crosslinkable NAD+ analog PC-NAD+. (h), (i) MST binding curves showing the interaction of two target proteins with PAR polymers derived from PCCT-NAD+ (blue) and PC-NAD+ (orange). (h) AF1521 Macro binding to PCCT-NAD+-derived PAR (orange, Kd = 10,289.4 nmol•L−1) and PC-NAD+-derived PAR (blue, Kd = 3,714.35 nmol•L−1). (i) WWE domain binding to PCCT-NAD+-derived PAR (orange, Kd = 16,520.6 nmol•L−1) and PC-NAD+-derived PAR (blue, Kd = 8,004.11 nmol•L−1). Data are presented as mean ± SD, n = 3, ns, not significant, *** P < 0.001. P values were calculated using a two-tailed unpaired Student's t-test.
Construction and validation of a bead-immobilized photo-crosslinkable PAR platform
-
Upon confirming the substrate activity and chain-terminating function of PCCT-NAD+ in PARP5A-catalyzed PARylation, we proceeded to isolate the terminally functionalized PAR polymers. PARylated PARP5A, generated from a mixture of PCCT-NAD+ and native NAD+, was first biotinylated via click chemistry to label the terminal photo-clickable ADP-ribose units. The biotin-labeled PAR chains were then released from the protein scaffold by proteinase K digestion and subsequently immobilized on streptavidin-coupled magnetic beads (Fig. 2b and c). To characterize the chain length distribution, the released PAR polymers were analyzed directly by native polyacrylamide gel electrophoresis (native PAGE) with xylene cyanol, orange G, and bromophenol blue as co-migrating size markers[40]. As shown in Fig. 2d, the dominant PAR species migrated at positions corresponding to chains longer than 20 ADP-ribose units (≥ 20-mer), indicating that the PCCT-NAD+-based PARP5a-automodification predominantly yields relatively long PAR species under used conditions. The high-density presentation of PAR on beads mimics a phase-separated microenvironment, elevating the local PAR concentration and thereby enhancing the capture efficiency for low-affinity or low-abundance PAR-binding proteins. We next evaluated the ability of the resulting functionalized PAR-coated beads (PAR-beads) to covalently capture PAR-interacting proteins via photo-crosslinking. The PAR-beads or control streptavidin-beads were incubated with the GST-tagged WWE domain of RNF146 (GST-WWE), a well-characterized PAR model reader, with or without UV irradiation (Fig. 2e). Following pull-down, washing, and elution, immunoblotting against GST revealed that UV irradiation specifically promoted cross-linking of GST-WWE to PAR-beads, whereas no signal was observed in the non-irradiated control (Fig. 2e, top panel). Parallel negative-control experiments using a GST-ubiquitin fusion protein showed no detectable capture on either PAR-beads or streptavidin-beads, irrespective of UV exposure (Fig. 2e, bottom panel). These data demonstrate that the photo-crosslinking capture system operates in a manner strictly dependent on both the presence of functionalized PAR and UV-triggered activation, enabling efficient and highly specific covalent capture of PAR-binding proteins.
To further assess whether bead-immobilized functionalized PAR enhances the binding efficiency with reader proteins compared to its free counterpart, we incubated PAR-beads or an equivalent amount of soluble terminally functionalized PAR with GST-WWE under identical conditions. Following UV irradiation and pull-down, the amount of unbound WWE protein remaining in the supernatant was quantified as an indirect measure of binding capacity. Immunoblot analysis revealed markedly lower residual WWE levels in the supernatant of PAR-bead samples, indicating superior capture efficiency for the bead-immobilized system (Fig. 2f). This enhancement likely resulted from the elevated local PAR concentration and optimized spatial presentation at the solid–liquid interface, which collectively promote more effective PAR-reader interactions.
To evaluate the performance of PCCT-NAD+, we directly compared it with a previously reported bifunctional NAD+ (PC-NAD+, Fig. 2g) that was used in combination with native NAD+ to generate randomly modified PAR probes. Two canonical PAR-binding domains, AF1521 (macrodomain) and the WWE domain, were then expressed as EGFP fusion proteins to measure the binding affinity against PAR polymers generated from each probe using microscale thermophoresis (MST). As shown in Fig. 2h and i, PCCT-NAD+-derived PAR bound to AF1521 Macro with a Kd of 3.7 μmol•L−1 and to WWE with a Kd of 8.0 μmol•L−1, whereas the PC-NAD+-derived PAR exhibited Kd values of 10.3 μmol•L−1 and 16.5 μmol•L−1, respectively, representing approximately 2.8-fold and 2.1-fold higher affinity for the PCCT-NAD+ probe. These results demonstrate that PCCT-NAD+-derived PAR polymers exhibit significantly stronger binding to both recognition domains, supporting the superiority of our PCCT-NAD+ probe design strategy.
Collectively, these findings establish bead-immobilized photo-crosslinkable PAR derived from PCCT-NAD+ as a robust and efficient platform for the capture and systematic identification of PAR-binding proteins.
Proteomic profiling of the PAR interactome under oxidative stress
-
Following validation of the PAR-beads platform for its specificity and efficiency in photo-crosslinking PAR-binding proteins, we sought to systematically characterize remodeling of the PAR-associated interactome under oxidative stress, a condition known to induce protein PARylation and trigger multifaceted PARylation-dependent cellular responses[41−43]. To this end, we employed UV-assisted photo-crosslinking with PAR-beads to enrich PAR-binding proteins from cell lysates treated with or without H2O2, followed by quantitative proteomic analysis (Fig. 3a; Supplementary Table S2). After stringent washing, enriched proteins were subjected to on-bead tryptic digestion and analyzed by Data-Independent Acquisition Mass Spectrometry (DIA-MS). Comparative quantification identified 1,576 differentially enriched proteins in the H2O2-treated group relative to DMSO vehicle control (FC ≥ 1.2 or FC ≤ 0.83, adjusted * P < 0.05; Fig. 3b and c; Supplementary Table S2, Sheets 1 and 2), indicating extensive remodeling of the PAR-centered protein interaction network under oxidative stress.
