









-
The genus Ficus, a member of the Moraceae family, comprises over 800 species exhibiting diverse growth forms, including evergreen trees, shrubs, herbaceous plants, and vines. Predominantly distributed across tropical and subtropical regions, Ficus plays a crucial role in tropical rainforest ecosystems. The genus is classified into six subgenera, with Sycomorus, Ficus, Synoecia, and Sycidium being dioecious, while Urostigma and Pharmacosycea are monoecious[1]. Ficus species hold significant ecological and economic value, often serving as ornamental plants due to their large trunks and broad canopies. Additionally, their fruits and roots are rich in nutritional and medicinal compounds. In various cultural traditions and religious practices, Ficus trees symbolize longevity, prosperity, enlightenment, and wisdom[2].
F. altissima is a monoecious evergreen tree in the Ficus genus, thriving in sunny, hot, and humid climates. Highly adaptable, it is primarily distributed across India, southern China, Thailand, and other regions[3]. F. altissima possesses a well-developed buttress root system, which provides structural support by counterbalancing the weight of its large canopy and trunk while also aiding in moisture retention. These roots not only facilitate the absorption of water and nutrients but also enhance the tree's stability, enabling it to withstand strong winds and shifting sands. Due to its resilience and adaptability, F. altissima is commonly planted as a roadside tree in southern regions. In various traditional cultures, it symbolizes longevity and happiness and is often found near temples and shrines.
F. hirta is a dioecious shrub or small tree within the Ficus subgenus Ficus, known for its medicinal properties. Its palmately lobed leaves, resembling the five fingers of a hand, and its peach-like fruits have earned it the nickname 'five-finger peach'. Unlike many other Ficus species, F. hirta lacks an aerial root system; instead, it has a relatively simple root structure composed mainly of underground roots. These roots are rich in bioactive compounds, including coumarins, flavonoids, terpenes, and phenolic compounds[4], and are commonly used in traditional medicine to treat conditions such as cough, phlegm, bacterial infections, rheumatism, and tuberculosis. In addition to its medicinal uses, F. hirta is a popular ingredient in soups in southern China.
Genomic research on the Ficus genus is still in its early stages. Zhang et al. sequenced the genomes of Ficus microcarpa and Ficus hispida. The genome of F. microcarpa has 2n = 26 and a size of approximately 423 Mb, while F. hispida has 2n = 28 and a genome size of about 359 Mb[1]. This study revealed dynamic karyotypic variations linked to adaptive evolution, including the amplification of auxin-related gene copies and increased auxin production, both associated with the development of aerial roots in F. microcarpa. The study also identified candidate genes for sex determination in F. hispida and explored the coevolution between Ficus species and Ficus fig wasps[1]. Additionally, Chakraborty et al. sequenced the genomes of Ficus benghalensis and Ficus religiosa, uncovering the longevity mechanisms of these two species[5]. Usai et al. and sequenced the genome of Ficus carica, with a chromosome number of 2n = 26 and a size of approximately 333 Mb. This study revealed the relationship between breeding, epigenetic changes, and phenotypic traits in F. carica[2].
Auxin is a central hormone in plant growth and development, playing a crucial role, particularly in promoting root growth, and increasing root biomass[6]. The auxin efflux proteins encoded by the PIN gene family mediate the polar transport of auxin within the plant, which is essential for root growth and development. Studies have shown that auxin is transported polarally through the localized positioning of PIN family proteins, influencing cell division in the root apical meristem and thereby affecting root growth[7]. In the roots, polar auxin transport relies on the polar localization of PIN proteins. Auxin is transported in an 'umbrella-like' pattern at the root tip, resulting in varying auxin concentrations across different regions of the root tip[8]. This concentration gradient influences cell division in the root apical meristem, subsequently impacting root growth. Therefore, the PIN gene family plays a central role in regulating polar auxin transport, promoting root growth, and enhancing root biomass, making it a key regulatory factor in plant growth and development.
F. hirta contains an important bioactive compound, psoralen, which occurs naturally. Chemically, psoralen consists of a coumarin structure fused with a furan ring, forming a furanocoumarin structure[9]. The furan ring can fuse in different ways, resulting in several isomers. Among these, psoralen is the predominant form, forming the linear furanocoumarin core structure[10]. The distribution of psoralen in plants has garnered increasing attention. Comprehensive studies have been conducted on the bioactivity and pharmacological effects of psoralen. Research has shown that psoralen exhibits a variety of biological activities, including photosensitivity, antibacterial, anti-inflammatory, anticancer, and immunosuppressive effects. These properties give psoralen broad potential applications in the pharmaceutical field, particularly in phototherapy, infection control, and cancer treatment.
-
Tender leaves from two wild banyan tree species, F. altissima, native to Chengmai County, Hainan Province, China, and F. hirta, native to Ledong Li Autonomous County, Hainan Province, China, were used in this study. High-quality genomic DNA was extracted from frozen leaf tissues using the cetyltrimethylammonium bromide (CTAB) method. The DNA quality was assessed through agarose gel electrophoresis and quantified using a Nanodrop spectrophotometer. Ultra-long ONT sequencing was performed with the SQK-LSK114 kit from Oxford Nanopore Technologies (ONT). Additionally, Hi-C and transcriptome sequencing were conducted using the BGI DNBSEQ platform.
