









-
Panax notoginseng, a valuable traditional Chinese medicinal herb, is renowned for its notable pharmacological properties, including promoting blood circulation, removing blood stasis, hemostasis, and relieving pain; it is widely used in the prevention and treatment of cardiovascular and cerebrovascular diseases[1]. However, continuous monoculture of P. notoginseng has caused a high incidence of root rot disease, which results in substantial yield losses and degradation of root quality. Root rot has become a major limiting factor for the sustainable development of its cultivation[2]. Current studies have identified Fusarium oxysporum as the primary causative agent of P. notoginseng root rot[3], F. oxysporum and other Fusarium species are highly destructive, producing a wide range of toxic secondary metabolites[4]. Traditional disease management practices in agricultural production largely rely on chemical fungicides[5,6]. Although these approaches can provide short-term control, they often entail significant drawbacks, including disruption to soil microbial communities and ecological balance[7], as well as the emergence of fungicide-resistant pathogens[8]. These limitations highlight the urgent need for alternative control strategies that are both effective and environmentally sustainable. In recent years, biological control has emerged as a promising approach for managing soil-borne diseases due to its strong antagonistic activity against pathogens and environmentally friendly characteristics[9−11]. Numerous studies have demonstrated that beneficial microorganisms can directly suppress pathogen colonization and spread, and modulate the rhizosphere microbial community, thereby enhancing plant immunity and overall health[12,13]. Notably, genera such as Trichoderma and Bacillus are widely recognized as effective biocontrol agents due to their ability to induce systemic resistance in plants and exert direct antifungal effects[14,15]. Moreover, increasing evidence indicates that plants under pathogen attack actively recruit plant growth-promoting rhizobacteria (PGPR) via root exudates to trigger systemic defenses and enhance resistance to pathogens[16,17]. These beneficial microbes not only contribute to disease suppression but also facilitate nutrient uptake, enhance plant growth, and trigger host systemic responses[18]. Although bacterial biocontrol agents have been widely studied, fungal biocontrol agents—particularly their antagonistic mechanisms and bioactive metabolites—remain relatively underexplored. Therefore, the isolation and characterization of fungal strains exhibiting robust antagonistic activity from the rhizosphere are of paramount importance for the development of effective and sustainable biocontrol strategies. Among these, the genus Trichoderma has garnered increasing attention for its broad-spectrum antifungal activity and ability to promote plant growth, making it an ideal candidate for green disease management in agriculture[19,20].
The genus Trichoderma, belonging to the order Hypocreales within the phylum Ascomycota, is among the most widely distributed beneficial fungi in soil ecosystems and represent key members of plant growth-promoting rhizosphere microbiota[21]. These fungi are pivotal in controlling fungal plant pathogens, mainly by synthesizing and secreting a diverse array of secondary metabolites that inhibit the growth and development of pathogens effectively[22]. In addition to their biocontrol capabilities, certain Trichoderma strains also exhibit nutrient-mobilizing properties, such as phosphate solubilization, potassium release, and nitrogen fixation, thus promoting nutrient uptake and plant growth while concurrently suppressing pathogens[23,24]. Trichoderma are closely associated with soil microbial communities. They typically colonize the rhizosphere through the production of abundant conidia and establish mutualistic interactions with host plants[25]. Their multifaceted modes of action include the secretion of hydrolytic enzymes such as cellulases, chitinases, and glucanases that degrade complex polysaccharides in plant cell walls, thereby contributing to soil organic matter decomposition and the carbon cycle[26]. In addition, they exhibit strong antagonistic effects against various plant pathogens through mechanisms such as competition for nutrients and space, direct mycoparasitism, and antibiosis, making them widely used in biological control strategies[27−29]. Moreover, Trichoderma spp. can induce systemic resistance (ISR) in plants and produce phytohormones such as indole-3-acetic acid (IAA), enhancing plant resilience to biotic and abiotic stresses and promoting overall crop health[30]. Numerous studies have confirmed the potent antagonistic activity of Trichoderma strains against F. oxysporum[31,32]. For instance, Trichoderma citrinoviride HT-1 was reported by Chen et al. to inhibit F. oxysporum with an efficacy of 71.85%, demonstrating its promising potential for managing soil-borne diseases[33]. Similarly, Feng et al. showed that T. hamatum achieved an inhibition rate of 68.07% against F. oxysporum[3]. However, the antagonistic efficacy of different Trichoderma strains varies significantly depending on the Fusarium species. For example, T. viride showed an inhibition rate of 80.17% against F. proliferatum, but only 70.46% against F. verticillioides[34]. These findings indicate that the antagonistic mechanisms employed by Trichoderma against Fusarium pathogens remain insufficiently understood and warrant further investigation.
In recent years, numerous studies have demonstrated that various Trichoderma species are highly effective in the biocontrol of soil-borne plant diseases[35,36]. Despite belonging to the same genus, these strains exhibit considerable diversity in genetic background, phenotypic traits, and developmental characteristics, largely due to interspecific and genome-level variations. Whole-genome sequencing provides an opportunity to dissect the functional genes and regulatory networks of Trichoderma strains at the molecular level, offering a more comprehensive understanding of their roles in biological control[37]. With the advancement of omics technologies, researchers have increasingly explored the genomic landscape of Trichoderma, revealing gene regulatory mechanisms and interactions with plant hosts that underpin their biocontrol capabilities. These insights lay the theoretical foundation for the broader application of Trichoderma in agricultural disease management[38,39]. For instance, whole-genome sequencing of T. koningiopsis strains UKM-M-UW RA5, RA6, and RA3a revealed the presence of secondary metabolic pathways potentially involved in antimicrobial activity, highlighting their promise in synthetic biology and the elucidation of novel antifungal mechanisms[40]. Similarly, genomic studies of T. harzianum strains IOC-3844 and CBMAI-0179 have enhanced our understanding of the evolutionary relationships within the Trichoderma genus and contributed to a more complete phylogenetic framework[41]. Notably, He et al. performed whole-genome sequencing of the endophytic Fusarium sp VM-40, identifying multiple genes involved in secondary metabolism. By combining genome mining using the antiSMASH platform with non-targeted metabolomic analysis, the biosynthetic potential of this strain was further elucidated[42]. Therefore, systematic genomic analyses of Trichoderma strains not only deepen understanding of their genetic attributes and functional mechanisms but also offer theoretical support for their application in the control of soil-borne pathogens. A detailed understanding of Trichoderma genome structure and function will help clarify their antagonistic mechanisms against Fusarium species that cause root rot in P. pseudoginseng. This study aims to provide a scientific and theoretical basis for the biocontrol of root rot disease in P. pseudoginseng.
Strain Z35 was isolated and purified from the rhizosphere soil of healthy P. pseudoginseng plants grown under long-term continuous cropping conditions. In vitro dual-culture assays demonstrated that Z35 exhibits significant inhibitory activity against F. oxysporum. To further explore its biocontrol potential, the antagonistic activity of Z35 against additional Fusarium species was assessed, including F. lateritium, F. clavum, and F. foetens. The results revealed that Z35 possesses broad-spectrum antifungal activity, indicating its potential applicability in controlling a wider range of Fusarium-induced plant diseases (Supplementary Fig. S1). Our previous field experiments further confirmed the biocontrol potential of Z35, showing that it not only suppresses pathogen infection effectively but also promotes plant growth and enhances stress resistance in P. pseudoginseng. However, the lack of genomic information and the unidentified profile of secondary metabolites have limited a deeper understanding of its biocontrol mechanisms and hindered its potential for industrial application. In this study, genomic and metabolomic approaches were employed to investigate the antagonistic mechanisms of strain Z35 against pathogenic fungi. These findings provide new insights into the functional basis of Z35-mediated biocontrol, offer theoretical support for the biological management of root rot disease in P. pseudoginseng, and lay a foundation for the exploration and application of beneficial microbial resources.
-
A fungal strain designated Z35 was isolated from the rhizosphere soil of healthy P. pseudoginseng plants cultivated in continuously cropped fields, and has been preserved at the National-Local Joint Engineering Research Center on Germplasm Innovation & Utilization of Chinese Medicinal Materials in Southwest China, Yunnan Agricultural University. The strain was first cultured on Potato Dextrose Agar (PDA) plates and incubated at 28 °C for 5–7 d to obtain mature colonies. Fresh mycelia were then transferred to Potato Dextrose Broth (PDB) and incubated in a rotary shaker at 28 °C and 200 rpm for 7 d. After incubation, the culture was filtered through sterile gauze to separate the mycelia from the fermentation broth, followed by centrifugation. The harvested mycelia were flash-frozen in liquid nitrogen and stored at –80 °C for subsequent experimental analyses.