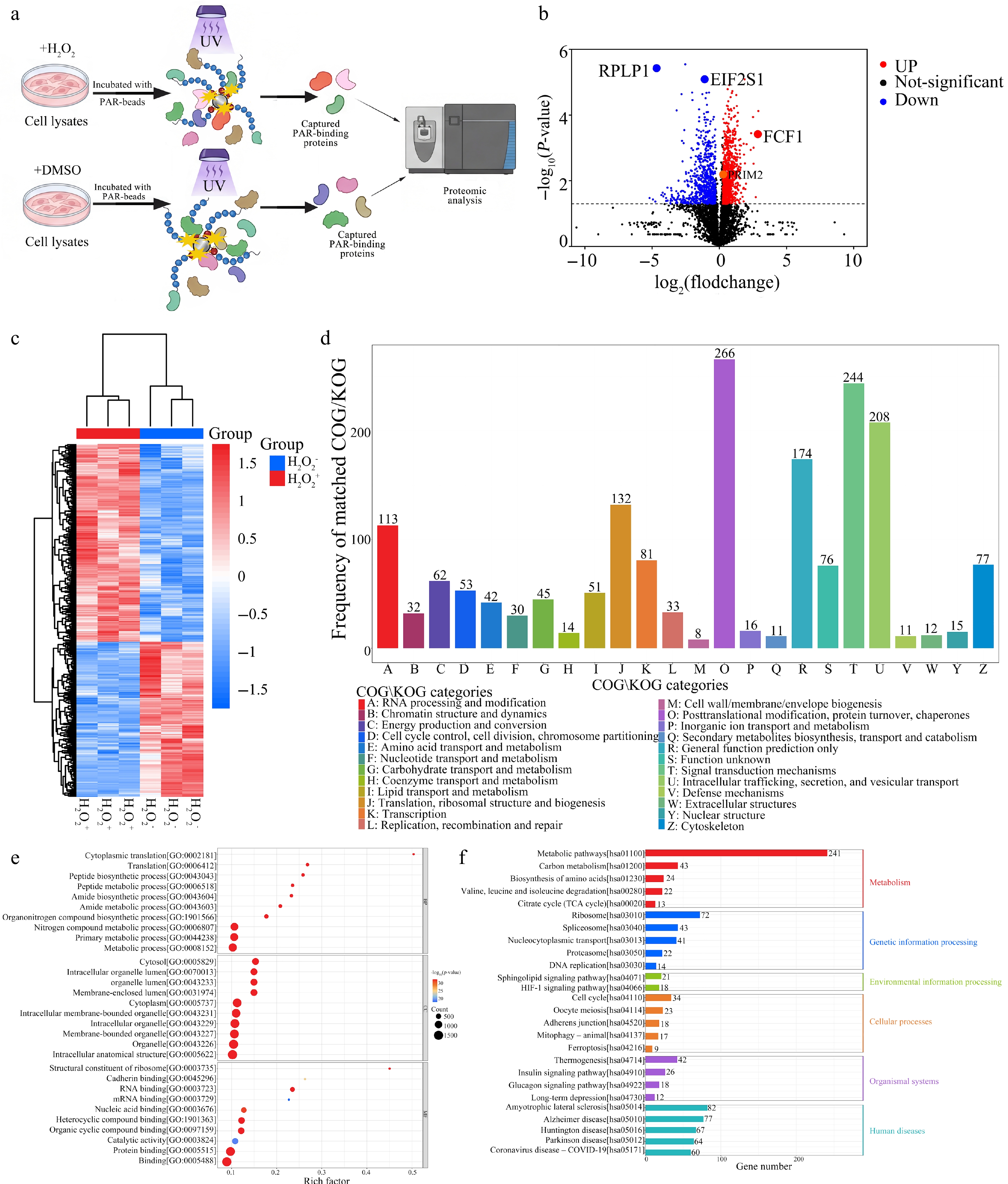
Figure 3.
Proteomic profiling of the PAR-associated interactome under oxidative stress reveals widespread remodeling of protein synthesis and proteostasis networks. (a) Schematic workflow for systematic characterization of PAR-associated interactome remodeling under oxidative stress. Cell lysates from H2O2- or DMSO-treated samples were incubated with PAR-coated beads, followed by UV-assisted photo-crosslinking, stringent washing, on-bead tryptic digestion, and quantitative proteomic analysis by DIA-MS. (b) Volcano plot showing differential enrichment of PAR-interacting proteins in H2O2-treated cells relative to DMSO vehicle control based on three independent biological replicates. Statistical cutoffs were set to FC ≥ 1.2 or FC ≤ 0.83 and adjusted P < 0.05 (multiple-testing corrected); selected candidates (RPLP1, EIF2S1, FCF1, PRIM2) are labeled. (c) Hierarchical clustering heatmap of significantly changed PAR-interacting proteins across control and H2O2 conditions, illustrating global remodeling of the PAR-associated interaction landscape. (d) COG/KOG functional classification of significantly changed PAR-interacting proteins. The bar chart displays the distribution of enriched proteins across major functional categories. (e) Gene Ontology (GO) enrichment analysis of significantly changed PAR-interacting proteins. Dot plots depict enriched terms in Biological Process (BP), Cellular Component (CC), and Molecular Function (MF) domains. (f) KEGG pathway enrichment analysis of significantly changed PAR-interacting proteins. Selected enriched pathways are shown.