Genome assembly and assessment
-
To reduce the error rate of the raw reads, we employed NanoPlot[11] software to correct the processed ONT data, and we used Fastp[12] software to correct the original Hi-C and original BGI data. ONT data were assembled using the NextDenovo[13] software with the following settings: genome size = 370 Mb, read cutoff = 30,000. Utilizing NextPolish[14] to correct base errors introduced during the initial assembly by combining second-generation BGI data. The initial assembly results were then enhanced by integrating Hi-C data using the 3D-DNA[15] software to scaffold F. altissima and F. hirta contigs separately.
Subsequently, the assembly results were refined using Juicebox[16], resulting in chromosome-level genomes. The completeness of the final chromosome-level assembly was assessed using the genome evaluation mode of Benchmarking Universal Single-Copy Orthologs (BUSCO v5.2.2)[17] and the Embryophyta_odb10 lineage dataset.
Transcriptome expression quantification
-
We aligned transcriptome data separately to the reference genomes of both F. hirta and F. altissima using HISAT2[18]. After sorting and compressing the alignment results, we quantified the expression levels of each gene in every sample based on the Fragments per Million Mapped reads (TMM) calculation formulas using the featureCounts[19] software. Heatmaps were plotted using DataColor[20].
Structural and functional annotation
-
We employed a combination of homology-based prediction and de novo prediction methods to identify repetitive sequences in the genomes of F. hirta and F. altissima. Homology-based prediction was carried out using RepeatMasker[21] to identify repetitive sequences based on sequence similarity, while de novo prediction was performed using RepeatModeler[22] to search for species-specific repetitive sequences.
Protein-coding genes in the genomes of F. hirta and F. altissima were predicted using a combination of de novo prediction, homology-based prediction, and RNA-seq-based methods. Finally, we used EVidenceModeler (EVM)[23] and BRAKER[24] to integrate all prediction results and generate the final gene models. Functional annotation of all predicted proteins was performed using the online version of EggNOG-Mapper[25].
Circos graph
-
The chromosomes were divided into 50 kb window regions using Circos[26] software. We analyzed gene density, repeat sequence content, overall genome GC content, Copia LTR-RT density, Gypsy LTR-RT density, and species-specific synteny. Additionally, we used Circos to visualize synteny between F. hirta and F. altissima.
Gene family clustering and phylogenetic evolutionary analysis
-
Using protein sequences from F. carica, F. hispida, F. erecta, F. hirta, F. altissima, and F. microcarpa along with the multiple sequence alignment results from OrthoFinder[27], we created a Venn diagram using online software available at
http://jvenn.toulouse.inra.fr/app/example.html [28]. We used OrthoFinder to identify and align orthogroups in the 11 species. The multiple sequence alignment results from OrthoFinder were used as input for RAxML[29] to construct a phylogenetic tree based on the maximum likelihood method[30]. Calibration times were determined using established divergence times from the TimeTree[31] website, and the divergence times of the 11 species were estimated using PAML's MCMCTree[32]. Gene family expansion and contraction analyses were performed using CAFE5[33] software, and evolutionary trees were generated using the iTOL[34] website. The dot plot and synteny analysis between species were generated using MCScanX[35] and JCVI[36] software.Identification and analysis of the PIN family members in Ficus
-
The PIN protein sequences of A. thaliana were downloaded from TAIR (
www.arabidopsis.org ). The domain HMM models were obtained from the Pfam[37] database, and BLAST[38] was used to identify the corresponding protein sequences in Ficus trees. Motifs were predicted using MEME[39], while cis-acting regulatory elements were analyzed with PlantCARE (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/ )[40]. The results were visualized using TBtools[41].Metabolite extraction, qualitative, and quantitative analysis
-
In this study, three samples were selected and categorized into three groups for metabolic analysis. Biological samples were vacuum freeze-dried and finely ground into powder using a grinding instrument (MM400, Retsch) at 30 Hz for 1.5 min. A precisely weighed 50 mg of sample powder was mixed with 1,200 μL of pre-cooled (−20 °C) 70% methanol aqueous internal standard extraction solution. The mixture was vortexed six times, followed by centrifugation at 12,000 rpm for 3 min. The supernatant was collected and filtered through a microporous membrane before analysis using a UPLC-MS/MS platform and an in-house metabolite database.
Metabolite identification was performed based on secondary mass spectrometry (MS²) data using the in-house Metware Database (MWDB). During data processing, isotopic signals, redundant signals containing K+, Na+, and NH4+ ions, as well as fragment ion signals originating from larger molecular weight compounds, were systematically removed to ensure accurate metabolite identification.
-
In the genome sequencing of F. altissima and F. hirta, we employed a combination of three different technologies: Oxford Nanopore, BGISEQ, and high-throughput chromosome conformation capture (Hi-C) platforms (Fig. 1a, b). Genome size estimation using flow cytometry indicated that the genome sizes of F. altissima and F. hirta are 430 and 300 Mb, respectively (Supplementary Table S1). The assembly process was carried out using Oxford Nanopore data, followed by assembly refinement with BGISEQ data, and contig anchoring using Hi-C data. We successfully constructed two genomes, with sizes of 355.9 and 323.7 Mb, respectively, each comprising 13 chromosomes (Fig. 1c, d). The largest contig lengths were 22.8 and 24.8 Mb, respectively. Assembly completeness was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO), with F. altissima achieving a complete BUSCO score of 98.2%, which includes 95.8% complete single-copy genes and 2.4% complete multi-copy genes. F. hirta also achieved a complete BUSCO score of 98.2%, including 94.7% complete single-copy genes and 3.5% complete multi-copy genes. Genome assembly completeness was further assessed using the K-mer-based Merqury software[42], with scores of 86.96 for F. altissima genome and 76.88 for F. hirta genome. Genome annotation was performed using de novo prediction, transcriptome data, and homologous protein evidence. The repeat content of F. altissima is 54.1%, with a GC content of 35.2%, and a total of 24,938 high-quality protein-coding genes were identified. The repeat content of F. hirta is 52.14%, with a GC content of 34.73%, and a total of 25,170 high-quality protein-coding genes were identified (Table 1).
