









-
For large-scale phylogenetics, especially involving angiosperms, the in-depth exploration of species relationships and evolutionary history often encounters deep phylogenetic incongruence. With the widespread application of high-throughput sequencing technology and multi-gene datasets, substantial progress has been made in constructing large phylogenetic trees, revealing the complex mechanisms underlying species diversification[1−3]. However, in certain plant groups, particularly in families like Gesneriaceae[4] and Solanaceae[5], phylogenetic relationships still exhibit considerable uncertainty due to highly inconsistent gene histories and ambiguous evolutionary signals. These conflicts often arise from factors such as incomplete lineage sorting (ILS)[6], gene flow best represented as evolutionary networks[7,8], and polyploidy[4]. All these processes leave reticulate genetic footprints in different genomic datasets, making it particularly challenging to infer evolutionary relationships between species using molecular data.
The evolutionary relationships within Malvaceae s.l., with its two major clades, Byttneriina and Malvadendrina, and significant morphological variation, have been difficult to resolve[9−11]. Subfamilies within these clades vary greatly in morphology, and molecular data suggest a complex and often conflicting evolutionary history[11−13]. The Byttneriina clade is composed of two subfamilies: Byttnerioideae and Grewioideae. Byttnerioideae mainly consists of shrubs, characterized by petals that extend to the base, typically lacking an epicalyx, and having a cup-shaped, curled margin (Fig. 1b). In contrast, Grewioideae includes trees and shrubs, featuring either separate or fused sepals and clawed petals (Fig. 1a). The Malvadendrina clade includes the remaining eight subfamilies, which display both morphological and molecular complexity and contentious relationships[10,11,14]. Helicteroideae mainly comprises trees and shrubs, characterized by fused sepals and clawed petals that are typically laterally constricted (Fig. 1c). The members of Sterculioideae comprise trees with petal-like sepals and prominently elongated stamens and pistils (Fig. 1d). Brownlowioideae is primarily composed of trees, with fused, bell-shaped sepals that irregularly split into two or three lobes (Fig. 1e). Dombeyoideae is characterized by a spiral leaf arrangement (Fig. 1f, g), persistent petals, and the presence of an epicalyx. Tilioideae exhibits a two-ranked leaf arrangement (Fig. 1h) and bears nectar glands on the abaxial side of its petals. The phylogenetic relationship of Dombeyoideae with the other subfamilies remains unresolved, with existing studies showing differing support for its sister-group relationship with Sterculioideae and Brownlowioideae[11,13], while some studies lean towards placing Dombeyoideae as a sister group to Tilioideae[15,16]. The subfamilies Bombacoideae and Malvoideae exhibit marked morphological differences; Bombacoideae primarily includes trees with palmate compound leaves, fused or absent sepals, and typically large seeds enclosed in capsules (Fig. 1i, j). In contrast, Malvoideae is predominantly composed of shrubs or herbaceous plants, characterized by five petals and typically forming schizocarps or capsules (Fig. 1k, l). Molecular evidence strongly supports the sister-group relationship between Malvoideae and Bombacoideae[17] (Fig. 1, Supplementary Fig. S1).

Figure 1.
Diversity of flowers and fruits of the Malvaceae Juss. (a) Grewia biloba var. parviflora (Bunge) Hand. -Mazz. Contributed by Maple, (b) Waltheria indica L., (c) Helicteres angustifolia L., (d) Sterculia brevissima H.H. Hsue ex Y. Tang, M.G. Gilbert & Dorrcontributed by Malvaceae, (e) Diplodiscus trichospermus (Merr.) Y.Tang, M.G. Gilbert & Dorr, (f) Melhania hamiltoniana Wall., contributed by janstudio, (g) Dombeya acutangula Cav., (h) Craigia yunnanensis W.W.Sm. & W.E.Evans, (i) and (j) Bombax ceiba L., (k) Urena lobata L., (l) Abutilon indicum (L.) Sweet.
Resolving these phylogenetic uncertainties is further complicated by the presence of conflicting signals across loci. Such discordance may reflect evolutionary processes including ILS, gene flow, and hybridization, which often obscure true species relationships[18]. Distinguishing whether observed discordance is due to ILS, introgression, or both remains challenging, particularly in lineages with complex coalescent histories. Dense taxon sampling and advanced analytical methods are therefore critical to disentangle these processes. Within Malvaceae s.l., the phylogenetic relationships among several subfamilies, including Tilioideae, Sterculioideae, Dombeyoideae, and Brownlowioideae, remain unresolved[13]. This provides an important opportunity to investigate how incomplete lineage sorting (ILS) and historical gene flow have influenced phylogenetic inference. In this study, the recently recognized subfamily Matisioideae is incorporated to re-examine the placement of all ten currently accepted subfamilies[19]. Additionally, divergence times and the reconstructed ancestral geographic ranges are estimated to explore the spatial and temporal patterns underlying the diversification of the family.
The specific objectives of this study are: (1) To analyze the phylogenetic relationships among the ten subfamilies of Malvaceae s.l. based on nuclear and plastid genomic data, using both concatenation and coalescence-based approaches. (2) To investigate the causes of phylogenetic discordance among the ten subfamilies of Malvaceae s.l. based on nuclear genomic data, by distinguishing the relative contributions of incomplete lineage sorting (ILS) and introgression using the quartet-based methods Phytop and QuIBL. (3) To reconstruct ancestral geographic distributions and examine biogeographic history and geographic expansion patterns of Malvaceae s.l. through analyses based on both nuclear and plastid phylogenies.
-
This study focused on genus-level sampling within Malvaceae s.l. A total of 134 species were examined, representing 62 genera (ca. 25% of the 248 genera currently recognized in the family), along with two outgroup species. Specifically, the sampling includes 11 genera from Byttnerioideae (representing 50% of all recognized genera within this subfamily), six genera from Grewioideae (25%), five genera from Helicteroideae (41.67%), three genera from Brownlowioideae (27.27%), two genera from Tilioideae (66.67%), five genera from Dombeyoideae (31.25%), seven genera from Sterculioideae (53.85%), five genera from Bombacoideae (27.78%), one genus from Matisioideae (33.33%), and 17 genera from Malvoideae (13.49%). Additionally, two non-Malvaceae angiosperms were included as outgroups: Anthoshorea assamica P.S. Ashton & J. Heck (Dipterocarpaceae, Malvales) and Mangifera indica L. (Anacardiaceae, Sapindales). Samples included both newly collected individuals for de novo sequencing and species with publicly available genomic data retrieved from the NCBI database (Supplementary Table S1). Newly collected samples were obtained in compliance with local rules and regulations.
Genomic DNA was extracted from fresh leaf material using a modified cetyltrimethylammonium bromide (CTAB) protocol[20]. The DNA concentration was quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA), with samples exceeding a total amount of 0.8 µg selected for subsequent library construction. Whole-genome sequencing libraries were prepared using the VAHTS Universal DNA Library Prep Kit (Vazyme Biotech Co., Ltd., Nanjing, China), and sequencing was performed on the DNBSEQ-T7 platform by BGI Life Sciences Co., Ltd. (Hainan Province, China), generating approximately 10 Gb of raw data per sample. Quality control and filtering of the raw data were conducted using SOAPfilter v2.2[21]; reads containing more than 10% ambiguous bases or low-quality bases (≤ 10) were removed to ensure high-quality data for downstream analyses.