To delineate the functional architecture of this stress-responsive PAR interactome, we performed COG/KOG functional category analysis (Fig. 3d; Supplementary Table S2, Sheet 3). This revealed a marked functional reprogramming of PAR-associated proteins under oxidative stress. Strikingly, proteins assigned to DNA replication, recombination, and repair (Category L) were modestly represented (33 proteins), contrasting with the well-documented role of PARylation in canonical DNA damage response. This observation suggests that under acute oxidative stress, PAR may preferentially recruit downstream signaling and effector modules over core DNA repair enzymes, potentially facilitating rapid transcriptional and translational reprogramming as an immediate cellular priority. Conversely, the interactome was overwhelmingly dominated by proteins involved in translation, ribosomal structure and biogenesis (Category J, 132 proteins) and post-translational modification, protein turnover, and chaperones (Category O, 266 proteins). The pronounced enrichment of these functionally linked categories indicates that orchestration of cellular proteostasis represents a primary downstream mission of PARylation under oxidative challenge. We propose that PAR functions as a stress-inducible molecular scaffold that directly engages ribosomal proteins, translation factors, ubiquitin-proteasome system components, and molecular chaperones to coordinate both the clearance of oxidatively damaged proteins and the prioritized synthesis of stress-adaptive proteins. Further functional classification revealed substantial enrichment of proteins associated with signal transduction (Category T, 244 proteins), intracellular trafficking and secretion (Category U, 208 proteins), RNA processing and modification (Category A, 113 proteins), transcription (Category K, 81 proteins), and cytoskeletal dynamics (Category Z, 77 proteins). This multi-category enrichment pattern delineates a hierarchical and spatially coordinated stress response network, wherein PAR integrates upstream signals (T), directs nuclear and cytoplasmic gene expression programs (A, K, J), mobilizes proteostasis machineries (O), and engages subcellular trafficking systems (U, Z) for efficient execution of adaptive programs. Additionally, a substantial fraction of PAR-associated proteins fell into Category S (function unknown, 76 proteins), representing an unexplored reservoir of putative PAR-dependent stress regulators. Collectively, these data reframe the functional paradigm of PARylation: rather than serving primarily as a DNA damage repair signal, PAR operates as a rapid-response molecular scaffold that amplifies stress signals and orchestrates a proteome-wide shift from homeostatic maintenance to stress defense.
To complement and extend the COG/KOG analysis with high-resolution functional annotation, we performed Gene Ontology (GO) enrichment analysis on the 1,576 differentially enriched PAR-binding proteins. The analysis revealed a highly polarized functional landscape (Fig. 3e; Supplementary Table S2, Sheets 4, 5 and 6). Within the biological process (BP) domain, the most significantly and specifically enriched terms were overwhelmingly associated with protein synthesis: cytoplasmic translation, translation, and peptide biosynthetic process ranked at the top. Cellular component (CC) analysis unambiguously localized these proteins to the cytosol and ribosome-associated compartments, while molecular function (MF) terms were dominated by structural constituents of ribosome and RNA/mRNA binding. The convergence between COG/KOG and GO analyses is striking: both orthogonal classification systems independently pinpoint cytosolic translation and ribosome-directed functions as the most specialized and dominant functional output of the oxidative stress PAR interactome. This cross-validation provides compelling evidence that PARylation rapidly reprograms cellular proteostasis by directly engaging the translational machinery, effecting a paradigm shift in our understanding of PAR from a predominantly nuclear DNA repair mediator to a cytoplasmic stress-response orchestrator.
To map PAR-associated proteins onto specific biological pathways, we performed KEGG enrichment analysis (Fig. 3f; Supplementary Table S2, Sheet 7). This analysis uncovered a multi-layered pathway architecture that both validates and substantially extends the COG/KOG and GO findings. Consistent with COG/KOG and GO results, the ribosome (hsa03010) emerged as one of the most significantly enriched pathways, with 72 PAR-associated proteins, providing independent pathway-level validation that the translational machinery is a primary direct target of PAR signaling under oxidative stress. Additional genetic information processing pathways were also robustly enriched, collectively confirming that PAR coordinates both protein synthesis and protein degradation, as well as RNA processing and subcellular trafficking. Multiple signal transduction pathways were significantly enriched, including sphingolipid signaling, HIF-1 signaling, insulin/Glucagon signaling, and thermogenesis. These findings align with COG/KOG Category T enrichment and reinforce the concept of PAR as a stress signal integrator that interfaces with diverse signaling cascades to coordinate context-appropriate cellular responses. Most notably, KEGG analysis revealed massive and highly significant enrichment of neurodegenerative disease and viral infection pathways[44,45]. The convergence of these disease pathways is not coincidental. Their shared pathological hallmarks, including proteostasis collapse, oxidative stress, mitochondrial dysfunction, and aberrant translation[44−46], are precisely the functional nodes we identified as being under PAR control. The striking enrichment of PAR-binding proteins in these pathways suggests that PARylation may represent a common molecular hub underlying the pathogenesis of diverse protein aggregation disorders and viral infections. This positions the PAR interactome as a previously unrecognized yet therapeutically actionable platform for intervention across a broad spectrum of human diseases.