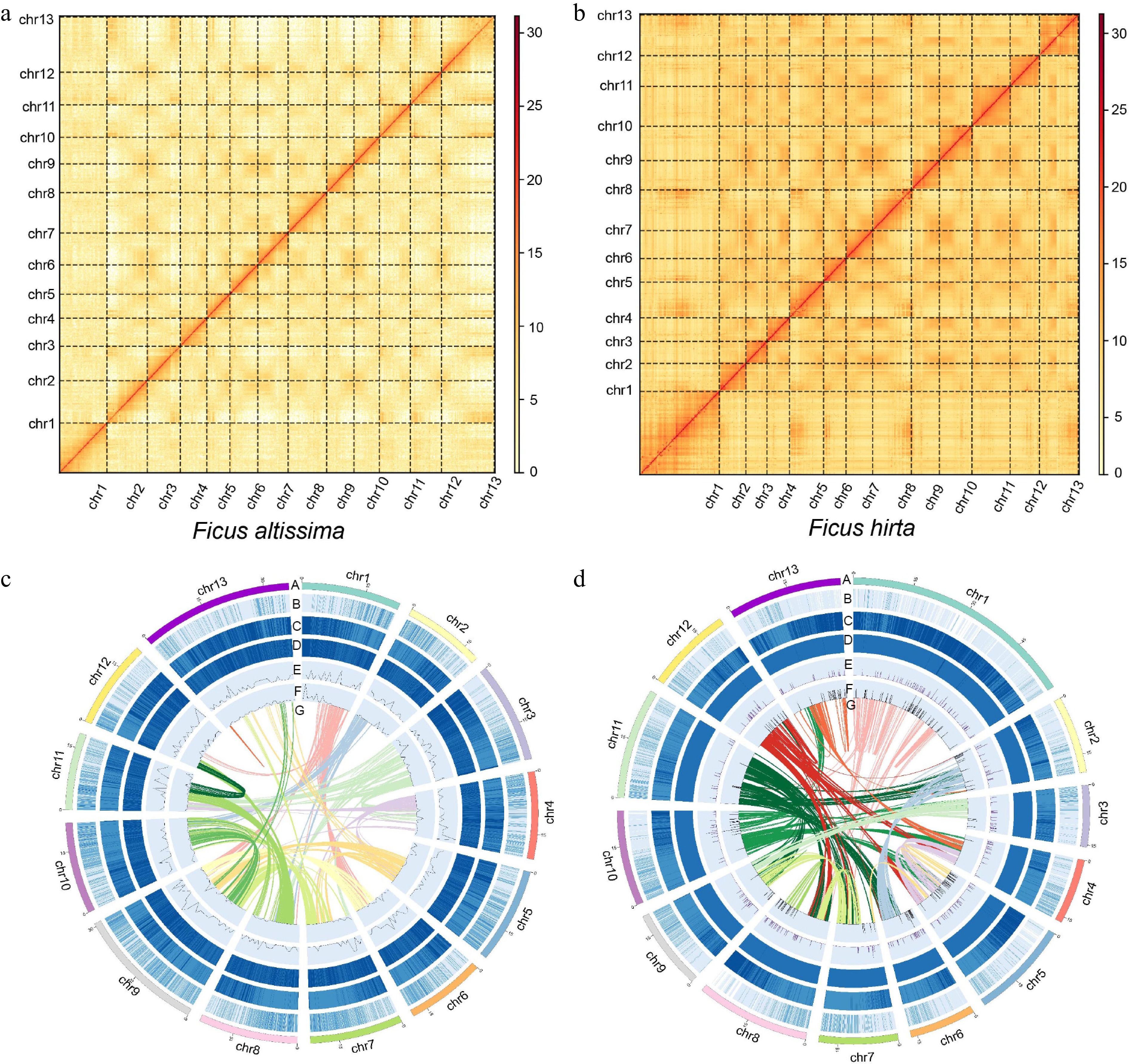
Figure 1.
The Hi-C results and genomic characteristics of Ficus. (a) Hi-C results of F. altissima. (b) Hi-C results of F. hirta. (c) The Circos plot showing the genome details of F. altissima: A, thirteen chromosomes of F. altissima; B, gene density; C, repeat sequence content; D, GC content density; E, density of Copia LTR–RTs; F, density of Gypsy LTR–RTs; G, syntenic blocks (all window sizes = 50 kb). (d) The Circos plot showing the genome details of F. hirta. A, B, C, D, E, F, G: A, thirteen chromosomes of F. hirta; B, gene density; C, repeat sequence content; D, GC content density; E, density of Copia LTR–RTs; F, density of Gypsy LTR–RTs; G, syntenic blocks (all window sizes = 50 kb).
Table 1. Basic genomic information of F. altissima and F. hirta.