Plastid genome generation and analysis
-
Plastid genomes were assembled from raw sequencing data using GetOrganelle v1.7.7.0[22] with k-mer sizes set to 21, 45, 65, 85, and 105 to optimize assembly across both conserved and variable regions. Assembly graphs were visualized and assessed with Bandage v0.9.02[23], and the parameters --max-kmer-coverage and --min-kmer-coverage were adjusted as needed to reduce coverage artifacts and low-depth noise. Assemblies failing to produce circular genomes were reprocessed by incrementally lowering --max-kmer-coverage values (e.g., 100, 50, 20) and re-evaluated for completeness and structure. Among the multiple FASTA outputs, the most complete and well-resolved circular genome—verified via Bandage visualization and Mauve v2.4.0[24] comparison against GetOrganelle's embplant_pt reference database—was selected for each sample. Genome annotation was performed using GeSeq[25], with gene features including protein-coding genes (CDS), rRNAs, and tRNAs transferred from reference plastomes based on Mauve alignments. Annotations were then exported in GenBank format and validated using the GB2Sequin tool (
https://chlorobox.mpimp-golm.mpg.de/GenBank2Sequin.html ). Detected annotation errors were manually corrected in Geneious Prime v22.1.1[26]. Final plastome statistics are summarized in Supplementary Tables S2 and S3.For phylogenetic analysis, complete plastid genome sequences were aligned using MAFFT v7.505[27] implemented in PhyloSuite v1.2.3[28]. One copy of the inverted repeat (IR) region was removed prior to alignment to avoid redundancy in branch length estimation. The final alignment was manually partitioned in Geneious Prime according to structural regions (LSC, SSC, IR) and functional categories (coding vs non-coding). Maximum likelihood (ML) trees were inferred using IQ-TREE v2.2.6[29], with best-fit models selected using ModelFinder[30] under the Bayesian Information Criterion (BIC). Branch support was assessed with 1,000 ultrafast bootstrap replicates[31].
Nuclear dataset assembly and analysis
-
To reconstruct the nuclear phylogeny of Malvaceae s.l., a dataset is assembled using Easy353 v2.0.1[32] to extract 353 conserved nuclear loci of angiosperms from shallow whole-genome sequencing data[33,34]. A two-step procedure was employed: first, representative sequences were extracted from selected taxa of different subfamilies to serve as reference sequences; then, target gene assembly was guided via read mapping. This process ultimately yielded approximately 350 nuclear loci for downstream analyses.
Sequences for each locus were aligned using MAFFT v7.520, and individual gene trees were inferred in IQ-TREE v2.2.6 under their corresponding best-fit substitution models as determined by ModelFinder using the Bayesian Information Criterion. A species tree was reconstructed from the set of 353 nuclear gene trees using ASTRAL III v5.7.1[35]. Branch lengths in the ASTRAL species tree are expressed in coalescent units rather than substitutions per site or absolute time and were therefore not interpreted as estimates of divergence time or genetic distance. Given the potential for incomplete lineage sorting and introgression, the ASTRAL species tree was adopted as the principal framework for downstream evolutionary and biogeographic analyses. For comparison, a concatenated nuclear supermatrix was assembled in PhyloSuite v1.2.3, with partitions defined by individual loci. Maximum likelihood phylogenetic inference was conducted in IQ-TREE v2.2.6 under a partitioned scheme. with the optimal partitioning strategy and the best fitting nucleotide substitution model for each partition selected by ModelFinder according to the Bayesian Information Criterion, and branch support evaluated with 1,000 ultrafast bootstrap replicates. Due to its susceptibility to gene-tree discordance, the concatenated topology was used only as a supplementary reference.
Phytop analysis
-
To quantify the sources of nuclear phylogenetic discordance and to distinguish patterns attributable to incomplete lineage sorting (ILS) from those compatible with introgression or hybridization (IH), Phytop v0.3.2 is used[36]. Phytop is a quartet-based method that takes as input an ASTRAL species tree with its associated quartet support and, for each internal branch, partitions the supporting gene tree quartets into the three possible unrooted topologies (q1, q2, and q3) and uses their relative frequencies to compute branch-specific summary indices of ILS and IH. In this study, Phytop is applied to the ASTRAL species tree inferred from the 353 nuclear loci, extracted and retained the per-branch ILS and IH indices for subsequent interpretation of nuclear gene tree discordance patterns, and examined the default graphical output, including pie charts that depict the proportions of q1, q2, and q3 at each internal branch.
QuIBL analysis
-
To further investigate whether the observed phylogenetic discordance among species was due solely to incomplete lineage sorting (ILS) or also involved introgression, the QuIBL (Quantifying Introgression via Branch Lengths) method[37] is employed. QuIBL fits two models to the branch length distributions: one assuming ILS alone and another incorporating both ILS and introgression. Model comparisons are conducted using the Bayesian Information Criterion (BIC), with a ΔBIC > 10 favoring the ILS-only model and a ΔBIC < –10 supporting the ILS plus introgression model[38]. For analysis, gene trees constructed from non-overlapping 2-kb genomic windows spaced every 20 kb across the genome were utilized, a strategy designed to minimize the impact of intralocus recombination[37]. Only gene trees containing at least five parsimony-informative sites were retained for analysis. All gene trees were rooted using the designated outgroup, Anthoshorea assamica. QuIBL was executed with default parameters, and the resulting ΔBIC values were used to infer the predominant evolutionary processes contributing to phylogenetic discordance among species[39].
Divergence time estimation
-
To obtain a subset of loci suitable for divergence time estimation while keeping the analysis computationally tractable, SortaDate pipeline[40] is used to rank nuclear loci according to their degree of clock-likeness, overall tree length, and topological congruence with the ASTRAL species tree. The 25th highest-ranking loci were then selected, which combined near clock-like rate constancy with moderate evolutionary rates and low levels of gene-tree conflict. This selection strategy reduced both the computational burden and the impact of incomplete lineage sorting and other sources of model misspecification in relaxed-clock analyses.
Divergence times were estimated in BEAST v2.7.7[41] under a Birth-Death tree prior and an Optimized Relaxed Clock (ORC) model of branch-rate variation. A total of nine temporal calibrations (one secondary calibration and eight fossil constraints) were applied. A secondary calibration on the crown node of Malvales was implemented as a uniform prior between 110.48 and 138.33 Ma, following Ramirez-Barahona et al.[42]. Eight fossil calibrations were assigned to the crown nodes of the major Malvaceae subfamilies. For all fossils, the lower bound of the uniform prior corresponded to the youngest limit of the stratigraphic interval reported for each fossil, whereas the upper bound was fixed at 138.33 Ma. The fossil constraints followed Hernandez-Gutierrez et al.[13] and comprised Bombacoxylon langstoni (Malvaceae, 72.1 Ma), Bombax-type pollen (Bombacoideae, 66 Ma), Discoidites borneensis (Brownlowioideae, 56 Ma), Sphinxia ovalis (Dombeyoideae, 47.8 Ma), Grewioxylon indicum (Grewioideae, 33.9 Ma), Malvaciphyllum macondicus (Malvoideae, 56 Ma), Sterculiaephyllum australis (Sterculioideae, 66 Ma), and Craigia oregonensis (Tilioideae, 47.8 Ma).
Nine independent MCMC chains were run for 100 million generations, sampling every 1,000 generations. Tracer v.1.7[43] was used to check for effective sample sizes (ESS > 200) with the first 25% discarded as burn-in. All the runs were combined using LogCombiner v2.7.7 after discarding the first 25% of trees of each as burn-in. TreeAnnotator v2.7.7 was used to generate the maximum clade credibility tree, displaying mean divergence time estimates with 95% highest posterior density (HPD) intervals. FigTree v.1.4.0 was used for tree visualization.