To assess the robustness of our findings under more stringent criteria, we re-analyzed the data using a threshold of FC ≥ 2.0 and P < 0.01. This reduced the number of differentially enriched proteins to approximately 300, but the major functional enrichment pathways (e.g., translation regulation, ribosome biogenesis, and proteostasis) remained largely consistent with the original analysis (Supplementary Table S2). Thus, our core conclusions are not sensitive to the choice of a lenient cutoff, and the 1.2-fold threshold allows comprehensive capture of biologically relevant moderate to low magnitude changes in PAR binding under oxidative stress.
Validation of representative PAR-interacting proteins
-
We next compared the PAR interactome identified in this study with two previously reported PAR-centric proteomic datasets[26,27]. The number of PAR-associated proteins captured by our approach substantially exceeded that of either prior study, with considerable overlap across datasets. Specifically, 60 proteins were commonly identified in all three studies, and 415 proteins had been previously reported[20,26], demonstrating strong concordance between our dataset and existing literature (Fig. 4a; Supplementary Table S2, Sheet 8). Notably, our strategy also uncovered a large set of PAR-interacting proteins uniquely detected in this work, underscoring the enhanced coverage and detection sensitivity afforded by our platform.
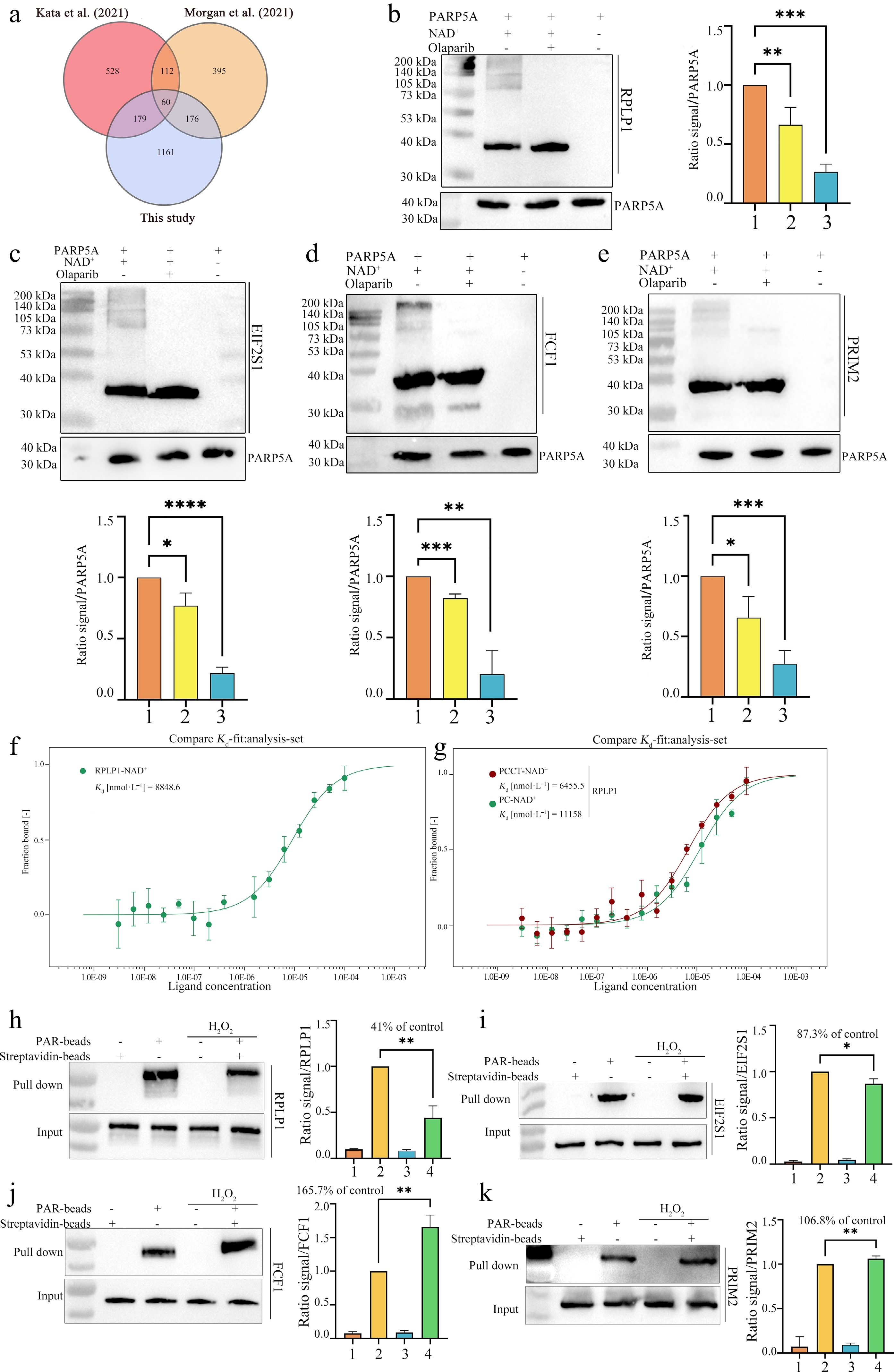
Figure 4.