Determination of bacterial inhibition rate of strain Z35
-
The antagonistic activity of strain Z35 against F. oxysporum was assessed using a modified dual-culture assay based on the method described by Chen et al.[43]. A five-point confrontation assay was performed on Potato Dextrose Agar (PDA) plates to evaluate the inhibitory effect of Z35. PDA plates inoculated only with F. oxysporum served as negative controls. Each treatment was conducted in triplicate. Plates were incubated at 25 °C for 2–3 d, after which the diameters of the F. oxysporum colonies were measured. The inhibition rate was calculated using the following formula: Inhibition rate (%) = [(Dc – Dt)/Dc] × 100, where Dc is the colony diameter of F. oxysporum in the control group, and Dt is the colony diameter in the treatment group.
Genome sequencing and assembly
-
High-quality genomic DNA of strain Z35 was extracted using a modified cetyltrimethylammonium bromide (CTAB) method. DNA purity, concentration, and integrity were assessed to ensure they met the requirements for library preparation. The fragment size of the constructed library was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Single Molecule Real-Time (SMRT) sequencing was then performed on the PacBio Sequel II platform. Raw reads were subjected to quality control using SOAPnuke to remove low-quality sequences and obtain clean reads. The genome size and heterozygosity were preliminarily estimated based on k-mer analysis. High-quality reads (length > 1,000 bp) were selected using SMRT Link software (v10.1.0) for downstream analysis. De novo genome assembly was carried out using Flye (v2.8.3), resulting in contig-level genomic sequences. To assess mapping metrics, including alignment rate, genome coverage, and sequencing depth distribution, raw reads were aligned back to the assembled genome using BWA (v0.7.17) and minimap2 (v2.21). Genome completeness was evaluated with BUSCO (v4.0.3) by analyzing the proportions of complete, fragmented, and missing conserved genes. Finally, the overall quality of the genome assembly was comprehensively assessed using Merqury (v1.3), and LTR_retriever (v2.9.0).
Genome component prediction
-
Genome annotation of strain Z35 included the identification of repeat sequences, gene structure prediction, functional gene annotation, non-coding RNA detection, and biosynthetic gene clusters (BGCs) analysis. Protein-coding genes were predicted using Glimmer (v3.02). Repetitive sequences were annotated based on the RepBase database (
www.girinst.org/repbase ) using a combination of RepeatMasker (v4.1.2), RepeatProteinMask (v1.36), and Tandem Repeats Finder (TRF, v4.09). Gene structure prediction integrated both homology-based and de novo approaches. Exonerate (v2.4.0), PASA (v2.4.1), and GeneMark (v4.65) were used for preliminary predictions, and results were integrated using MAKER (v3.01.03) to produce a high-confidence gene set after filtering out transposable elements and potential pseudogenes. For non-coding RNA annotation, tRNA genes were identified using tRNAscan-SE (v2.0.9), rRNA genes were predicted with RNAmmer (v1.2), and miRNAs and snRNAs were annotated using INFERNAL (v1.1.4) with reference to the Rfam database. To explore the biosynthetic potential of strain Z35, the genome sequence was analyzed using the antiSMASH platform (v7.0,https://antismash.secondarymetabolites.org/#!/start ) for the prediction of secondary metabolite biosynthetic gene clusters.Genome functional annotation and analysis
-
Functional annotation of predicted genes was performed using multiple public databases, including the Non-Redundant Protein Database (NR), Kyoto Encyclopedia of Genes and Genomes (KEGG), Eukaryotic Orthologous Groups (KOG), Gene Ontology (GO), and SwissProt. The genome-encoded protein sequences were aligned to the NR, SwissProt, KOG, and KEGG databases using BLASTp (v2.9.0). GO functional annotation was carried out using InterProScan (v5.32-71.0), which integrates diverse protein signature databases to assign Gene Ontology terms.
Phylogenomic and comparative genomic analysis
-
Phylogenetic analysis and orthologous gene identification were conducted between strain Z35 and ten representative fungal strains. The ITS sequences and genome assemblies of all reference strains were retrieved from the National Center for Biotechnology Information (NCBI) database. A maximum likelihood phylogenetic tree based on ITS sequences was constructed using MEGA (v10.0.5), with bootstrap values calculated from 1,000 replicates. Divergence time estimation was performed using r8s (v1.81) and PAML (v4.10.0). The most recent common ancestor divergence times were obtained and calibrated using data from the TimeTree database.
Secondary metabolite analysis and identification
-
A single colony of strain Z35 was inoculated into potato dextrose broth (PDB) and incubated at 28 °C with shaking at 200 rpm for 7 d. After cultivation, the fungal mycelia and fermentation broth were separated by filtration through sterile gauze followed by centrifugation. The fermentation supernatant was mixed with 50% glycerol and stored at –80 °C for subsequent analysis. Samples were then sent to Frasergen Bioinformatics Co., Ltd. (Wuhan, China) for non-targeted metabolomic profiling. Liquid chromatography was performed using an LC-30A system (Shimadzu, Japan) equipped with a Waters ACQUITY Premier HSS T3 column (1.8 µm, 2.1 mm × 100 mm). The mobile phases consisted of 0.1% formic acid in water (A), and 0.1% formic acid in acetonitrile (B). The chromatographic conditions were as follows: column temperature, 40 °C; flow rate, 0.4 mL/min; injection volume, 4 μL. The gradient program was: 0–2 min, 95%–80% A; 2–5 min, 80%–40% A; 5–6 min, 40%–1% A; 6–7.5 min, 99% B; 7.5–7.6 min, 99%–5% B; 7.6–10 min, 5% B. Mass spectrometry was carried out using a TripleTOF 6600+ instrument (SCIEX, Foster City, CA, USA) with an electrospray ionization (ESI) source. The MS parameters were set as follows: ion source temperature, 550 °C; ion spray voltage, 5,000 V; curtain gas, 35 psi; nebulizer gas (GS1), 50 psi; auxiliary gas (GS2), 60 psi; declustering potential, 60 V; collision energy for MS1, 10 V; and for MS2, 30 V.
-
Strain Z35 exhibited rapid mycelial growth, forming fluffy, white, cotton-like colonies. In dual-culture confrontation assays with F. oxysporum, a distinct inhibition zone was observed around the periphery of the Z35 colony, suggesting the secretion of diffusible secondary metabolites that may suppress the growth of the pathogen. Using the five-point confrontation method on PDA plates incubated at 28 °C, Z35 demonstrated strong antagonistic activity against F. oxysporum after 2 d of co-culture, with an inhibition rate of up to 75%, indicating a pronounced biocontrol potential (Fig. 1). To further elucidate its antagonistic mechanism, whole-genome sequencing and untargeted metabolomic analyses were conducted on strain Z35, aiming to uncover its antimicrobial genetic background and the underlying bioactive metabolites at both the genomic and metabolic levels.