Genomic feature F. altissima F. hirta Genome size (Mb) 355.9 323.7 Number of chromosomes 13 13 Scaffold N50 length (Mb) 22.8 24.8 GC content (%) 35.21 34.73 Genomic heterozygous (%) 1.33 1.02 Repeat sequence content (%) 54.11 52.14 The number of protein-coding genes 24,938 25,170 Genome BUSCOs (%) 98.2 98.2 Merqury completeness 86.96 76.88 Phylogenetic relationships and WGD analysis of the Ficus genus
-
To explore the evolutionary relationships between F. altissima, F. hirta, and other species of the Ficus genus, we constructed a phylogenetic tree using 11 species, including six Ficus species and Vitis vinifera as the outgroup. The results indicated that the Ficus genus diverged from the Moraceae family approximately 50 Mya, while the divergence between Ficus species occurred around 41 Mya (Fig. 2a). The expansion and contraction of gene families are closely associated with the adaptive evolution of species, suggesting that during the evolutionary process, Ficus species underwent varying degrees of gene family expansion and contraction. Compared to other Ficus species, F. altissima and F. hirta have fewer gene families that underwent expansion, but a greater number of gene families were lost during contraction.
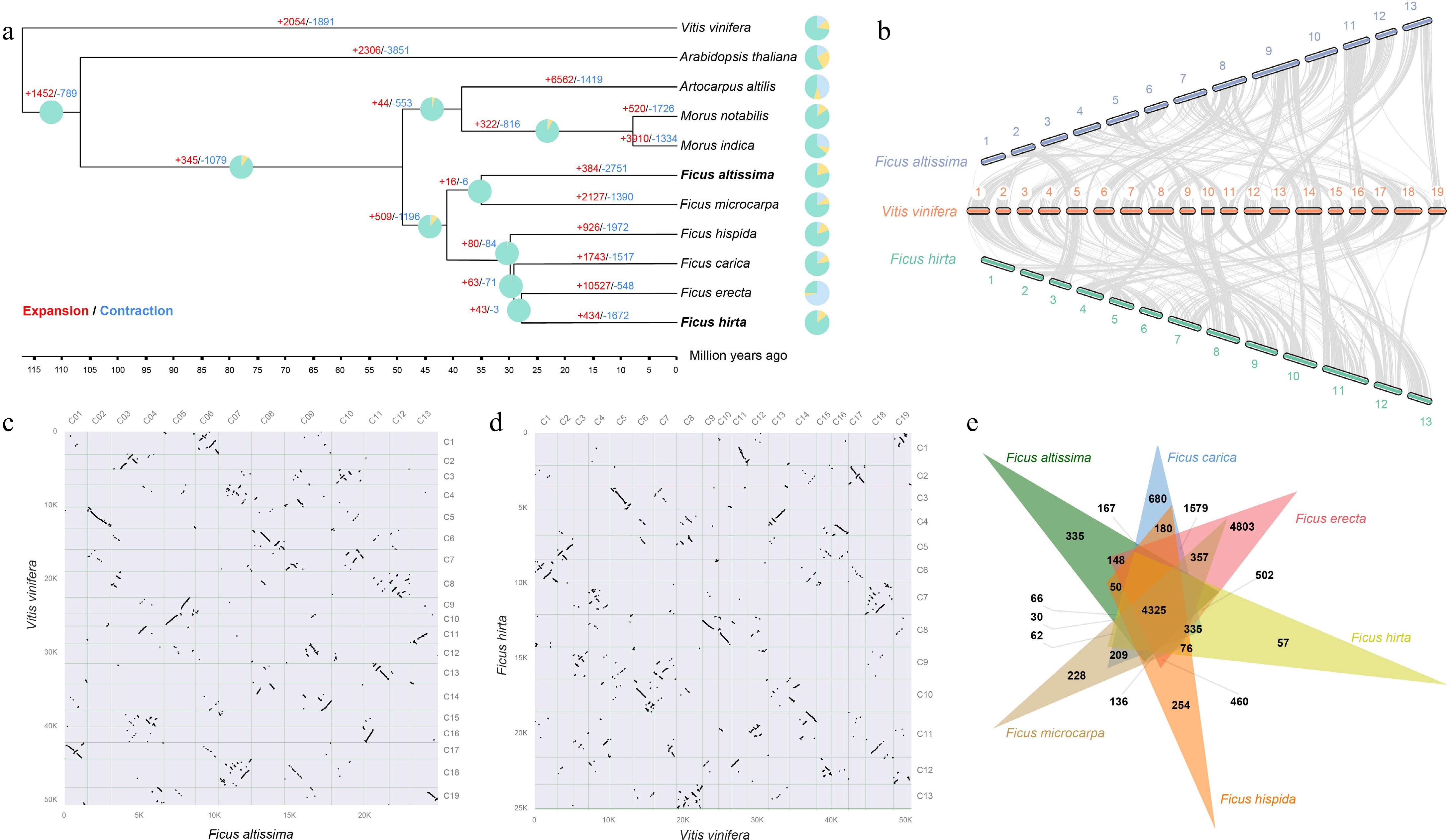
Figure 2.
Evolutionary analysis of the Ficus genus and the genomes of F. altissima and F. hirta. (a) Phylogenetic tree showing F. altissima, F. hirta, and nine other species, along with divergence times and the expansion and contraction of gene families. Green represents gene retention, yellow represents gene contraction, and blue represents gene expansion. (b) Synteny analysis between F. altissima, F. hirta, and Vitis vinifera. (c) Dot plot of F. altissima and V. vinifera. (d) Dot plot of F. hirta and V. vinifera. (e) Venn diagram showing the shared orthologous gene groups among the genomes of the Ficus species.