Validation of representative PAR-interacting proteins identified by the bead-immobilized photo-crosslinkable PAR platform. (a) Venn diagram comparing PAR-interacting proteins identified in this study with those reported in two previously published PAR-centric proteomic datasets (Kata et al.[26]; Morgan et al.[27]). (b)−(e) In vitro PAR overlay assays validating the direct PAR-binding capacity of selected candidates. PAR polymers were synthesized using recombinant PARP5A (10 μmol•L−1) with NAD+ (1 mmol•L−1) in the absence (lane 1) or presence (lane 2) of the PARP inhibitor olaparib (100 μmol•L−1); PARP5A alone served as a negative control (lane 3). Reaction products were resolved by SDS-PAGE, transferred to membranes, and incubated with purified candidate proteins (20 μmol•L−1). Bound proteins were detected by immunoblotting using antibodies against (b) RPLP1 or (c) GST-tag for GST-tagged fusion proteins EIF2S1-GST, (d) FCF1-GST, and (e) PRIM2-GST. PARP5A inputs are shown as loading controls (lower panels). Bottom, densitometric quantification of overlay signals normalized to loading controls. (f), (g) Microscale thermophoresis (MST) binding assays of recombinant RPLP1 with PAR derived from (f) native NAD+ and (g) NAD+ analogs. (f) Binding curve of RPLP1 to PAR derived from native NAD+, with a measured dissociation constant (Kd) of 8,848.6 nmol•L−1. (g) Binding curves of RPLP1 to PAR derived from PCCT-NAD+ (red, Kd = 6,455.5 nmol•L−1) and PC-NAD+ (green, Kd = 11,158 nmol•L−1). (h)–(k) PAR-beads pull-down assays assessing the direct PAR-binding activity of endogenous RPLP1, EIF2S1, FCF1, and PRIM2 under oxidative stress. Lysates from control and H2O2-treated cells were incubated with PAR-coated beads or control streptavidin beads. Bound proteins were detected by immunoblotting, with input samples representing total protein. Bar graphs show densitometric quantification of pull-down signals normalized to the corresponding input, expressed as relative binding activity compared to the untreated control (set to 100%). The remaining PAR-binding activity after H2O2 treatment is indicated. Data are presented as mean ± SD, n = 3. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001, one-way ANOVA followed by Dunnett's post hoc test.
Based on differential enrichment significance, we selected FCF1 (ribosome biogenesis factor), PRIM2 (DNA primase), RPLP1 (ribosomal protein, most downregulated), and EIF2S1 (translation initiation factor) as representative candidates for validation. To assess their direct binding to PAR, we performed in vitro PAR overlay assays using recombinant PARP5A-catalyzed PAR polymers generated in the presence or absence of the PARP inhibitor olaparib. Immunoblotting revealed robust binding signals for each candidate, whereas these signals were completely abolished in the presence of olaparib (Fig. 4b−e). As a stringent negative control, ubiquitin (Ub), a protein known not to bind PAR, was tested under identical PAR overlay conditions and produced no detectable signal (Supplementary Fig. S2), confirming that the observed binding of our candidate proteins is specific and not attributable to non-specific interactions or simply a consequence of high protein concentration. To further confirm that RPLP1 is a genuine PAR-interacting protein, we constructed and expressed an RPLP1-EGFP fusion protein and measured its binding affinity against native PAR and PAR polymers derived from different NAD+ analogs using MST. As shown in Fig. 4f and g, RPLP1 bound to native PAR with a Kd of 8.8 μmol•L−1, to PCCT-NAD+-derived PAR with a Kd of 6.4 μmol•L−1, and to the control probe-derived PAR with a Kd of 11.2 μmol•L−1. These results demonstrate that RPLP1 is a bona fide PAR-interacting protein and that PCCT-NAD+-derived PAR exhibits superior binding affinity compared to the randomly modified control probe. To further validate these interactions at the endogenous level, we performed pull-down assays using PAR-conjugated beads (with unconjugated beads as a negative control) on HeLa cell lysates treated with or without H2O2, followed by western blotting with specific antibodies against RPLP1, FCF1, PRIM2, and EIF2S1. All four candidate proteins were specifically enriched by PAR-beads, whereas no significant signals were detected in the negative control groups (Fig. 4h−k). These results recapitulated the differential enrichment trends observed in the mass spectrometry data, demonstrating that the PAR-coated beads efficiently captured endogenous candidate proteins from cell lysates and supporting the physiological relevance of these interactions. Collectively, these results further substantiate that our bead-immobilized, photo-crosslinkable PAR platform enables efficient and specific identification of bona fide PAR-interacting proteins.
Both protein abundance and PAR-binding affinity alterations under oxidative stress contribute to differential enrichment of proteins
-
To determine whether the observed differential enrichment reflects changes in total protein abundance or genuine alterations in PAR-binding affinity, we performed immunoblot analysis of whole cell lysates to quantify total protein levels of the above four candidate proteins under control and H2O2-treated conditions, and directly compared these changes with the differential enrichment log2FC values from the PAR pull-down proteomics (Fig. 5a−d). Under H2O2 treatment, the total protein level of RPLP1 decreased markedly to 54% of the control level, while its PAR pull-down signal dropped to approximately 4% (log2FC = −4.68), indicating a combination of reduced abundance and reduced binding. For FCF1, total protein increased slightly to 124%, yet its PAR pull-down signal increased dramatically to about 730% (log2FC = 2.86), revealing a genuine increase in PAR-binding affinity. PRIM2 total protein remained essentially unchanged (105%), while its PAR pull-down signal increased to approximately 180% (log2FC = 0.88), demonstrating a true increase in binding affinity. EIF2S1 total protein was unchanged (97%), whereas its PAR pull-down signal decreased to about 47% (log2FC = −1.09), indicating a genuine decrease in PAR-binding affinity. These data demonstrate that the observed differential enrichment reflects a combination of changes in protein abundance and genuine alterations in PAR-binding behavior.