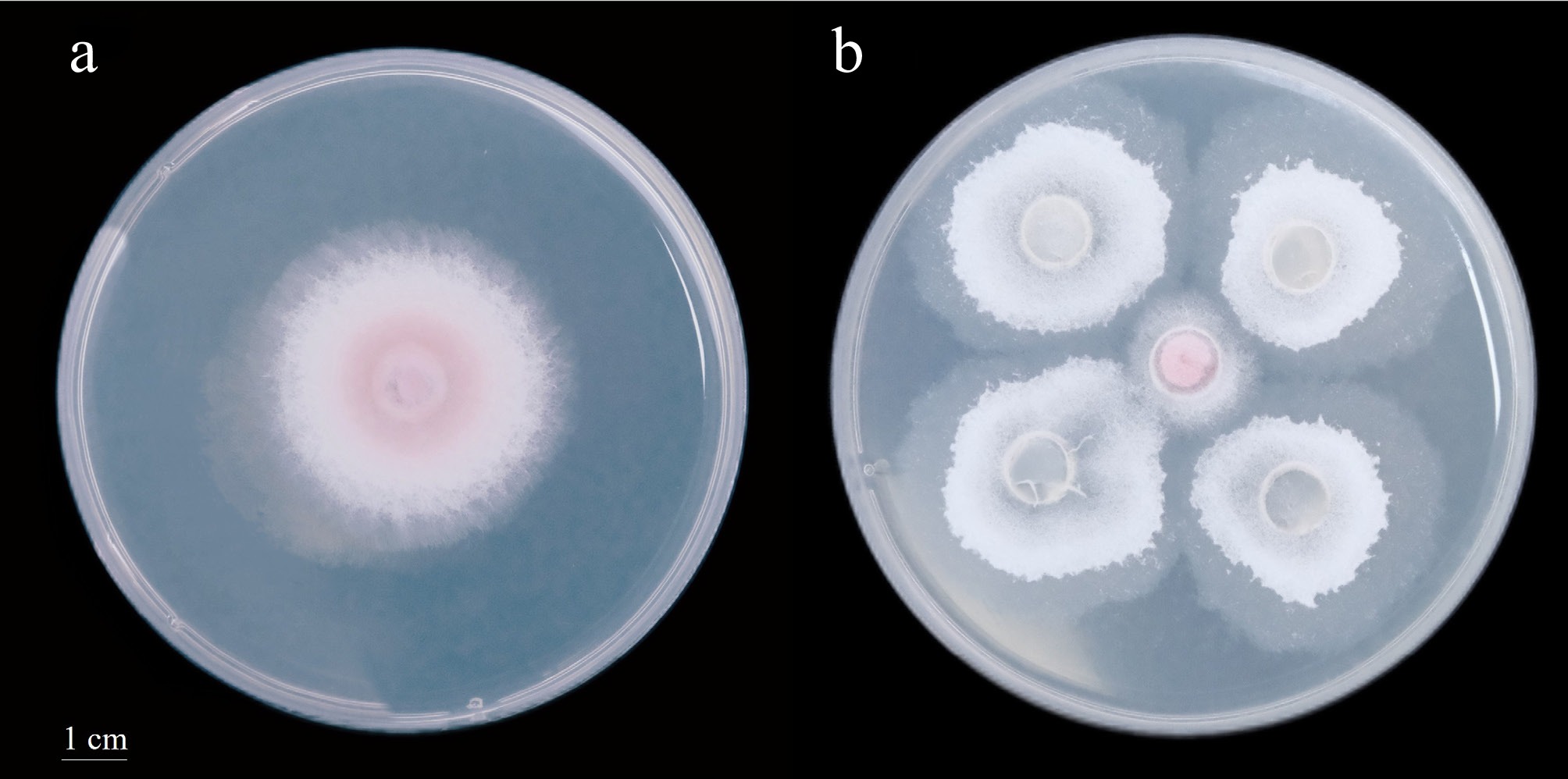
Figure 1.
Dual culture assay between strain Z35 and F. oxysporum after 2 d of incubation (the diameter of the petri dish is 9 cm).
(a) F. oxysporum cultured alone as the control. (b) Antagonistic interaction between Z35 and F. oxysporum using the five-point confrontation method on PDA medium.Genome assembly, evaluation, and annotation of Z35
-
To characterize the genomic features of strain Z35, SMRT sequencing was performed using the PacBio Sequel II platform. A total of 5,853,172 clean reads were obtained, yielding 38,649,990 bp of sequence data. The genome was assembled into seven contigs and one scaffold, with a total size of 38.65 Mb (Fig. 2), a contig N50 of 5.85 Mb, and a GC content of 47.48%. To assess assembly accuracy, Illumina short reads were mapped to the assembled genome using BWA, resulting in a high mapping rate of 99.82%, with an average sequencing depth of 308.32× and 100% coverage, indicating excellent sequence consistency. Additionally, third-generation reads were aligned to the polished genome using Minimap2, achieving an average depth of 141.96× and a mapping rate of 99.87%. Genome completeness was further evaluated using BUSCO, which identified 99.9% of the expected conserved single-copy orthologs, suggesting a high level of assembly completeness (Supplementary Table S1).
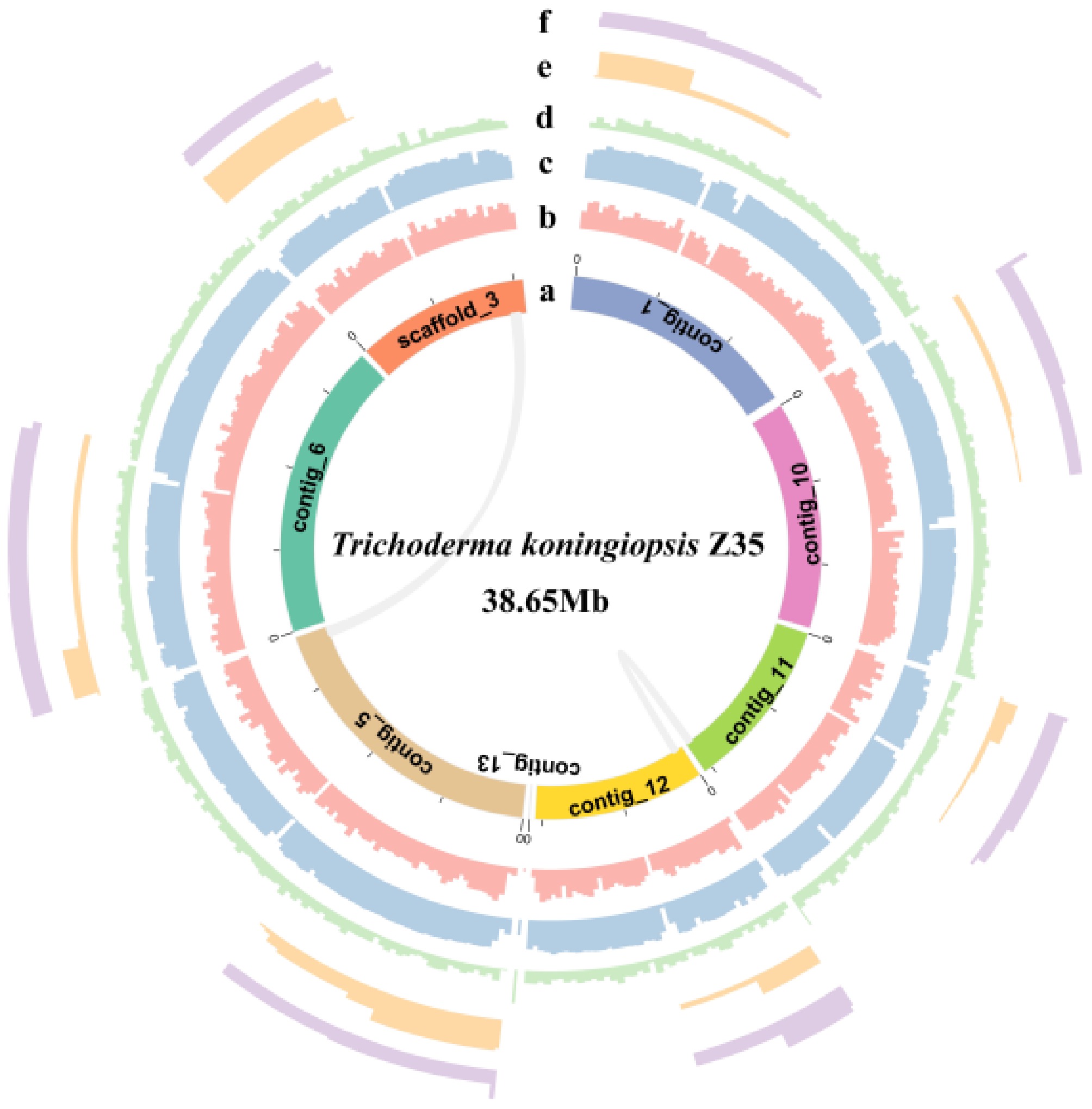
Figure 2.
Circos plot of the genome of strain Z35. From the innermost to the outermost ring, the plot displays the scaffold, gene density, GC content, GC skew, Copia, and Gypsy elements.