Collinearity analysis showed a nearly one-to-one correspondence of collinear blocks between F. altissima and F. hirta, and V. vinifera (Fig. 2b). Dot plot analysis further confirmed a 1:1 collinearity ratio between the two, suggesting that Ficus and Vitis shared an ancient whole-genome triplication event, after which no independent whole-genome duplication events occurred (Fig. 2c, d). Comparative analysis revealed that the six Ficus species shared 4,325 orthogroups, with F. altissima containing 335 unique gene families and F. hirta containing 57 unique gene families (Fig. 2e).
PIN-FORMED (PIN) gene family in F. altissima
-
Zhang et al. emphasized that the formation of aerial roots in fig trees is primarily influenced by the PIN gene family. F. altissima possesses a well-developed buttress root system[1]. To investigate the molecular mechanisms underlying its root development, we analyzed transcriptome data and quantified the expression levels of auxin pathway-related genes. The results revealed that the PIN gene family is highly expressed in the buttress roots of F. altissima (Fig. 3a), suggesting a key role in underground root system development. To further explore this, we compared the protein sequences of the PIN gene family in Arabidopsis and the domain models of PIN with those in F. altissima, identifying a total of 14 PIN family members. Structural analysis showed that the PIN genes in F. altissima contain multiple introns and exons. Conserved motif analysis revealed that PIN1 to PIN8 share a set of conserved motifs, including Motif1, Motif2, Motif3, Motif4, Motif5, Motif7, Motif8, and Motif9, while other PIN proteins possess a distinct combination of motifs, such as Motif6, Motif8, and Motif10 (Fig. 3b). These findings provide valuable insights into the functional conservation and divergence of the PIN gene family and their potential roles in regulating root development in F. altissima.
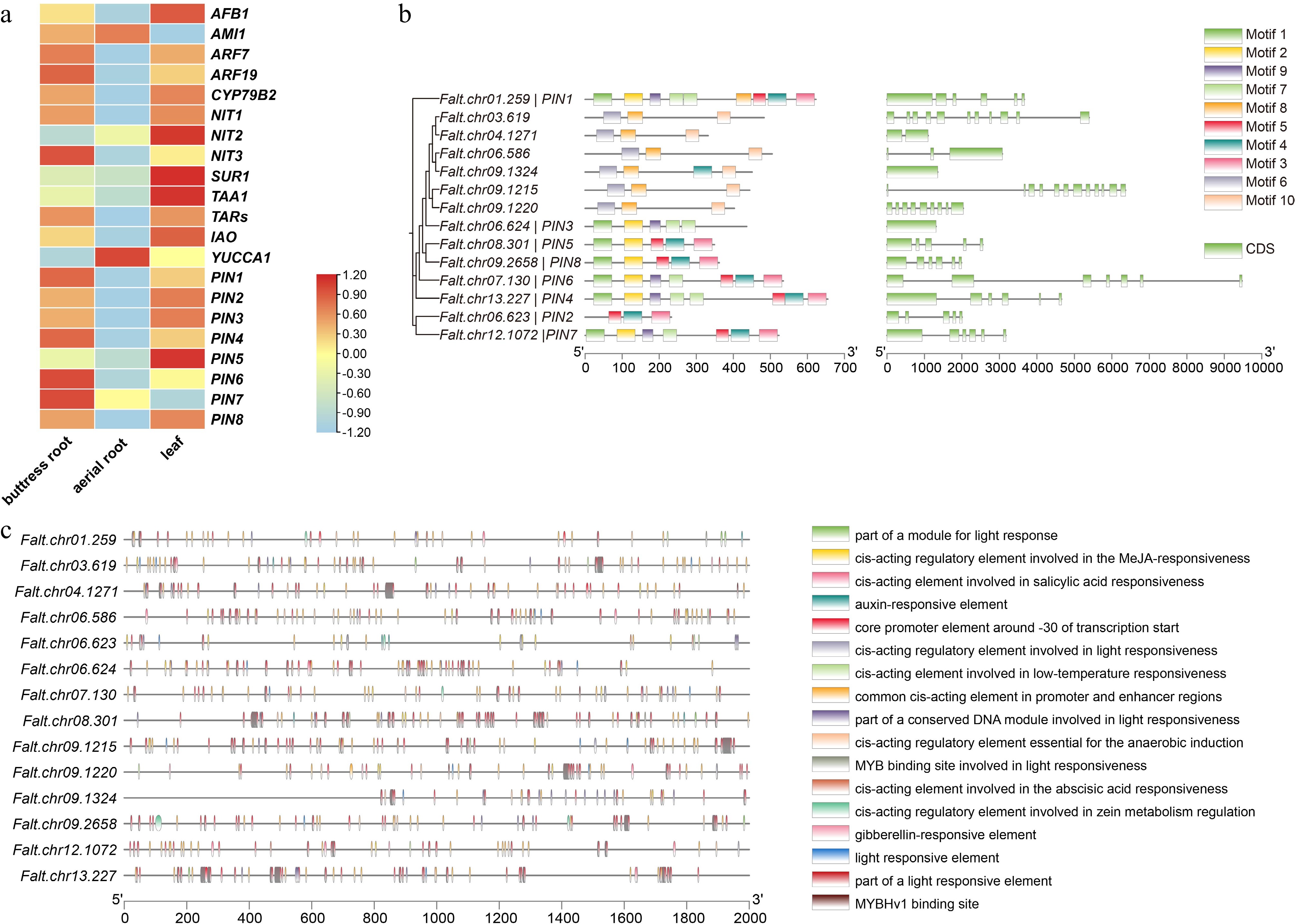
Figure 3.