Ancestral area reconstructions
-
Occurrence data were downloaded from the GBIF database (DOI:
10.15468/dl.frwh2g ). Raw data were cleaned using the rgbif package v3.5.2[44] in R 4.4.0 to remove erroneous records and ensure consistency in species identification and geographic coordinates. The cleaning process involved filtering out records with missing or obviously incorrect coordinates. After data cleaning, the R package BioGeoBEARS v1.1.3 is used to estimate ancestral geographic ranges under a maximum likelihood framework[45]. The time-calibrated divergence tree generated from BEAST v.2.7.7 was used as input for this analysis. BioGeoBEARS was chosen for its capability to compare various biogeographic models and to incorporate complex processes, such as dispersal, extinction, and founder-event speciation, into the analysis. Six different models—DEC, DEC + J, DIVALIKE, DIVALIKE + J, BAYAREALIKE, and BAYAREALIKE + J were applied—to reconstruct the ancestral distribution areas of Malvaceae s.l. To better reflect the biogeographic history of Malvaceae s.l., the global distribution is divided into six main regions: (A) Asia, (B) Africa, (C) North America, (D) South America, (E) Oceania, and (F) Europe. The regional division was based on the main endemic distribution areas and ecological–geographic characteristics of Malvaceae s.l.[46]. Because the taxon sampling includes only approximately one quarter of the genera of Malvaceae s.l., these ancestral range reconstructions should be regarded as hypotheses for the sampled lineages rather than as definitive family-wide scenarios. RASP v4.2 was used to reconstruct the ancestral distribution areas of Malvaceae s.l. using Statistical Dispersal–Vicariance Analysis (S-DIVA) and Bayesian Binary MCMC (BBM) analysis[47], providing additional insights into the historical biogeography of the group. The combination of BioGeoBEARS and RASP allowed us to cross-validate the results and ensure robustness by comparing different methodologies and models in reconstructing the ancestral areas. -
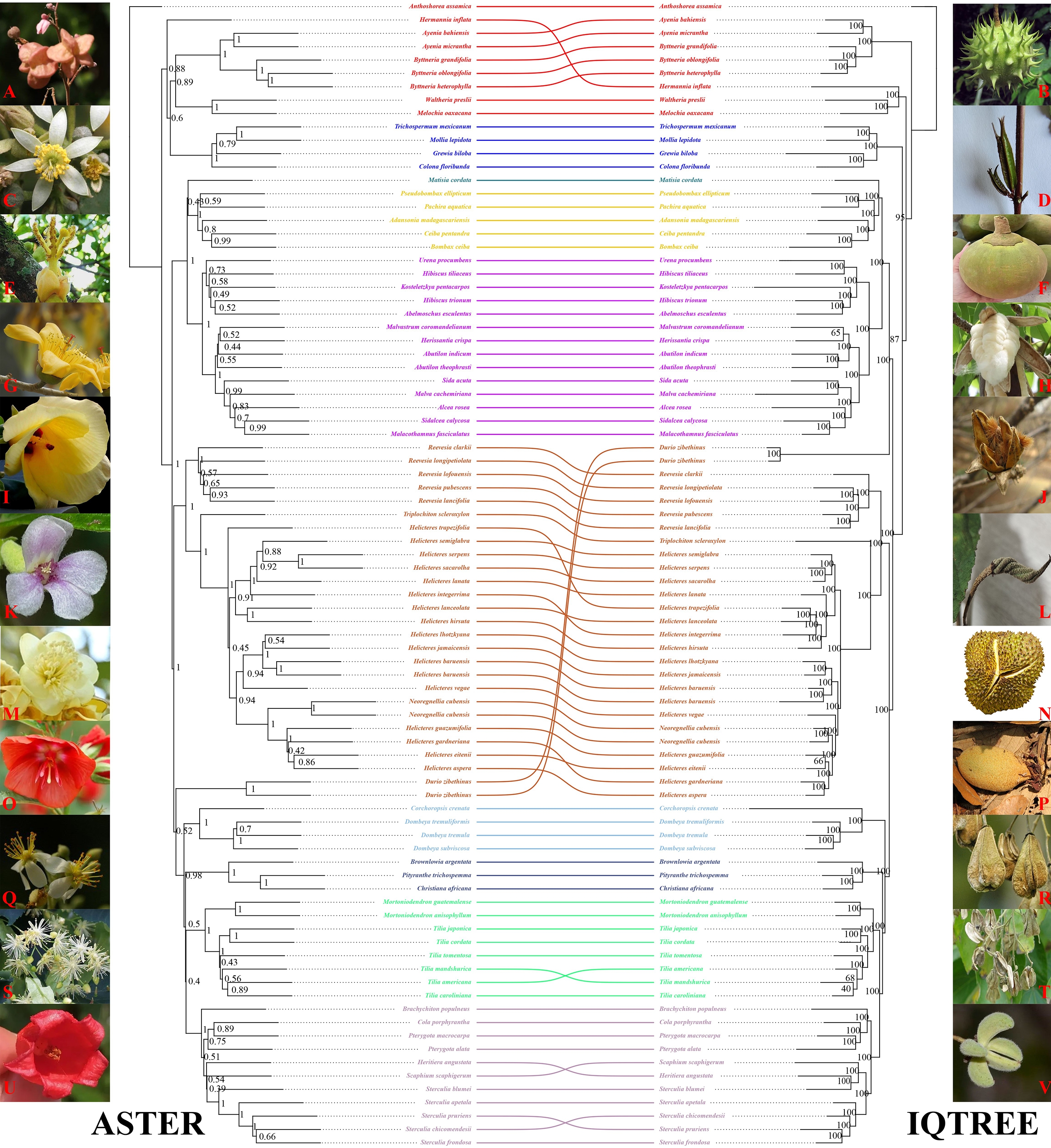
Both nuclear reconstructions, the ASTRAL species tree (Fig. 2) and the concatenated ML tree (Fig. 3), yielded a broadly congruent deep structure for Malvaceae s.l., with a primary split between Byttneriina and Malvadendrina and with most subfamilies supported as monophyletic. Within Malvadendrina, the backbone topology was largely congruent between the two analyses. Sterculioideae and Tilioideae formed a sister pair (ASTRAL: PP = 0.40; ML: BS = 100), and only minor differences were observed in the order of divergence among Pterygota, Cola, and Heritiera within Sterculioideae. Brownlowioideae was recovered as sister to the Sterculioideae + Tilioideae clade (PP = 0.50; BS = 100), and this inclusive lineage was, in turn, sister to Dombeyoideae (PP = 0.98; BS = 100). Bombacoideae and Matisioideae were inferred as sister groups (PP = 0.43; BS = 100), and this clade was sister to Malvoideae (PP = 1.00; BS = 100). The ASTRAL and ML trees also agreed in recovering Helicteroideae (sensu lato) as non-monophyletic, splitting the sampled taxa traditionally assigned to Helicteroideae into two well-supported, phylogenetically distinct lineages, one including Durio and the other comprising the remaining genera. These lineages have at times been treated as separate families or segregate subfamilies in previous classifications, and results support the use of a broad Malvaceae s.l. concept while explicitly acknowledging the polyphyly of Helicteroideae s.l. The main topological incongruence within Malvadendrina concerns the placement of Durio: in the ASTRAL species tree, it is resolved near the base of the clade comprising taxa traditionally placed in Helicteroideae, whereas in the concatenated ML tree, it is positioned as the sister lineage to Malvaceae s.s. By contrast, topological conflict is more pronounced within Byttneriina, where the relationships between Grewioideae and Byttnerioideae differ between the two analyses, indicating substantial gene tree discordance in this part of the tree.