A total of 13,153 protein-coding genes were predicted in the Z35 genome, exceeding the number identified in three closely related species. The average gene length was 1,158.61 bp (Supplementary Fig. S2). On average, each gene contained 2.32 exons, with an average exon length of 500.33 bp and an average intron length of 229.25 bp (Supplementary Table S2). Transposable elements (TEs) were annotated using RepeatMasker, identifying 331, 631 bp of repetitive sequences, accounting for 0.86% of the entire genome. Among these, long terminal repeat (LTR) elements were the most abundant, comprising 0.44%, followed by DNA transposons (0.29%) and long interspersed nuclear elements (LINEs) (0.22%). Unclassified elements accounted for only 0.01%. In addition, 305 non-coding RNAs were predicted, including 231 tRNAs, 52 rRNAs, and 22 snRNAs, comprising approximately 0.12% of the genome (Supplementary Table S3). Functional annotation of protein-coding genes was performed using the Nr, GO, KEGG, and SwissProt databases. In total, 11,192 genes were successfully annotated (Supplementary Table S4). Among the 8,264 genes classified by the GO database, those related to nucleic acid catalytic activity were the most enriched in the Molecular Function category, while genes involved in metabolic processes and cellular processes dominated the Biological Process category (Supplementary Fig. S3a). KOG annotation assigned 4,950 genes to four major categories: Cellular Processes and Signaling, Information Storage and Processing, Metabolism, and Poorly Characterized, with the majority falling into Cellular Processes and Signaling (Supplementary Fig. S3b). KEGG analysis annotated 10,754 genes, which were mapped to five major pathways. The largest group of genes participated in metabolic pathways, particularly carbohydrate metabolism and amino acid metabolism, which were the most enriched functional categories (Supplementary Fig. S3c).
Taxonomic identification and comparative genomic analysis of strain Z35
-
Phylogenetic analysis based on ITS sequences was conducted for nine Trichoderma strains (Fig. 3). The ITS-based phylogenetic tree divided the strains into two major clades. Strain Z35 clustered closely with T. koningiopsis DF239, indicating a close evolutionary relationship. Therefore, strain Z35 was identified as T. koningiopsis.
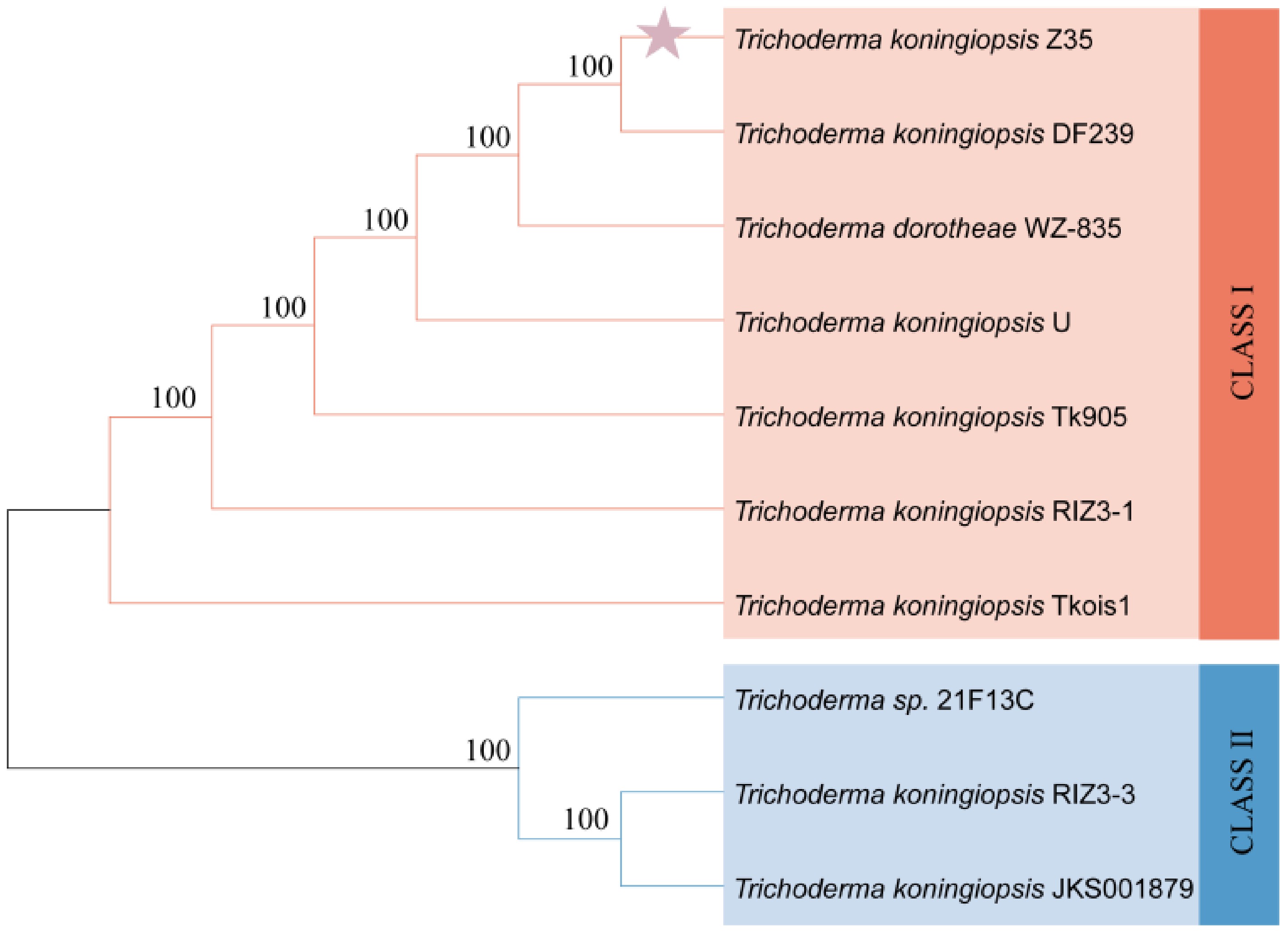
Figure 3.
Phylogenetic tree of strain Z35 based on ITS sequence alignment.
The tree was constructed using ITS sequences from nine Trichoderma strains.
A comparative genomic analysis was performed between Trichoderma strain Z35 and five other publicly available Trichoderma genomes (Supplementary Table S5). The genome size of Z35 was comparable to that of strain T51. Notably, Z35 exhibited the highest contig N50 value (5.9 Mb), indicating the best assembly continuity among the compared genomes. In addition, the GC content of Z35 showed minimal variation compared with the other three strains, suggesting a relatively conserved nucleotide composition across these Trichoderma species.
Phylogenomic and evolutionary analyses of strain Z35
-
Single-copy orthologous genes from seven Trichoderma species and three outgroup fungi were aligned to infer the evolutionary relationships among the ten species. Divergence time estimation suggested that Thermothelomyces thermophilus, representing the Thermothelomyces outgroup, diverged approximately 247.5 million years ago (MYA). The two other outgroups, F. oxysporum and Paramyrothecium foliicola, were estimated to have diverged at 136.4 MYA and 126.2 MYA, respectively. Within the Trichoderma clade, divergence from Metarhizium anisopliae and Purpureocillium takamizusanense was estimated at around 113.4 MYA. The target strain of this study, T. koningiopsis Z35, was predicted to have diverged from T. asperellum approximately 6.7 MYA (Fig. 4). During the evolutionary history of the ten fungal species analyzed, gene family contractions occurred more frequently than expansions. Among Trichoderma species, the number of expanded gene families in T. asperellum, T. koningiopsis, T. citrinoviride, T. simmonsii, and T. harzianum was 117, 160, 51, 173, and 102, respectively. In contrast, the number of contracted gene families in these species was 215, 141, 840, 67, and 194, respectively—indicating a predominance of gene family contraction over expansion in the evolutionary process (Fig. 4).