Characteristics of the PIN gene family in F. altissima. (a) Heatmap of auxin-related genes in F. altissima. (b) Phylogenetic relationships, conserved motifs, and gene structure of the PIN gene family in F. altissima. (c) The role of cis-elements in the PIN gene family of F. altissima.
To examine the hormonal regulation of the PIN gene family in F. altissima, we predicted and analyzed cis-regulatory elements within the 2,000 bp upstream regions of their transcription start sites (Fig. 3c). The analysis revealed that the promoter regions of the 14 PIN family members are enriched with various hormone-responsive elements, including those responsive to light, gibberellin, salicylic acid, methyl jasmonate, abscisic acid, and hypoxia. These regulatory elements suggest that multiple hormonal pathways coordinate the expression of PIN genes, potentially influencing auxin-mediated root growth and adaptation in F. altissima.
Chemical components and psoralen synthesis in F. hirta roots
-
Using extensive targeted metabolomics, we analyzed the chemical composition of the thin, middle, and coarse roots of F. hirta and identified key metabolite categories, including flavonoids, phenolic acids, lipids, lignans and coumarins, terpenoids, and amino acids and their derivatives. A total of 1,238 metabolites were detected (Fig. 4a, Supplementary Table S2). Differential metabolite analysis was conducted through pairwise comparisons among the three root types. The results revealed 601 differentially abundant metabolites between thin and coarse roots, with 389 increased and 212 decreased; 354 different metabolites between thin and middle roots, with 261 increased and 93 decreased; and 439 different metabolites between middle and coarse roots, with 228 increased and 211 decreased (Supplementary Table S3).

Figure 4.
(a) Total metabolites extracted from the thin root, medium root, and course root. (b) Differentially expressed top 20 metabolites between thin root and course root, showing that psoralen is highly expressed in the course root.
Among the top 20 differentially accumulated metabolites between thin and coarse roots, psoralen was highly enriched in coarse roots (Fig. 4b). Psoralen, a member of the coumarin class, is primarily synthesized via the phenylalanine metabolic pathway[43]. To elucidate the biosynthetic mechanism of psoralen in F. hirta roots, we integrated metabolomic and transcriptomic analyses, identifying 11 highly expressed genes involved in its biosynthesis. These include phenylalanine ammonia-lyase (Genes include: Fhir2858, Fhir14383, Fhir10087, Fhir14382), phenylalanine/tyrosine ammonia-lyase (Fhir14383, Fhir10087, Fhir14382), 4-coumarate-CoA ligase (Fhir2815), feruloyl-CoA 6-hydroxylase (Fhir16658, Fhir22185), umbelliferone 6-dimethylallyltransferase (Fhir1941), marmesin synthase (Fhir18558), and psoralen synthase (Fhir12011, Fhir12014) (Fig. 5). Additionally, we found that Fhir14382 and Fhir14383 in phenylalanine ammonia-lyase and phenylalanine/tyrosine ammonia-lyase, Fhir18558 in marmesin synthase, and Fhir12011, Fhir12012, and Fhir12014 in psoralen synthase form gene clusters. These clustered genes appear to be co-regulated in response to specific biosynthetic regulatory signals. Their coordinated expression suggests an active role in promoting psoralen biosynthesis, highlighting their importance in the metabolic regulation of psoralen production in F. hirta roots. By participating in key enzymatic reactions, these genes not only influence psoralen yield but may also affect its bioactivity.
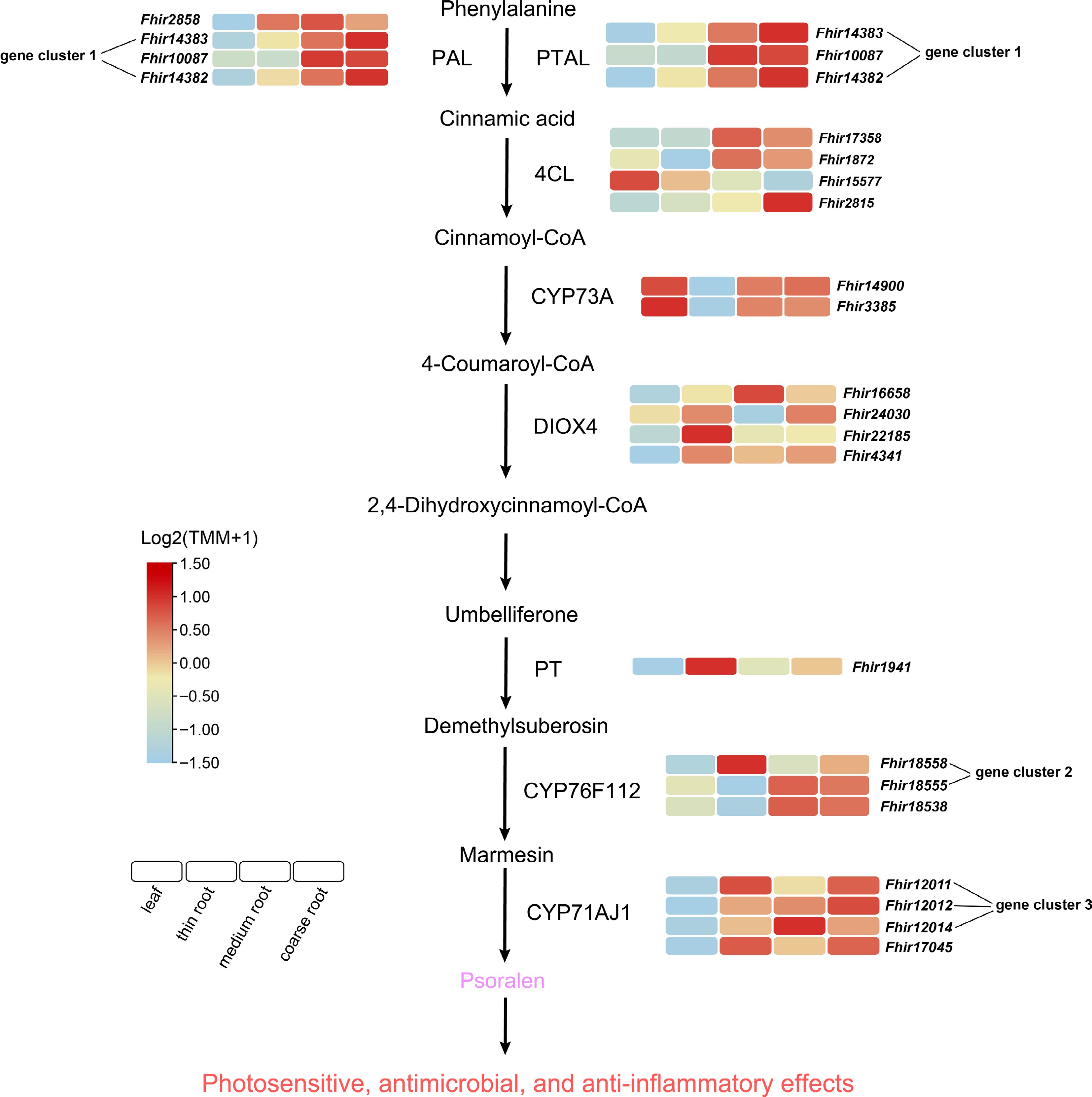
Figure 5.
Biosynthetic pathway of psoralen and gene expression in F. hirta. PT: phenylalanine ammonia-lyase, PTAL: phenylalanine/tyrosine ammonia-lyase, 4CL: 4-coumarate--CoA ligase, CYP73A: trans-cinnamate 4-monooxygenase, DIOX4: trans-4-coumaroyl-CoA 2-hydroxylase, PT: umbelliferone 6-dimethylallyltransferase, CYP76F112: marmesin synthase, CYP71AJ1: psoralen synthase.
-