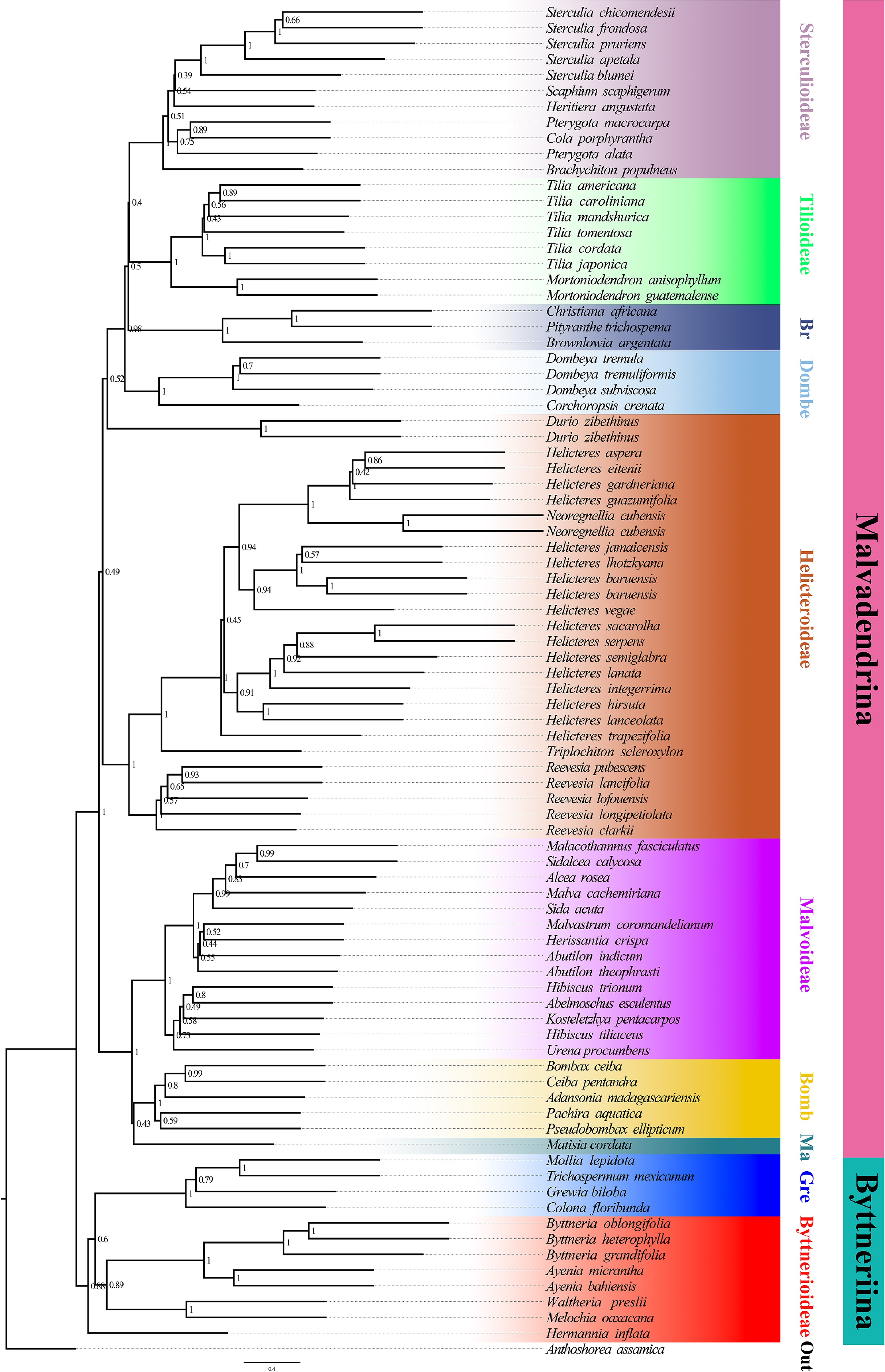
Figure 2.
Species tree inferred from the nuclear dataset using ASTRAL-III. Branch support is given by local posterior probabilities (PP). Major clades corresponding to currently recognized subfamilies are indicated by branch colours as visual references of relationships.
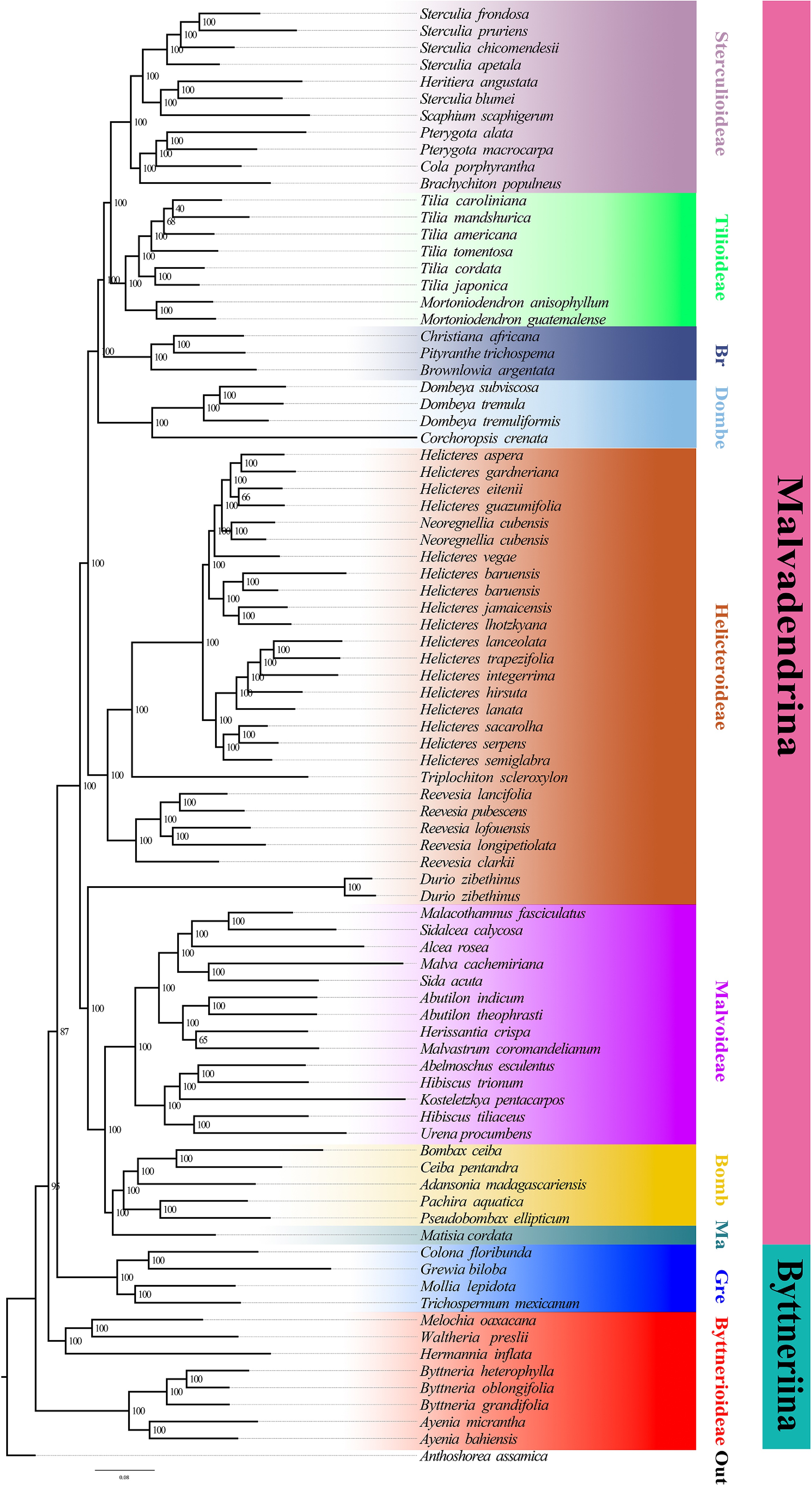
Figure 3.
Maximum-likelihood tree inferred from the concatenated nuclear dataset. Branch support is given by ultrafast bootstrap values (BS). Major clades or evolutionary lineages within the currently recognized subfamilies are indicated by branch colours as visual references of relationships.
Phylogenetic analysis based on plastid data
-
The phylogenetic tree inferred from whole plastid genomes also recovered the monophyly of the ten subfamilies of Malvaceae s.l. (Fig. 4). However, this topology exhibited incongruence with the nuclear-based trees. For instance, Dombeyoideae was placed as sister to Tilioideae in the plastid tree, whereas in the nuclear trees it was recovered as sister to a clade comprising Tilioideae, Brownlowioideae, and Sterculioideae. Given the history of hybridization and polyploidy in Malvaceae s.l., it is expected that plastid data may not accurately reflect the underlying species relationships.