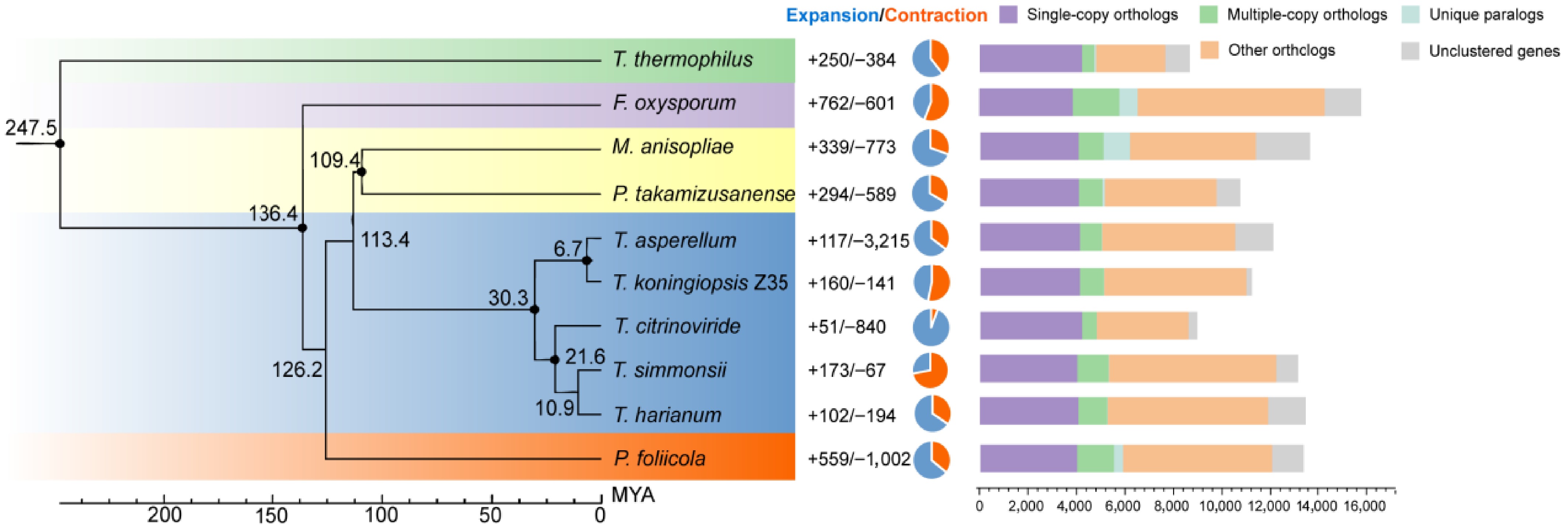
Figure 4.
Maximum likelihood phylogenetic tree based on single-copy orthologous genes from seven Trichoderma species and three outgroup taxa. All nodes are supported with 100% bootstrap values. The black numbers on the branches indicate estimated divergence times in MYA. Gene family expansions and contractions for each species are represented by blue and orange pie charts, respectively.
Analysis of secondary metabolite biosynthesis in strain Z35
-
To further investigate the biosynthetic potential and localization of secondary metabolite production in T. koningiopsis Z35, BGCs were predicted using the Antibiotics and Secondary Metabolite Analysis Shell (antiSMASH). A total of 49 BGCs were identified in the genome. Among these, 41 BGCs were located on seven contigs, while the remaining eight were distributed on the scaffold. The predicted BGCs included 17 T1PKS clusters, 10 NRPS clusters, nine NRPS-like clusters, eight terpene clusters, nine fungal RiPP-like clusters, three isocyanide clusters, and three hybrid isocyanide-NRPS clusters (Fig. 5).
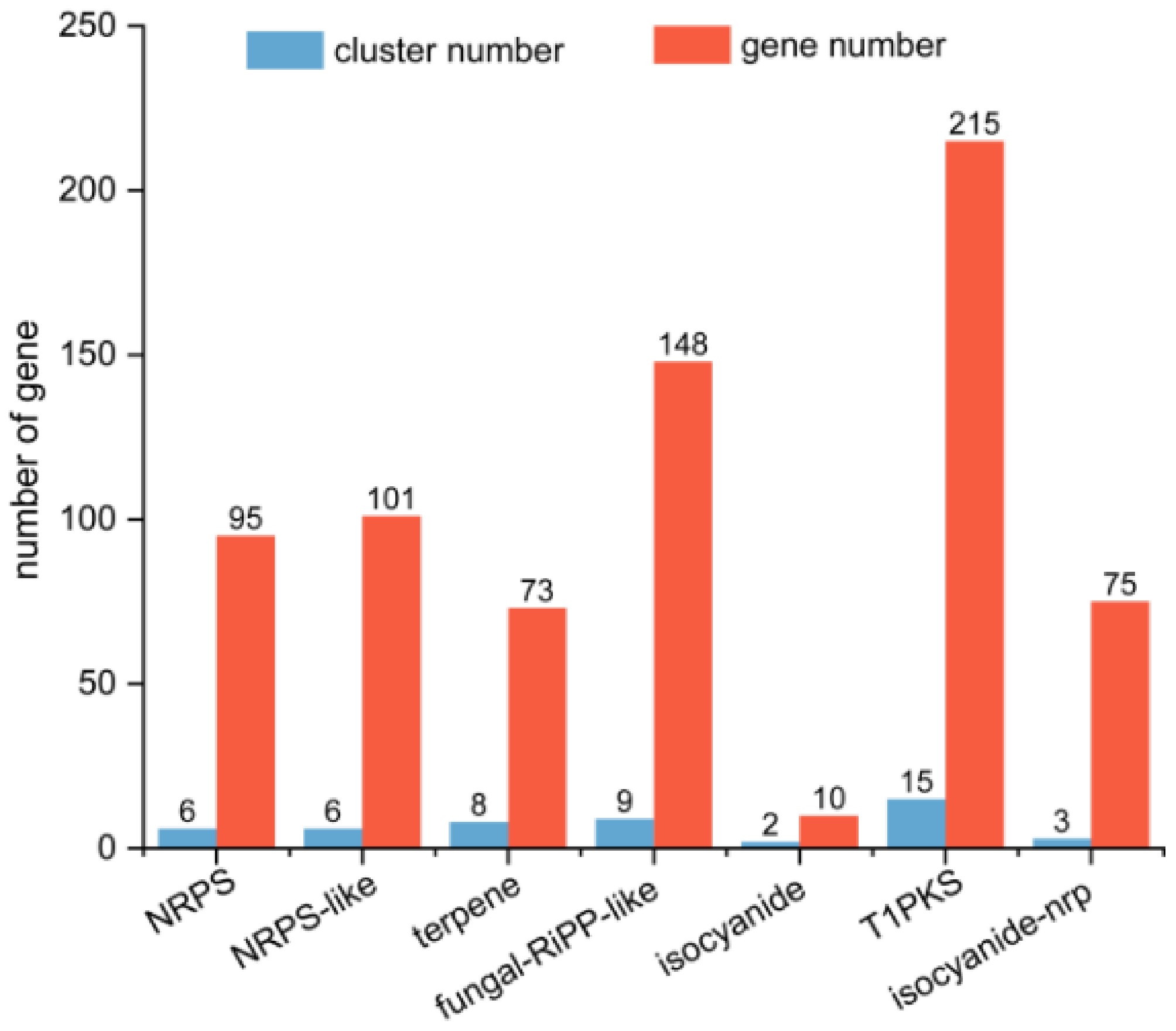
Figure 5.
Quantitative analysis of secondary metabolite biosynthetic gene clusters (BGCs) in T. koningiopsis Z35. Abbreviations: NRPS, NonRibosomal Peptide Synthetase; NRPS-like, NonRibosomal Peptide Synthetase-Like; T1PKS, Type I PolyKetide Synthase; Isocyanide-NRP, Isocyanide–NonRibosomal Peptide Synthetase.