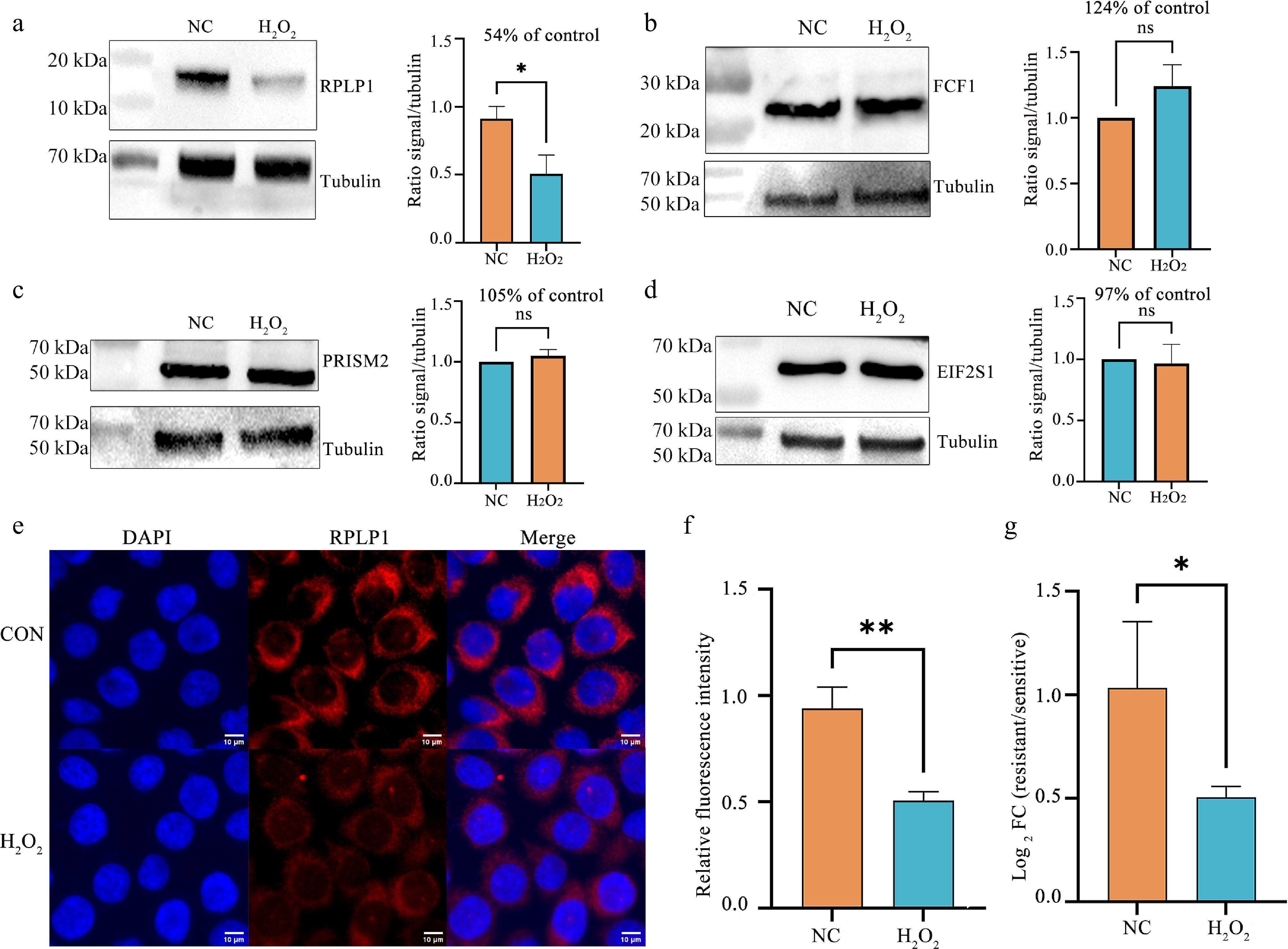
Figure 5.
Validation of oxidative stress-induced changes in selected PAR-interacting proteins and RPLP1 expression. (a)−(d) Immunoblot analysis of (a) RPLP1, (b) FCF1, (c) PRISM2, and (d) EIF2S1 protein levels in DMSO-treated control (NC) cells and cells exposed to H2O2 (500 μmol•L−1, 2 h). Tubulin was used as a loading control. Densitometric quantification of protein levels was normalized to tubulin. * P < 0.05 (two-tailed unpaired Student's t-test). (e) Confocal immunofluorescence images showing nuclear staining (DAPI, blue) and endogenous RPLP1 (red) in DMSO-treated control cells and cells exposed to H2O2 (500 μmol•L−1, 2 h). Scale bar, 10 μm. (f) Quantification of relative cellular RPLP1 fluorescence intensity from images in (a). Data are presented as mean ± SD (n ≥ 80 cells per condition). ** P < 0.01 (two-tailed unpaired Student's t-test). (g) RT-qPCR analysis of RPLP1 mRNA levels following H2O2 treatment (500 μmol•L−1, 2 h). Relative expression was calculated using the 2−ΔΔCᴛ method, with values normalized to the DMSO control group (set to 1.00). Data are presented as mean ± SD, n = 3. Statistical significance was assessed on ΔCᴛ values using a two-tailed unpaired Student's t-test (* P < 0.05).
Oxidative stress downregulates RPLP1 through transcriptional suppression
-
Given that RPLP1 exhibited the most pronounced change in our differential proteomic analysis (Fig. 3b) and that the precise biological functions of RPLP proteins remain incompletely understood[47,48], we validated its regulation under oxidative stress using two additional complementary approaches. Confocal immunofluorescence imaging revealed a global attenuation of intracellular RPLP1 signal following H2O2 treatment, with quantitative analysis confirming a significant reduction in fluorescence intensity (Fig. 5e and f). Immunoblot analysis has similarly demonstrated a marked decrease in RPLP1 protein levels after H2O2 exposure compared to control (Fig. 5a). RT-qPCR further showed that RPLP1 transcript levels were significantly reduced to approximately 0.55-fold of the control (Fig. 5g), indicating that oxidative stress primarily suppresses RPLP1 at the transcriptional level. Collectively, these results demonstrate that oxidative stress induces widespread downregulation of RPLP1, predominantly through transcriptional suppression. As a core component of the ribosomal P complex essential for translational elongation, RPLP1 downregulation may reflect an adaptive reduction in translational capacity under stress, conserving energy and minimizing mistranslation-associated proteotoxic damage. These findings establish RPLP1 as a potential sentinel of stress-induced translational remodeling and provide a foundation for exploring functional links between PAR-associated interaction networks and translational regulation.