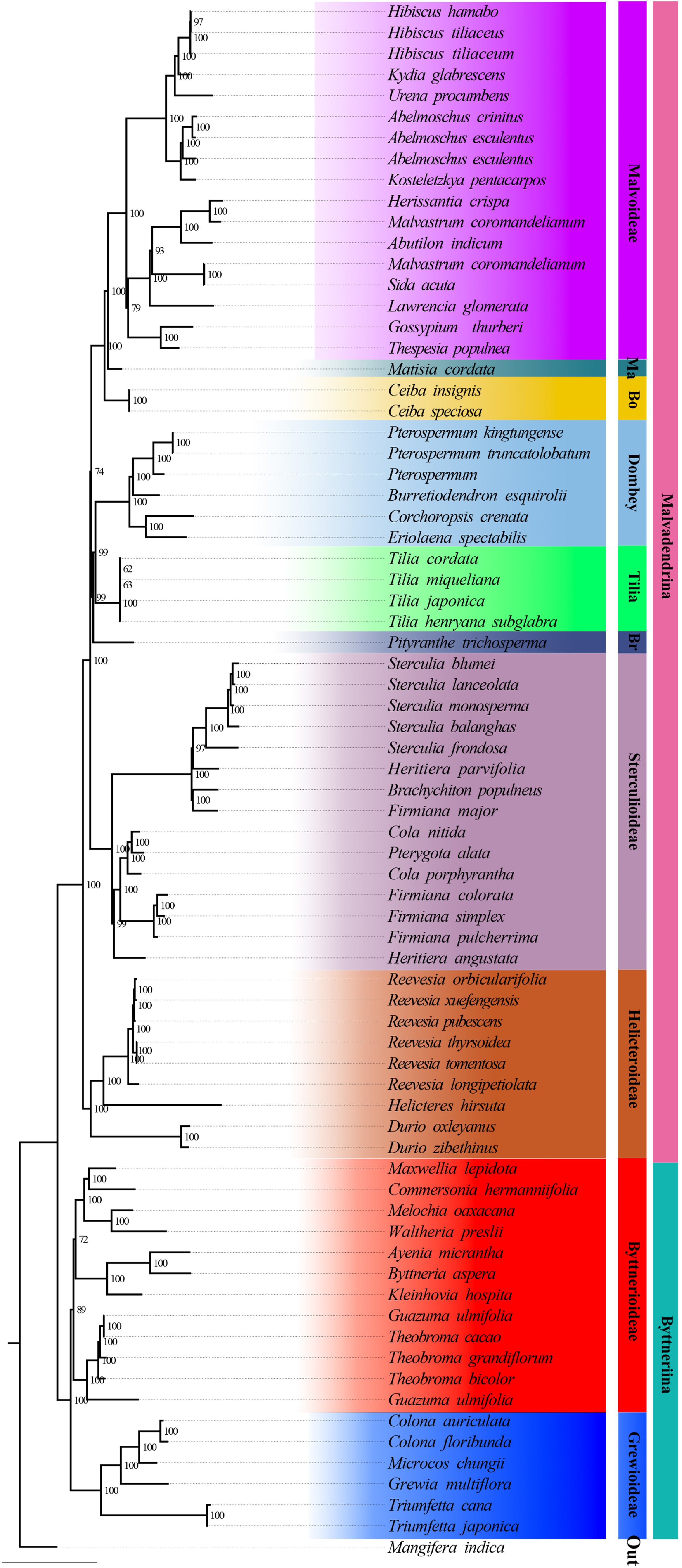
Figure 4.
Maximum-likelihood tree based on plastid data. Support values are displayed above the branches. Currently recognized major taxa or major clades are indicated by branch colours as visual references of relationships.
Phytop and QuIBL analysis
-
Phytop results indicated that, for most subfamily level branches within Malvadendrina, the species tree topology q1 accounted for the majority of supporting quartets at each node, typically with q1 clearly exceeding q2 and q3, and ILS indices ranging from approximately 30% to 60%, while IH indices were zero for most branches (Fig. 5). Within Byttneriina, several key nodes showed reduced q1 proportions and correspondingly higher contributions of q2 and q3, indicating stronger gene tree discordance than along the Malvadendrina backbone. Most of these nodes also had IH indices equal to zero. This pattern is consistent with a scenario dominated by ILS under rapid lineage diversification, and a strong node-level signals of introgression was not detected.
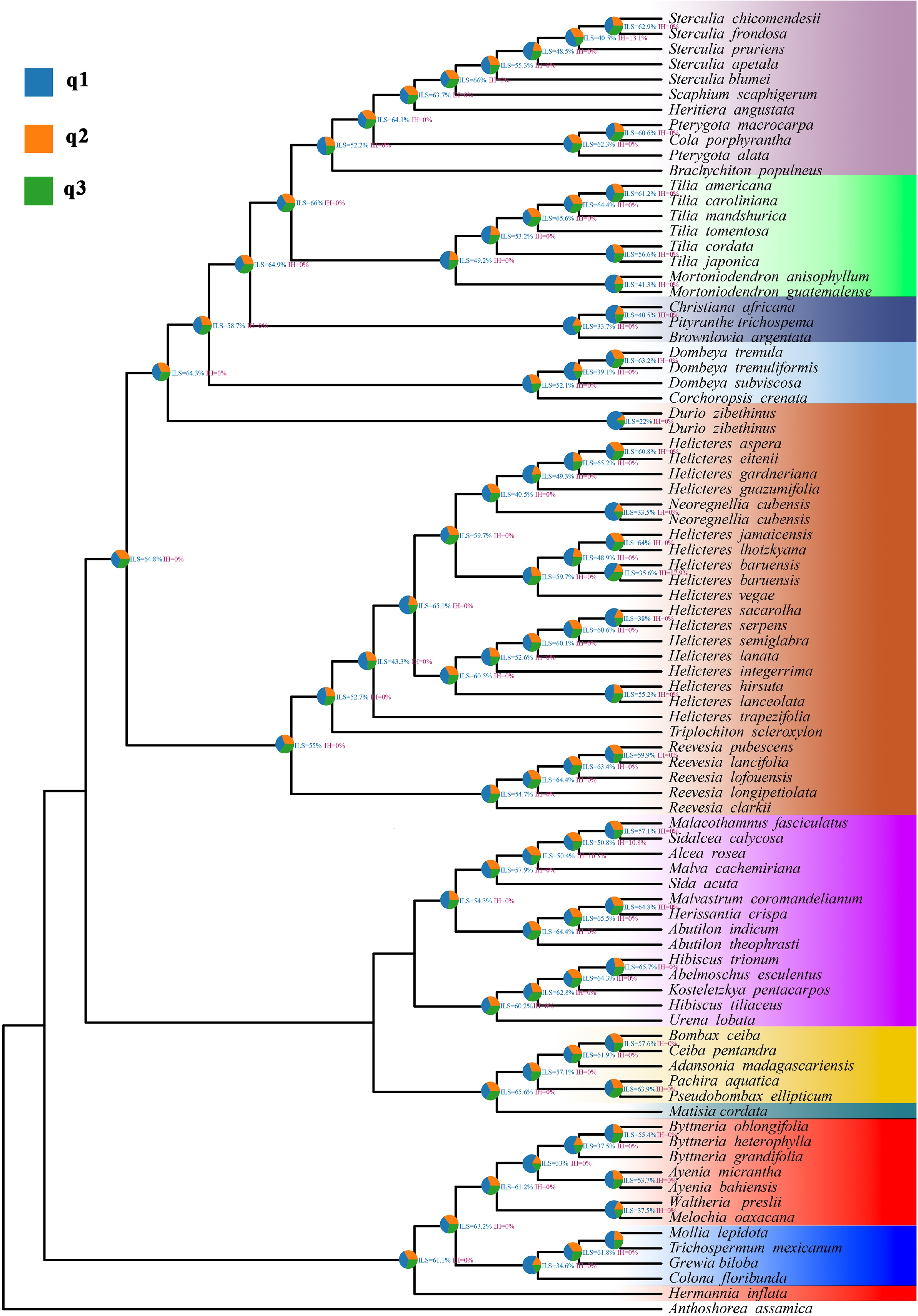
Figure 5.
Phytop analysis of quartet patterns on the ASTRAL species tree of Malvaceae s.l. Each internal node is annotated with a pie chart showing the relative frequencies of the three alternative quartet topologies (q1, q2, q3) around that branch, with blue indicating the topology concordant with the ASTRAL species tree and orange and green indicating the two discordant topologies. Numerical labels next to nodes give the ILS and IH indices inferred by Phytop.