Among the predicted BGCs, two clusters showed 100% amino acid sequence identity with known gene clusters, including one NRPS-like cluster, and one terpene cluster. These were predicted to be responsible for the biosynthesis of choline and clavaric acid, respectively. Choline serves as a precursor for phospholipid synthesis and plays a critical role in membrane formation. It may also indirectly inhibit pathogen invasion by inducing host systemic resistance. Further analysis of this cluster revealed the presence of core biosynthetic genes associated with the synthesis of SQCY-1, a nonribosomal peptide compound known for its inhibitory effects on pathogen growth and reproduction (Fig. 6a). Clavaric acid, a triterpenoid compound, was identified as a product of a terpene cluster. It acts as a farnesyltransferase (FTase) inhibitor. FTase is essential in post-translational protein modification, particularly in the farnesylation process during protein prenylation, which is crucial for membrane localization, signaling, and function. Inhibiting FTase can interfere with the membrane localization of key pathogenic proteins (e.g., Ras), thereby exerting antifungal effects. Additionally, the core biosynthetic genes in this cluster encode enzymes belonging to the α-amidase (α-AM-Amid) superfamily, which are involved in blocking the biosynthesis of pathogen-derived toxins (Fig. 6b). One NRPS cluster showed 75% amino acid identity to a known cluster and was predicted to produce a series of compounds, including metachelin C, metachelin A, metachelin A-CE, metachelin B, dimerumic acid 11-mannoside, and dimerumic acid. Structural analysis of this NRPS cluster also revealed the presence of polyketide synthase (PKS)-like domains, which may influence the dehydrogenation and reduction steps of polyketide backbones—potentially affecting fungal membrane-disrupting toxin biosynthesis, and promoting the production of antimicrobial compounds in biocontrol fungi (Fig. 6c). Furthermore, one NRPS-like cluster and one T1PKS cluster exhibited 50% sequence identity to known BGCs, and were predicted to synthesize ascochlorin and trichoxide. These clusters contained genes encoding several biosynthetic superfamilies, including PRK12316, C-NRPS-like, AcpA, SAT, and BD-FAE. These genes are known to be involved in the biosynthesis of nonribosomal peptide antibiotics with strong antifungal activity, regulation of resistance-related metabolites, and production of bioactive compounds such as lipopeptides and polyphenols (Fig. 6d, e). In addition, six other BGCs shared less than 50% amino acid sequence similarity with known clusters, while 38 BGCs had no detectable matches in any database (Supplementary Table S6). Five biosynthetic gene clusters (BGCs) were predicted from the genome of T. koningiopsis Z35 using antiSMASH. Each cluster was annotated based on the presence of conserved biosynthetic domains and sequence similarity to reference clusters in the MIBiG database. Contig11.2 shares 100% similarity with a known choline-producing BGC (MIBiG ID: BGC0002276.2), while Contig12.3 resembles the metachelin gene cluster (MIBiG ID:BGC0001248.3). Notably, antiSMASH analysis of T. koningiopsis Z35 predicted five biosynthetic gene clusters (BGCs) related to the biosynthesis of compounds such as choline, clavaric acid, metachelin, ascochlorin, and trichoxide. While these specific compounds were not detected in the untargeted metabolomic profiling, choline, a related quaternary ammonium compound, was consistently identified. Additionally, a variety of compounds belonging to the same structural classes—such as terpenoids, polyketides, and polyphenols—were detected. These results provide indirect evidence supporting the metabolic potential of the predicted BGCs.
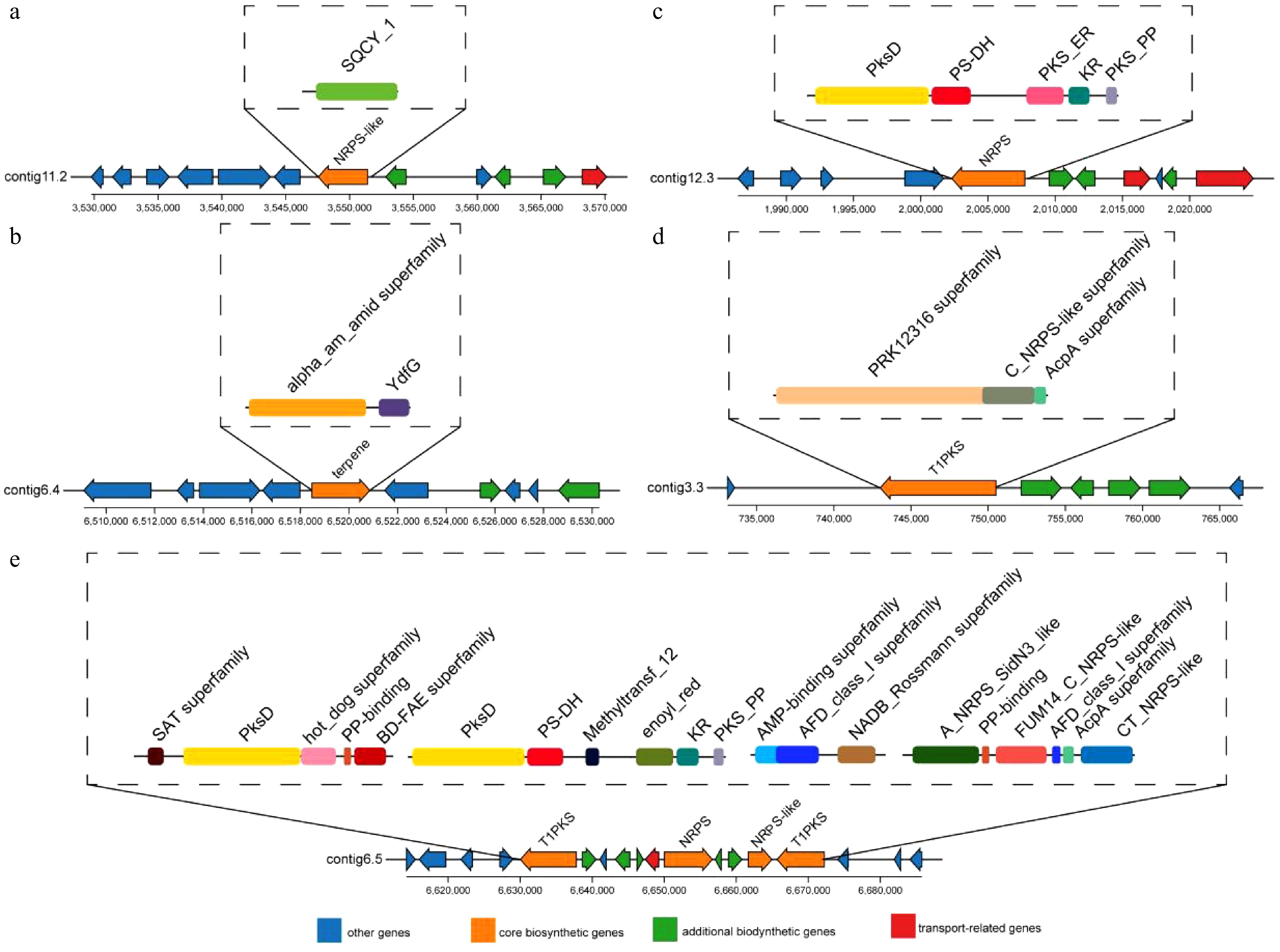
Figure 6.
Structural analysis of five BGCs identified in the T. koningiopsis Z35 genome using antiSMASH.
(a) Nonribosomal peptide synthetase-like (NRPS-like) BGC; (b) Terpene synthase BGC; (c) Nonribosomal peptide synthetase (NRPS) BGC; (d) Type I polyketide synthase (T1PKS) BGC; (e) T1PKS, NRPS, NRPS-like BGC.Analysis of secondary metabolites in the fermentation broth of strain Z35
-
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) was employed to analyze the fermentation broth of T. koningiopsis Z35. A total of 5,087 identifiable metabolites were detected and quantified. Among these, choline derivatives (e.g., methacholine) and triterpenoids (e.g., salannin, medicagenic acid) exhibited potential antifungal activity, while indole and its derivatives (e.g., indole, indole-3-acetamide) were associated with the promotion of plant growth and development (Supplementary Table S7). Under the positive ion mode, amino acids and their metabolites constituted the largest proportion of the detected compounds (31.74%), followed by benzene and substituted derivatives (15.65%). A similar trend was observed under the negative ion mode (Supplementary Fig. S4). Detailed metabolite classification is provided in Supplementary Table S7. Principal component analysis (PCA) was performed to assess the overall variation in metabolite profiles among the T. koningiopsis Z35 samples. The first two principal components, PC1 and PC2, accounted for 60.5% and 39.5% of the total variance, respectively, cumulatively explaining 100% of the variation in the dataset. Although some distributional differences were observed among the three biological replicates of Z35 in the PCA plot, the clustering was relatively tight, indicating good reproducibility and internal consistency of the metabolomic data (Fig. 7).
Figure 7.
Principal component analysis (PCA) of three biological replicates of T. koningiopsis Z35.
(a) PCA plot under positive ion mode; (b) PCA plot under negative ion mode. PC1 and PC2 represent the first and second principal components, respectively, with the percentage indicating the proportion of variance explained by each component. Each dot represents one sample, and samples from the same group are shown in the same color. 'Group' refers to the sample grouping. PCA was performed on three biological replicates of strain Z35 under identical conditions to assess data consistency. No inter-group comparison was applied.To further investigate the metabolic profiles of T. koningiopsis Z35, hierarchical clustering analysis based on heatmap visualization was performed under both positive and negative ion modes. The results revealed distinct metabolite expression patterns, with hundreds of metabolites detected across diverse categories, including amino acids, nucleotides, lipids, terpenoids, alkaloids, and flavonoids (Fig. 8). The three biological replicates (Z35-1, Z35-2, Z35-3) exhibited highly consistent metabolic profiles, indicating good reproducibility and experimental reliability. The predominant metabolite classes were amino acid derivatives, organic acids and their derivatives, phenolic acids, and flavonoids, suggesting that this strain harbors extensive secondary metabolic potential, which may contribute to its resistance traits, growth-promoting effects, or ecological adaptability. Moreover, several bioactive secondary metabolites were identified in Z35, including choline, alternariol, and triterpenes. These compounds are known to enhance plant resistance to abiotic and biotic stresses, modulate rhizosphere microbial communities, and improve plant immunity. Taken together, the diversity and function of the detected metabolites suggest that T. koningiopsis Z35 possesses promising capabilities in promoting plant growth and enhancing plant resilience against pathogens.