-
In this study, we developed and validated a photo-crosslinkable, bead-immobilized PAR capture platform for systematic profiling of the PAR interactome under oxidative stress. By integrating chemical probe design, affinity capture, quantitative proteomics, and biochemical validation, we delineated the stress-induced remodeling of PAR-mediated protein interaction networks and uncovered a prominent role for PARylation in coordinating translational control and ribosome homeostasis.
Platform advantages and methodological contributions
-
Our approach addresses key limitations of existing PAR interactome profiling methods. First, terminal functionalization preserves the structural integrity of internal ADP-ribose units, minimizing the risk of altered binding specificity associated with randomly modified probes. Second, solid-phase immobilization creates a high local-concentration microenvironment that stabilizes weak or transient interactions often lost in solution-based affinity purification. Third, photo-crosslinking enables covalent capture of interactors, allowing stringent washing conditions that reduce non-specific background. The substantial overlap with existing datasets validates the specificity of our platform, while the large number of uniquely identified proteins underscores its enhanced coverage and detection sensitivity.
Biological implications: PARylation as a regulator of translation and proteostasis
-
PARylation has long been established as a critical regulator of DNA damage repair and genome stability[49,50]. Our findings show that PAR-associated interactions extend beyond DNA repair and are strongly linked to translation-related processes under oxidative stress.
Among the 1,576 differentially enriched PAR-binding proteins identified under oxidative stress, a substantial fraction is involved in RNA metabolism, translation initiation, and ribosome biogenesis. This observation aligns with emerging evidence that oxidative stress triggers reprogramming of protein synthesis[51,52] and suggests that PARylation serves as a molecular bridge linking DNA damage signaling to the translational machinery. The energetic cost of protein synthesis is considerable. Thus, adaptive downregulation of translation under oxidative stress may represent a conserved cellular strategy to conserve energy and mitigate proteotoxic stress caused by misfolded protein accumulation[52]. Our data support a model wherein PARylation facilitates the assembly of protein complexes that modulate translation initiation and ribosomal function, enabling rapid and coordinated cellular adaptation.
RPLP1 as a sentinel of stress-induced translational remodeling
-
Among the identified candidates, RPLP1 exhibited the most pronounced downregulation under oxidative stress. Our multi-layer validation confirmed that this downregulation occurs primarily at the transcriptional level, establishing RPLP1 as a stress-responsive component of the ribosomal P complex. The ribosomal P complex (comprising RPLP1, RPLP2, and RPLP0) is essential for translational elongation and has been implicated in stress-induced autophagy and viral replication. Our findings suggest that RPLP1 downregulation may represent a checkpoint in stress-induced translational reprogramming, and its regulation by PARylation warrants further investigation. However, we acknowledge that the functional relevance of RPLP1 downregulation under oxidative stress remains to be fully determined. Future studies should investigate the functional consequences of RPLP1 modulation, including gain or loss-of-function experiments to assess its impact on translational adaptation and cell survival to further validate the physiological PAR-RPLP1 interaction.
Limitations and future directions
-
There are several points that need further study. First, our platform generates predominantly linear PAR chains, which, while enhancing structural controllability, may not fully recapitulate the complexity of endogenous PAR, including branching and heterogeneous chain lengths. This could lead to underrepresentation of structure-selective interactors.
The study here did not perform a global proteome analysis of input lysates to normalize our PAR pull-down enrichment data. Therefore, we cannot definitively distinguish whether all 1,576 differentially enriched proteins reflect genuine changes in PAR-binding affinity vs changes in total protein abundance. While our targeted validation of four candidates (Fig. 5a−d) suggests that both mechanisms contribute, a systematic normalization would be required to fully deconvolute these two factors for the entire dataset. Future studies integrating global proteomics with PAR pull-down will be necessary to address this question comprehensively.
Another potential concern is the possibility that endogenously generated PARylation in H2O2-treated cell lysates could theoretically compete with the exogenous PAR beads for binding to PAR-interacting proteins, thereby affecting capture efficiency. However, several features of our experimental design minimize this concern. First, the immobilized PAR on the beads is present in large molar excess relative to reported endogenous PAR levels. Second, typical PAR-binding motifs (e.g., PBZ, BRCT, and macrodomains) exhibit moderate affinities for PAR, making their binding highly dependent on local PAR concentration. Third, our workflow includes a pre-incubation step that allows the binding equilibrium to re-establish, enabling any proteins initially bound to endogenous PAR to exchange and rebind to the exogenous beads. Taken together, these considerations suggest that endogenously generated PARylation does not meaningfully interfere with our proteomic profiling under the conditions used. Nevertheless, we cannot completely rule out the possibility that trace amounts of free PAR oligomers or polymers in the lysates might cause minor interference, a limitation common to current PAR-based affinity enrichment approaches.