To further explore phylogenetic discordance across lineages, QuIBL analysis was employed. The resulting introgression matrix revealed that most high-probability introgression events occurred within subfamilies, whereas cross-subfamily comparisons consistently exhibited lower introgression proportions, typically in the range of 0.1–0.3 (Fig. 6a). This pattern implies that gene flow, if present, is likely restricted to closely related lineages. To evaluate the model support across gene trees, the distribution of loci favoring the ILS + introgression model was compared between discordant and true topologies (Fig. 6c, d). The discordant topologies showed a broad and continuous band of high introgression support, particularly concentrated at low Non-ILS C values (x < 10), with y-axis values spanning from ~0.125 to nearly 1.0. In contrast, true topologies exhibited a more fragmented distribution, with two distinct regions: a small cluster of high-support points (y ≈ 0.75–1.0) and a broader set around y ≈ 0.3–0.5. These patterns suggest that introgression contributes disproportionately to topological discordance, while loci supporting the species tree are more consistent with ILS or localized introgression. Finally, the distribution of loci classified under the ILS vs ILS + introgression models (Fig. 6b) confirmed that ILS dominates the evolutionary signal across the dataset. However, a subset of loci showed strong preference for the introgression model, underscoring the potential for lineage-specific gene flow events, even in the absence of broader subfamily-level signals.
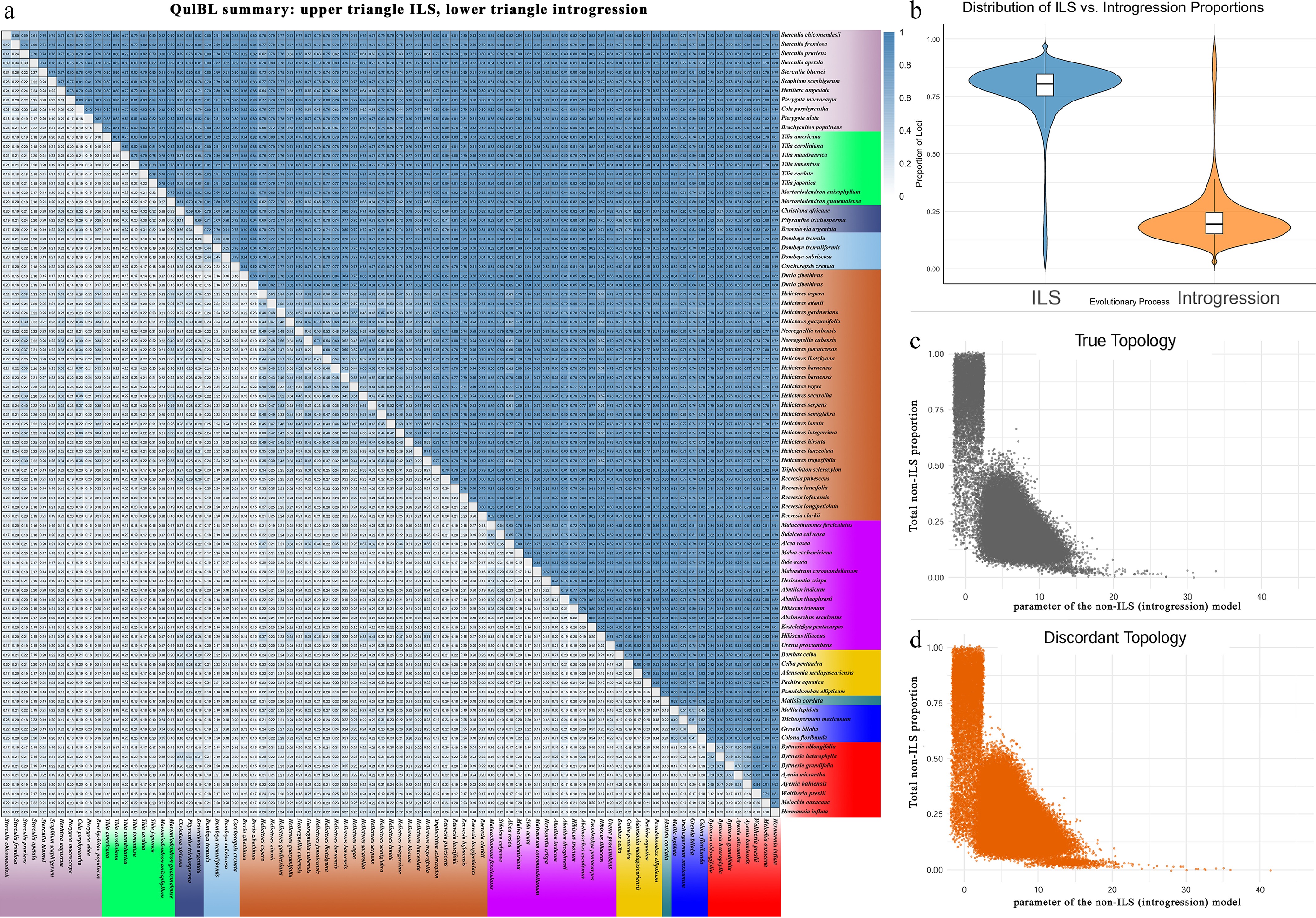
Figure 6.
Tests for introgression. (a) Heatmap summarizing QuIBL results for pairwise species comparisons. For each species pair, the upper triangle shows the mean proportion of loci attributed to ILS, and the lower triangle shows the mean proportion attributed to introgression (mixprop2). Colours indicate proportions from zero to one, and empty cells mark pairs for which no informative triplets were available. (b) Distribution of the proportion of loci that exhibit a history of ILS or introgression across all discordant topologies, respectively. (c) Relationship between internal branch length (in coalescent units) and the proportion of non-ILS loci for triplets matching the true topology. (d) Same as (c), but for triplets with discordant topologies.
Divergence time estimation
-
Based on the nuclear gene dataset, the crown age of Malvaceae s.l. was estimated at 119.38 Ma (95% HPD = 106.48–130.92 Ma), corresponding to the divergence between Byttneriina and Malvadendrina (Fig. 7). Within Malvaceae s.l., the stem lineage leading to Byttneriina dates back to 128.30 Ma, whereas the crown age of Byttneriina, marking the first diversification among the sampled lineages, was inferred at 96.47 Ma. The crown age of Malvadendrina was estimated at 110.66 Ma (95% HPD = 96.86–124.34 Ma). At the subfamily level, Bombacoideae began diversifying around 80.67 Ma (95% HPD = 66.01–93.94 Ma), Dombeyoideae around 54.68 Ma (95% HPD = 47.80–67.90 Ma), Tilioideae at approximately 72.79 Ma (95% HPD = 53.31–89.12 Ma), and Brownlowioideae at 61.62 Ma (95% HPD = 56.00–73.98 Ma). The crown age of the main Helicteroideae clade excluding Durio was estimated at 84.11 Ma (95% HPD = 64.07–04.03 Ma). Although Durio is traditionally classified within Helicteroideae, in the nuclear divergence time tree, it branches off earlier along the Malvadendrina backbone, with a stem age of 110.66 Ma. The crown age of the sampled Durio lineage is much younger, at 13.52 Ma (95% HPD = 5.30–25.45 Ma), indicating a long stem branch associated with relatively recent diversification within the genus.