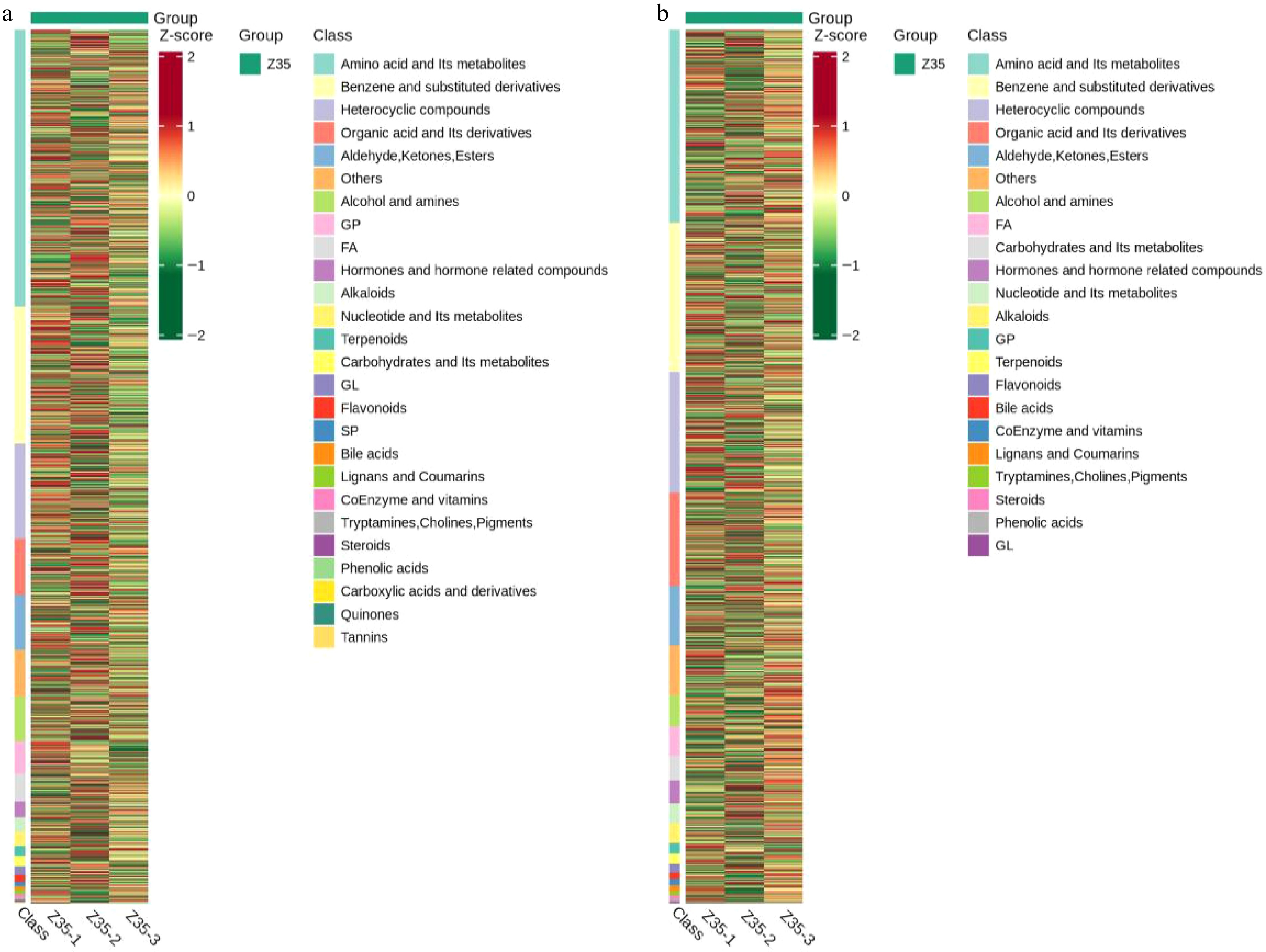
Figure 8.
Hierarchical clustering analysis of metabolites detected in the fermentation broth of Z35 under positive and negative ion mode.
(a) Positive ion mode, and (b) negative ion mode. The x-axis represents sample information, while the y-axis corresponds to metabolite identities. Groups indicate sample classifications. Colors reflect normalized relative metabolite abundance, with red indicating high levels and green indicating low levels. -
Biological control is recognized as an environmentally friendly and sustainable strategy for managing plant diseases, effectively reducing dependence on chemical pesticides and mitigating the ecological risks associated with their use, particularly the risk of promoting pathogen resistance[10,44]. In recent years, biocontrol technologies have been increasingly adopted in agricultural production systems[45]. However, P. notoginseng, under long-term continuous cropping conditions, is particularly vulnerable to root rot, which poses a major constraint to the sustainable development of the P. notoginseng industry[46]. In this study, a fungal strain, designated Z35, exhibiting significant antifungal activity was isolated from the rhizosphere soil of healthy P. notoginseng plants cultivated under continuous cropping conditions. The strain exhibited strong antagonistic effects against soil-borne pathogenic fungi, indicating its considerable potential as a biocontrol agent. Members of the genus Trichoderma, known for their broad ecological distribution and potent antagonistic properties, play a pivotal role in global plant disease control and are widely regarded as key microbial resources in the field of biocontrol research[47−49].
Whole-genome sequencing was performed to provide a comprehensive characterization of the genomic features of the biocontrol strain Z35. The assembled genome was 38.65 Mb in size (Fig. 2), with a GC content of 48.0%, comparable to other Trichoderma species and slightly larger in overall size (Supplementary Table S1). A total of 13,153 protein-coding genes were annotated in Z35, exceeding those of its three closest relatives (Supplementary Table S2), indicating greater coding potential and metabolic versatility. Phylogenetic analysis based on both ITS and whole-genome sequences confirmed that Z35 belongs to T. koningiopsis. Gene family evolution analysis revealed a higher frequency of contraction than expansion, suggesting possible gene rearrangements that may contribute to ecological adaptation and functional specialization. Notably, typical industrial Trichoderma strains typically possess fewer than 10,000 genes[50], whereas Z35 was predicted to encode 11,192 genes (Supplementary Table S8), thereby reinforcing its functional potential as a biocontrol agent.
Trichoderma species are well recognized for their diverse biocontrol mechanisms against plant pathogens, which include the production of antibiotics[51], mycoparasitism[28], niche competition, and plant immune modulation[52]. These defense strategies often rely on the coordinated expression of multiple functional genes[53]. In this study, confrontation assays demonstrated that strain Z35 exerts a strong inhibitory effect against F. oxysporum. The formation of a distinct inhibition zone indicates that Z35 may inhibit pathogen growth through the secretion of antifungal metabolites. Additionally, the abundant production of conidia by Trichoderma promotes rapid colonization and confers a competitive advantage in the rhizosphere, thereby enabling it to dominate this niche and restrict pathogen proliferation.