We acknowledge that some candidate proteins identified in this study, including RPLP1, are listed as common contaminants in the CRAPome database for conventional AP-MS experiments. To address this concern, we submitted our spectral count data (both H2O2-treated and NC groups) to the CRAPome analysis platform, using our own experimentally matched NC samples as the sole negative controls. This analysis yielded FC-B and SAINT probability (SP) scores for all identified proteins (Supplementary Table S2, sheet 9). The above four representative candidates all achieved the maximum SAINT score (SP = 1), with FC-B scores of 11.74, 1.93, 0.74, and 0.06 for FCF1, PRIM2, EIF2S1, and RPLP1, respectively. Notably, although RPLP1 showed a relatively low FC-B score, its SAINT score of 1, together with the in vitro PAR overlay (Fig. 4b), MST assay (Fig. 4f and g), and endogenous pull-down validation data (Fig. 4h), robustly confirms it as a bona fide PAR-binding protein. These results also demonstrate the reliability of our PCCT-NAD+-based proteomic platform for identifying genuine PAR interactor.
Moreover, oxidative stress responses are inherently dynamic; time-resolved analysis would help establish the temporal ordering of PAR-driven recruitment events vs subsequent transcriptional reprogramming, particularly for early-response factors such as EIF2S1 and RPLP1. Finally, extending this approach to cancer-relevant models and therapy-induced stress contexts will be essential to assess whether the PAR-translation connections identified here contribute to ROS tolerance, translational adaptation, and differential sensitivity to PARP inhibition.
-
In conclusion, this work establishes a versatile chemical proteomics platform for profiling PAR-associated proteins and reveals oxidative stress-driven remodeling of the PAR interactome with pronounced enrichment of translation-related modules. Our findings position PARylation as a coordinator linking stress signaling to translational control, with potential implications for targeting tumor stress adaptation and PARP inhibitor resistance. The platform described herein is broadly applicable to other stress conditions, cell types, and disease models, providing a foundation for systematic exploration of PAR-dependent signaling networks.
-
The data used in this study were generated by the authors through original experiments/measurements, and no external datasets or secondary sources were utilized. Therefore, no ethics committee approval was required for this study.
-
The authors confirm contributions to the work as follows: study conception and design: Gao L, Tu Z, Zhou Y, Li M, Zhang XN; data collection: Gao L, Tu Z, Zhou Y, Xu Y; analysis and interpretation of results: Gao L, Tu Z, Zhang XN; draft manuscript preparation: Gao L, Tu Z, Zhou Y, Zhang XN; providing assistance with protein expression: Sun Y, Dai T; providing assistance with protein purification: Zhan J. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this manuscript and its supplementary information files. Any additional data supporting the findings of this study are available from the corresponding author upon reasonable request.
-
This work was supported by the National Natural Science Foundation of China (22377146), and the China Pharmaceutical University Start-Up Fund for New Faculty. The authors would like to thank the Analytical Testing Center of China Pharmaceutical University, Hangzhou Hikvision Digital Technology Co., Ltd; Acchrom Technologies Co., Ltd; and Wooking Scientific Instruments (Shanghai) Co., Ltd for providing instrument support.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/targetome-0026-0030.
-
#Authors contributed equally: Lijuan Gao, Zhenlin Tu, Yangyang Zhou
- Supplementary Table S1 The amino acid sequences of various expressed recombinant protein.
- Supplementary Table S2 Quantitative mass spectrometry analysis results.
- Supplementary Fig. S1 Purification of recombinant proteins.
- Supplementary Fig. S2 In vitro PAR overlay assay validating the direct PAR-binding capacity of GST-Ub.
- Supplementary File 1 Supplementary methods to this study.
- Supplementary Scheme 1 Chemical synthesis of PCCT-NAD+.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gao L, Tu Z, Zhou Y, Xu Y, Sun Y, et al. 2026. A photo-clickable, chain-terminating NAD+ analogue enables systematic profiling of the PAR-interacting proteome under oxidative stress. Targetome 2(4): e031 doi: 10.48130/targetome-0026-0030 

A photo-clickable, chain-terminating NAD+ analogue enables systematic profiling of the PAR-interacting proteome under oxidative stress
- Received: 31 March 2026
- Revised: 24 May 2026
- Accepted: 17 June 2026
- Published online: 15 July 2026
Abstract: Poly(ADP-ribosyl)ation (PARylation) is a critical post-translational modification that orchestrates diverse cellular processes through the dynamic assembly of PAR-dependent interaction networks. However, systematic profiling of PAR-interacting proteins under physiological and pathological conditions remains technically challenging due to limitations in probe design and interactome capture strategies. Here, we report a photo-clickable, chain-terminating NAD+ analogue (PCCT-NAD+) that enables the generation of PAR polymers bearing a terminal photo-clickable ADP-ribose. Leveraging this probe, we developed a bead-immobilized, photo-crosslinkable PAR platform for high-efficiency capture of PAR-interacting proteins. Quantitative proteomics under oxidative stress identified 1,576 differentially enriched PAR-binding proteins, revealing extensive remodeling of the PAR interactome. Functional enrichment analysis revealed pronounced reprogramming toward translation and proteostasis pathways, with canonical DNA repair proteins being modestly represented, suggesting a broader role for PARylation in coordinating protein synthesis and turnover beyond its established functions in genome maintenance. Among the candidates, RPLP1, a core component of the ribosomal P complex, exhibited the most pronounced downregulation, which we validated at both protein and transcript levels, establishing oxidative stress-induced transcriptional suppression of RPLP1. Overall, this work provides a versatile chemical proteomics approach for studying PAR-associated networks and supports a role for PARylation in linking stress signaling to translational control.
-
Key words:
- PAR interactome /
- NAD+ analogue /
- ADP-ribosylation /
- Poly(ADP-ribose) /
- Oxidative stress