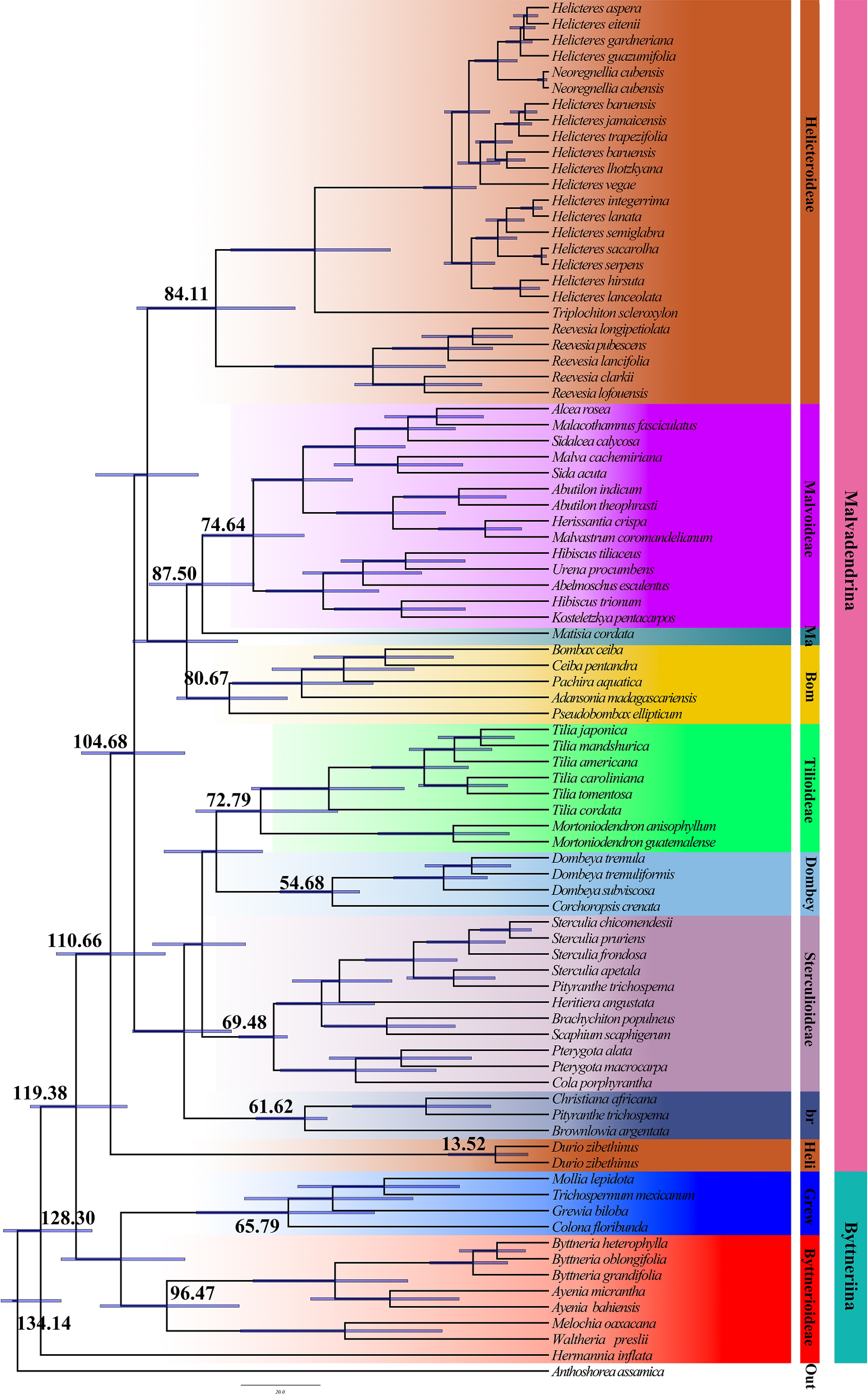
Figure 7.
Divergence time analysis based on nuclear data using BEAST. Each major branch provides mean divergence times and 95% highest posterior density (HPD) intervals. A complete visualization of the 95% HPD intervals across all nodes is provided in Supplementary Fig. S1.
Ancestral area reconstruction
-
Ancestral area reconstructions were performed using RASP and BioGeoBEARS (Fig. 8). The DEC + J and DIVA-like + J models in BioGeoBEARS produced very similar results, with a ΔAIC difference of just 0.8 (Supplementary Table S4). Both models consistently supported that Malvaceae s.l. most likely originated in Africa (B). Under the DEC + J model, the root ancestral area of Malvaceae s.l. was reconstructed as a combination of Africa and South America (B + D), whereas the DIVA-like + J model inferred a single origin in Africa (B). Apart from this difference at the root, the Byttneriina clade was consistently reconstructed with a South American (D) origin in both models. Similarly, the Malvadendrina clade was predominantly reconstructed with an African (B) origin. Taken together, these results indicate that, although the choice of model slightly affects the precise reconstruction of the ancestral area of Malvaceae s.l., the broader biogeographical pattern is robust, with Africa and South America emerging as key regions in the diversification of the family.
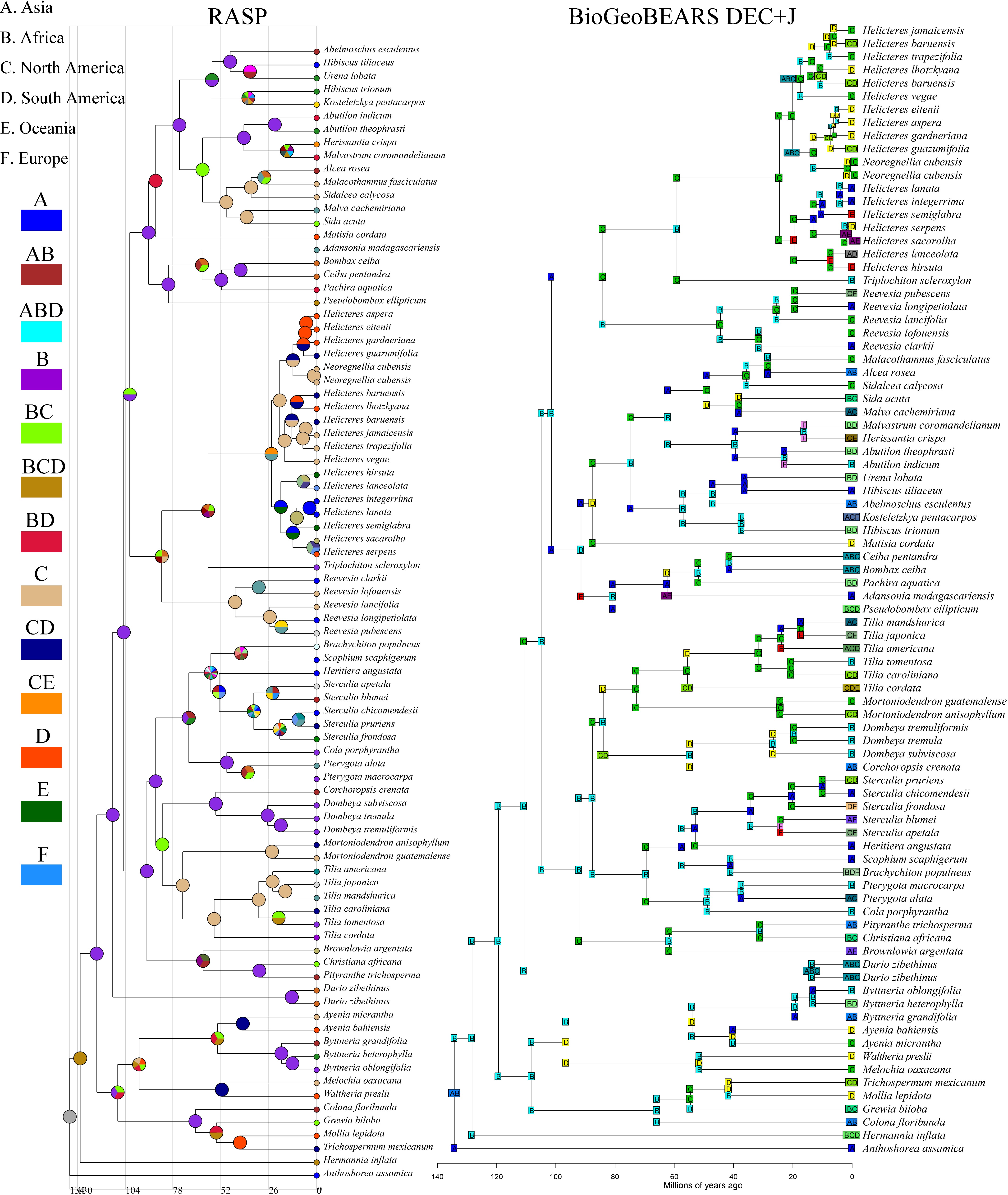
Figure 8.
The figure on the left is an estimate of the ancestral range of Malvaceae s.l. taxa in the RASP using the S-DIVA model. The figure on the right uses the BioGeoBEARS script to select the optimal output model based on six models to estimate the ancestral range of the Malvaceae s.l. groups, which are geographically distributed as (A) Asia, (B) Africa, (C) North America, (D) South America, (E) Oceania, and (F) Europe.