Fig trees are important landscape trees and hold significant ecological value for plant-insect interaction studies. Substantial progress has been made in fig genome research, including the decoding of the F. microcarpa and F. hispida genomes, the identification of key genes regulating aerial root formation, studies on fig sex chromosomes[1], and the Telomere-to-Telomere (T2T) genome assembly of F. hispida, among others[44]. This study successfully sequenced, assembled, and annotated the genomes of two Ficus species, F. altissima and F. hirta, by integrating Oxford Nanopore, BGI, and Hi-C sequencing technologies. High-quality genome assemblies were obtained, with sizes of 355.9 and 323.7 Mb, respectively, each spanning 13 chromosomes. These assemblies provide a comprehensive reference for future genomic studies of Ficus species. The consistency between genome size estimates obtained through flow cytometry and our assembled genomes further validates the accuracy of our approach. BUSCO evaluation revealed that both genomes exhibit over 98% completeness, highlighting the robustness of our assembly and annotation pipeline. These high-quality genome assemblies provide a solid foundation for comparative genomics, evolutionary biology, and functional genomics research within the Ficus genus and beyond.
Phylogenetic analysis revealed the evolutionary positions of F. altissima and F. hirta within the Ficus genus. Our findings showed notable differences in gene family expansion and contraction between F. altissima, F. hirta, and other Ficus species. Specifically, F. altissima and F. hirta exhibit fewer gene family expansions but more contractions compared to other Ficus species. This suggests that these two species may have followed distinct evolutionary paths in response to their unique ecological environments, offering new insights into the adaptive evolution of Ficus species. Synteny analysis further confirmed that F. altissima and F. hirta share an ancient whole-genome triplication event with V. vinifera, providing additional evidence for the evolutionary history of Ficus species, as observed in previous studies of other plant species[1].
Auxin is a critical regulator of plant growth and development, particularly in root formation[45,46]. Our comprehensive analysis of the PIN gene family in F. altissima provides new insights into its role in root development. We identified 14 PIN family members, which are highly conserved both in number and structure. Their elevated expression in buttress roots suggests a fundamental role in auxin-mediated root architecture development. Furthermore, motif analysis revealed conserved domains across different PIN proteins, indicating functional conservation and potentially suggesting subfunctionalization[47]. Additionally, our study predicts and analyzes the response elements of the PIN gene family in F. altissima. We found that the promoters of these genes predominantly contain cis-acting elements related to light response, plant hormone response, and stress response. This provides important clues for further understanding the role of PIN genes in plant growth, development, and environmental adaptation.
Through integrated metabolomic and transcriptomic analyses, we explored the chemical composition of F. hirta roots, with a particular focus on the biosynthesis of psoralen, a bioactive coumarin with medicinal properties. Our metabolomic profiling identified 1,238 metabolites, revealing distinct metabolic profiles between the thin, middle, and coarse roots. Notably, psoralen was highly enriched in coarse roots, prompting a detailed investigation into its biosynthetic pathway. We identified 11 key genes involved in psoralen biosynthesis, and their clustered organization suggests co-regulation under specific biosynthetic regulatory signals. This clustering may enhance metabolic flux through the pathway, ensuring the efficient production of psoralen[48]. The identification of these genes offers new targets for metabolic engineering and potential biotechnological applications in medicinal plant research. Future studies could explore the regulation of these biosynthetic genes under various environmental and hormonal conditions to optimize psoralen production.