Secondary metabolites synthesized by fungi through BGCs play crucial roles in microbial interactions and environmental adaptation[39]. Previous studies have demonstrated that the antifungal capabilities of certain fungal strains are primarily attributed to the synthesis and secretion of bioactive secondary metabolites capable of interfering with pathogen development[54,55]. In this study, antiSMASH analysis of the Z35 genome predicted 49 putative biosynthetic gene clusters (BGCs). Notably, several clusters were predicted to be involved in the biosynthesis of compounds such as choline and clavaric acid, both known for their antimicrobial properties. Although several BGCs and their putative associated metabolites were identified, additional transcriptomic analyses (e.g., RNA-seq or qRT-PCR) will be necessary to validate the expression of these clusters under antagonistic conditions. This will be a key focus of future studies. Untargeted metabolomic profiling of Z35 fermentation extracts revealed a diverse array of bioactive compounds, including cholines, triterpenoids, indole derivatives, and related metabolites. Metabolomic analyses in this study followed a robust LC–MS/MS workflow, in line with recent advances such as MetaboAnalystR 4.0, that emphasize accuracy and reproducibility in metabolite quantification[56]. These compounds are associated not only with antifungal activity but also with plant growth promotion, widely reported to occur through modulation of root development and hormone signaling pathways. For instance, studies have demonstrated that indole and its derivatives can enhance root formation and biomass accumulation in various plant species[57]. Although we did not directly measure growth promotion effects in this study, the presence of these compounds suggests that T. koningiopsis Z35 may potentially enhance plant growth. Future work is needed to experimentally validate this hypothesis. Although specific predicted secondary metabolites were not detected, choline, an important primary metabolite, was consistently present. The presence of metabolite classes such as terpenoids and polyphenols further suggests the biosynthetic capacity of T. koningiopsis Z35. The absence of some predicted compounds may be due to low expression or non-inductive conditions, highlighting the need for further targeted metabolomics and transcriptomic validation. In addition, this study employed a metabologenomics strategy that combines BGC prediction with metabolomics to reveal biosynthetic potential, even when specific compounds are not detected. Similar approaches have been successfully used to connect cryptic BGCs with metabolite profiles in other microorganisms[58−60]. The integration of genomic and metabolomic data revealed that Z35 harbors multiple active biosynthetic pathways—such as NRPS-like, terpene, NRPS, and T1PKS—responsible for the production of a broad spectrum of bioactive metabolites, including choline, metachelin C, metachelin A, metachelin A-CE, metachelin B, dimerumic acid 11-mannoside, dimerumic acid, clavaric acid, ascochlorin, and trichoxide. These findings suggest that Z35 mediates its biocontrol effects via the coordinated action of multiple secondary metabolic pathways and underscore its potential as a valuable microbial resource for sustainable plant disease management. While the in vitro analyses demonstrated the promising antagonistic activity of T. koningiopsis Z35, its biocontrol efficacy under actual plant growth conditions requires further evaluation. Considering the significance of continuous cropping systems in agriculture, future studies will prioritize evaluation of Z35 performance in greenhouse and field trials under continuous cropping conditions to validate its practical application.
-
To comprehensively characterize the genetic characteristics and biocontrol potential of strain Z35, a systematic investigation was undertaken on a fungal isolate derived from the rhizosphere soil of P. notoginseng under long-term continuous cropping. This study included assessments of antagonistic activity, whole-genome sequencing, functional annotation, confrontation bioassays, and prediction and analysis of biosynthetic gene clusters (BGCs) involved in secondary metabolite production. In dual-culture plate assays, strain Z35 demonstrated strong inhibition against F. oxysporum, with an inhibition rate of up to 75%, indicating its potent biocontrol capacity. Untargeted metabolomic analysis of fermentation extracts further revealed secretion of several bioactive metabolites—including methacholine, salannin, medicagenic acid, indole, and indole-3-acetamide—that are associated with both pathogen suppression and plant growth promotion. Phylogenetic analysis based on ITS and single-copy gene sequences identified strain Z35 as T. koningiopsis. Whole-genome sequencing revealed a range of BGCs potentially involved in the regulation of induced systemic resistance and the direct or indirect inhibition of pathogen infection (Fig. 5a). antiSMASH analysis identified a total of 49 BGCs and 717 genes potentially associated with secondary metabolite biosynthesis, encompassing functional families including the SAT superfamily, alpha_am_amid superfamily, and PRK12316 superfamily. Key clusters, including NRPS-like, terpene, NRPS, and T1PKS, were predicted to biosynthesize antimicrobial metabolites such as cholines (e.g., choline), triterpenoids (e.g., clavaric acid), the metachelin family, dimerumic acid, ascochlorin, and trichoxide—all of which may contribute to the antagonistic activity against F. oxysporum. Metabolomic profiling identified 5,087 metabolites, including several indole derivatives associated with plant growth regulation (e.g., indole-3-acetamide, indole, and 5-methoxyindoleacetate), as well as secondary metabolites with known antifungal properties such as salannin, ganoderic acid F, and methacholine. Collectively, this integrated genomic and metabolomic analysis provides novel insights into the biosynthetic potential and metabolic versatility of T. koningiopsis Z35, offering a theoretical foundation for its future application as a promising biocontrol agent in sustainable agriculture.
This work was supported by the National Key Research and Development (2021YFD1601003); the Project of The Sino-Vietnamese International Joint Laboratory for Characteristic & Cash Crops Green Development of Yunnan Province (202403AP140013). 'Development and Application of Personalized Cigarette Products' (2023CP02).
-
The authors confirm contribution to the paper as follows: study conception and design: Liu T, Dong Y; draft manuscript preparation: Zhang H, Zhang Q; manuscript revision and editing: Jiang J, Dong Y, Liu T, Yang S, Tiên L, Tín H; soil sample collection: Feng Y, Zhou Y, Yang N; data collection: Zhang H, Shi C. All authors reviewed the results and approved the final version of the manuscript.
-
These data can be accessed through the NCBI BioProject PRJNA1163317, and BioSample SAMN43845572.
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Honglin Zhang, Qing Zhang
- Supplementary Table S1 Integrity results of gene regions in whole assembled genome results assessed by BUSCO.
- Supplementary Table S2 Annotation Statistics of coding gene from the T. koningiopsis Z35 and homo species genome.
- Supplementary Table S3 Genetic Structure.
- Supplementary Table S4 Statistics of T. koningiopsis Z35 protein-coding gene annotation.
- Supplementary Table S5 Comparative analysis of sequencing, assembly metrics, and genome quality of T. koningiopsis Z35 and related Trichoderma strains.
- Supplementary Table S6 T.
- Supplementary Table S7 Identified metabolites in Trichoderma koningiopsis Z35.
- Supplementary Table S8 Gene family clustering of T. koningiopsis Z35 with other species is shown in the species clustering statistics table and the gene family clustering classification statistics.
- Supplementary Fig. S1 In vitro antagonistic activity of strain Z35 against various Fusarium species.
- Supplementary Fig. S2 Comparing the genetic characteristics of sequenced species with those of related species, the red one is the self-assembled species Z35.
- Supplementary Fig. S3 (a) Statistical map of functional annotation classification based on GO database. (b) KOG Functional Annotated Classification. (c) KEGG Pathway Functional Classification Chart.
- Supplementary Fig. S4 (a) Statistical graph of metabolite classes detected in positive ion mode (ESI+); (b) Statistical graph of metabolite classes detected in negative ion mode (ESI-). Different colors represent different chemical classes, and the values in the graphs are the relative proportions (%) of metabolites in each class.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Yunnan Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang H, Zhang Q, Jiang J, Yang N, Zhou Y, et al. 2025. Integrated genomic and metabolomic insights into the secondary metabolism of Trichoderma koningiopsis Z35. Agrobiodiversity 2(3): 62−72 doi: 10.48130/abd-0025-0009 

Integrated genomic and metabolomic insights into the secondary metabolism of Trichoderma koningiopsis Z35
- Received: 03 June 2025
- Revised: 12 August 2025
- Accepted: 05 September 2025
- Published online: 29 September 2025
Abstract: Strain Z35, isolated from the rhizosphere of healthy Panax pseudoginseng under long-term continuous cropping, exhibits strong antagonistic activity against Fusarium oxysporum and shows potential as a biocontrol agent. However, the genetic and metabolic basis of its antimicrobial and plant growth-promoting functions remain unclear. In this study, plate confrontation assays confirmed that Z35 inhibited F. oxysporum by up to 75%. Whole-genome sequencing using the PacBio platform revealed a 38.65 Mb genome with seven contigs and one scaffold, a GC content of 47.48%, and 99.9% completeness (BUSCO). Phylogenetic analysis identified Z35 as Trichoderma koningiopsis. Genome annotation uncovered 49 secondary metabolite biosynthetic gene clusters and 717 functionally relevant genes, including clusters related to nonribosomal peptide synthetases (NRPS), polyketide synthases (PKS), and terpenes. These clusters are potentially responsible for producing bioactive compounds such as clavaric acid, the metachelin series, dimerumic acid, ascochlorin, and trichoxide. Non-targeted metabolomic profiling validated the presence of these metabolites in Z35 fermentation products, supporting their role in antifungal activity and plant-beneficial effects. This study provides insights into the genetic and metabolic mechanisms underlying Z35's biocontrol potential, offering a theoretical basis for its further development and application in sustainable agriculture.