In conclusion, this study leveraged high-quality genomic and metabolomic data to comprehensively explore the genomic structure, gene family evolution, and the mechanisms underlying medicinal compound synthesis in F. altissima and F. hirta. We identified significant differences between the two species in terms of adaptive evolution, emphasized the critical role of PIN genes in the root system development of F. altissima, and elucidated the genetic basis of psoralen biosynthesis in F. hirta. These findings not only deepen our understanding of Ficus biology but also provide valuable resources for future research on species adaptation, ecological interactions, and the medicinal potential of Ficus species. Further investigations integrating functional genomics, ecological adaptation studies, and metabolic engineering have the potential to unlock new applications of Ficus species in forestry, agriculture, and medicine.
This work was supported by the Hainan Province Science and Technology Special Fund (ZDYF2023XDNY050), Hainan Provincial Natural Science Foundation of China (324RC452), National Natural Science Foundation of China (32172614), and supported by the Project of National Key Laboratory for Tropical Crop Breeding (Grant No. NKLTCB202337).
-
The authors confirm contribution to the paper as follows: study design: Wang W, Chen F; plant samples collection: Zhang J, Guo G, Wang S, Liang Y; performing experiments and analyses: Zhang J, Wang D, Zhang J; writing the manuscript: Zhang J. All authors reviewed the results and approved the final version of the manuscript.
-
Raw data High-Fidelity sequencing, Illumina, Hi-C data, genome files are available online at the National Genomics Data Center (https://ngdc.cncb.ac.cn/) with the project ID PRJCA033645. The genome and annotation files have been uploaded to the public database (Figshare), with the specific webpage address: https://figshare.com/articles/dataset/_i_Ficus_altissima_i_and_i_Ficus_hirta_i_genome/28328081.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0025-0014)
-
Received 15 December 2024; Accepted 27 February 2025; Published online 30 June 2025
-
We generated high-quality genome assemblies for Ficus altissima (355.9 Mb) and Ficus hirta (323.7 Mb), each spanning 13 chromosomes, providing a valuable reference for Ficus genomic research.
Phylogenetic and synteny analyses confirmed that F. altissima and F. hirta share an ancient whole-genome triplication event with Vitis vinifera. Their distinct gene family expansion and contraction patterns suggest unique evolutionary trajectories.
Comprehensive analysis of the PIN gene family in F. altissima revealed 14 conserved members with high expression in buttress roots, indicating a crucial role in auxin-mediated root development and environmental adaptation.
Metabolomic profiling of F. hirta roots identified 1,238 metabolites, with psoralen highly enriched in coarse roots. Additionally, we identified 11 key genes involved in psoralen biosynthesis, whose clustered organization may enhance biosynthetic efficiency.
These findings advance our understanding of Ficus genome evolution, root system development, and medicinal compound biosynthesis, offering new insights for applications in forestry, agriculture, and medicine.
-
# Authors contributed equally: Junyu Zhang, Dantong Wang
- Supplementary Table S1 Flow cytometry results.
- Supplementary Table S2 Metabolite categories in Ficsu hirta roots.
- Supplementary Table S3 Differential metabolite analysis in Ficsu hirta roots.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang J, Wang D, Guo G, Wang S, Zhang J, et al. 2025. Genomic and metabolomic analysis of Ficus altissima and Ficus hirta: evolutionary divergence and medicinal implications. Tropical Plants 4: e022 doi: 10.48130/tp-0025-0014 

Genomic and metabolomic analysis of Ficus altissima and Ficus hirta: evolutionary divergence and medicinal implications
- Received: 15 December 2024
- Revised: 11 February 2025
- Accepted: 27 February 2025
- Published online: 30 June 2025
Abstract: Ficus altissima is an evergreen tree characterized by a distinctive buttress root system, which plays a crucial role in its ecosystem. Ficus hirta, a shrub to small tree, is well known for the medicinal properties of its roots. In this study, we sequenced the genomes of both species using third-generation Oxford Nanopore sequencing technology, revealing genome sizes of 355.9 Mb for F. altissima and 323.7 Mb for F. hirta. By integrating publicly available Ficus genome data, we constructed a phylogenetic tree and estimated that F. altissima and F. hirta diverged approximately 41 million years ago (Mya). In F. altissima, we identified genes involved in the auxin signaling pathway and found that members of the PIN gene family were highly expressed in buttress roots. To further investigate their role, we conducted a comprehensive analysis of the PIN gene family, including phylogenetic relationships, gene structure, conserved motifs, and protein domains. Promoter analysis revealed that the regulatory regions of 14 PIN genes contain cis-acting elements associated with light response, hormone signaling, and stress response. Additionally, through extensive targeted metabolomic analysis, we used high-performance liquid chromatography (HPLC) to detect a diverse range of secondary metabolites in the roots of F. hirta. By integrating these findings with transcriptomic data, we identified key genes involved in the psoralen biosynthetic pathway. The high-quality reference genomes and metabolomic data generated in this study provide valuable resources for advancing research on Ficus root biology and its bioactive compounds.
-
Key words:
- Ficus altissima /
- Ficus hirta /
- PIN gene family /
- Psoralen biosynthetic pathway