-
Phylogenetic incongruence arises from various complex factors, including genetic drift, selective pressures, gene flow, hybridization events, and variation in evolutionary rates. Particularly in Malvaceae s.l., hybridization has been identified as a key factor contributing to phylogenetic inconsistency, although other evolutionary mechanisms also play significant roles[13]. For instance, genetic drift in small populations can lead to random fluctuations in allele frequencies, resulting in divergences in the evolutionary trajectories of the nuclear and plastid genomes[48,49]. Moreover, differing selective pressures may cause nuclear and chloroplast genomes to respond independently to environmental conditions, thereby intensifying phylogenetic discordance between them[50]. In flowering plants, the nuclear genome evolves at a higher rate than the plastid genome, which tends to be more conserved. This disparity can lead to distinct evolutionary trajectories between the two genomes, thereby amplifying phylogenetic discordance[51,52]. Resolving these discrepancies is essential for accurately reconstructing the evolutionary history of complex plant families, such as Malvaceae s.l.
Many previous studies of Malvaceae s.l. relied on plastid genomic data[10,53]. However, as Hernandez-Gutierrez et al.[13] pointed out, nuclear genomic data offer unique advantages in addressing complex issues and provide a more comprehensive view of the evolutionary relationships within Malvaceae s.l. While plastid data are widely used, findings further highlight the necessity of prioritizing nuclear genomic data in phylogenetic analyses. The relatively rapid evolutionary rate of the nuclear genome further exacerbates the phylogenetic incongruence between nuclear and plastid data[52]. In Malvaceae s.l., nuclear and plastid phylogenetic trees exhibit marked topological discordance across multiple subfamilies[13,53]. Except for the sister group relationship between Grewioideae and Byttnerioideae, which is consistently recovered in both nuclear and plastid trees[13,19], the other eight subfamilies display distinct topologies in the two types of phylogenetic trees. This points to a broad and systematic discordance between nuclear and plastid signals. A major part of this discordance likely reflects the different modes of inheritance and evolutionary dynamics of the two genomes. Localized plastid incongruence is also observed for a small number of taxa, but these cases do not affect the higher-level relationships inferred in this study. The plastid genome is usually maternally inherited, has a much smaller effective population size than the nuclear genome, and generally evolves more conservatively, with lower substitution rates and fewer phylogenetically informative sites at deep nodes[54,55]. It was long regarded as essentially non-recombining, but recent studies have shown that plastids can, in fact, undergo recombination[56]. In particular, plastid capture is a frequent phenomenon in taxa with a history of hybridization, which can bias phylogenetic inferences by misrepresenting underlying species relationships[53,57]. In contrast, the nuclear genome is biparentally inherited and undergoes recombination, enabling it to integrate genetic information from both parental lineages and to reflect nuclear-specific evolutionary processes such as whole-genome duplication (WGD)[58]. While plastid data remain informative at certain phylogenetic levels, nuclear gene trees are generally considered more robust for inferring species-level relationships in lineages with complex evolutionary histories[11,13,59].
Even when based on the same multilocus nuclear dataset, different phylogenetic methods can in principle yield incongruent topologies, particularly in lineages that have experienced rapid diversification or gene exchange[13,18]. In this study, the species tree inferred under the multispecies coalescent in ASTRAL and the concatenated ML tree were largely congruent, recovering the same deep split between Byttneriina and Malvadendrina and identical placements for most subfamilies. Residual discrepancies were confined to a small number of branches, most notably the alternative positions of Durio within Malvadendrina and the internal relationships among Grewioideae and Byttnerioideae in Byttneriina. These remaining conflicts are best interpreted as the outcome of discordant signals among individual nuclear loci generated by pervasive incomplete lineage sorting and localized introgression, as suggested by the Phytop and QuIBL analyses. In this context, the multispecies coalescent framework implemented in ASTRAL, which explicitly accommodates gene tree heterogeneity due to ILS, provides a more appropriate summary of the nuclear phylogenomic signal than concatenated ML approaches that assume a single underlying history for all loci[18,60].
Phylogenetic divergence and speciation complexity in Malvaceae s.l.
-
Resolving phylogenetic relationships among subfamilies within Malvaceae s.l. has long been hampered by pervasive incongruence among nuclear gene trees, particularly in groups such as Sterculioideae, Tilioideae, and Brownlowioideae. This phenomenon epitomizes a central challenge in phylogenomics, namely, how to distinguish discordance generated by incomplete lineage sorting from discordance caused by introgression resulting from historical gene flow and hybridization[57]. The results show a pattern in which a comparatively stable deep backbone contrasts with much more complex relationships among several shallow lineages. The nuclear genomic species tree recovers the primary split between Byttneriina and Malvadendrina and supports the monophyly of most subfamilies, a result broadly consistent with recent classification frameworks[13,61]. Compared with earlier studies, the inclusion of large-scale nuclear genomic data combined with explicit conflict analyses enables a more precise localization of discordance, particularly among intergeneric relationships within Byttneriina and in the placement of recalcitrant lineages in Malvadendrina such as Durio.
The results of Phytop and QuIBL provide further quantitative evidence for the relative roles of incomplete lineage sorting and introgression. PhyTop indicates that, for most internal nodes within Malvadendrina, the species tree topology q1 clearly predominates among quartet frequencies, the ILS index is at intermediate levels, and the IH index is close to zero. QuIBL model comparisons likewise show that, across most species combinations, the ILS component generally concentrates around 70%–80%, whereas the introgression component rarely exceeds 30% and accounts for only a minority of loci (Fig. 5b). Together with the pairwise heatmap results, these analyses indicate that higher proportions of introgression are mainly restricted to closely related species within individual subfamilies (Fig. 5a). Within the assumptions of the models, the combined evidence thus points to a consistent conclusion: incomplete lineage sorting is the predominant source of nuclear gene tree discordance in Malvaceae s.l., whereas introgression exerts a much more limited, lineage specific, and spatially localized influence that does not alter the deep phylogenetic structure of the family. This pattern mirrors the findings of previous genomic studies, which likewise concluded that ILS, together with lineage-specific episodes of introgression, primarily drives local topological instability while exerting only modest effects on the deeper relationships of the family[56]. Recognizing the heterogeneity in the intensity and timing of ILS and introgression across lineages and evolutionary scales will be important for future attempts to integrate genomic, morphological, and ecological data when refining the classification of Malvaceae s.l., reconstructing trait evolution, and elucidating its biogeographic history.
Divergence time and ancestral distribution reconstruction
-
The analysis suggests that the stem age of Malvaceae s.l. dates to the Early Cretaceous, at approximately 134.14 Ma (95% HPD = 123.16−138.33 Ma), with the lineage likely originating in Africa. This timing coincides with significant geological events, such as the ongoing fragmentation of Gondwana. This geological reconfiguration created new environmental niches and isolated populations, facilitating the early diversification of Malvaceae s.l.[62]. A major phase of lineage diversification is inferred to have occurred between 119.38 and 54.68 Ma, likely promoted by continued continental drift and increasing regional biogeographic isolation[63]. During this time, the divergence of major clades such as Byttneriina and Malvadendrina highlights the role of geographic isolation in shaping evolutionary trajectories. The breakup of Gondwana likely facilitated this isolation by separating populations and leading to distinct evolutionary pathways. For example, the continued separation of Africa and South America during the Late Cretaceous provided opportunities for diversification driven by geographic and ecological factors[64].
The analyses also suggest that the early diversification of the sampled Malvaceae s.l. lineages most likely occurred in the tropical regions of northern Africa. The diversification of Malvaceae s.l. is consistent with the notion that early plant lineages diversified in tropical environments before dispersing to other regions via vicariance and long-distance dispersal[65]. Several evolutionary mechanisms may have contributed to the geographic spread and diversification of Malvaceae s.l. The breakup of Gondwana during the Late Jurassic and Cretaceous probably created both migration routes and geographic barriers among tropical landmasses, which may in turn have facilitated early range expansion and spatial structuring of the family[66]. Hybridization and introgression can also facilitate ecological adaptation and contribute to rapid diversification in many plant lineages[67]. These evolutionary processes, combined with geographic isolation, likely played a critical role in the rapid diversification observed during the Late Cretaceous[68]. The complex biogeography of Malvaceae s.l. can also be partly attributed to long-distance dispersal, which allowed certain lineages to reach geographically distant regions. These divergence time estimates and biogeographic inferences provide an initial framework for understanding the early history of Malvaceae s.l., although further improvements in fossil sampling, taxon representation, and model specification will be essential for refining the temporal and spatial context of its evolution.
-
By integrating plastid and nuclear genomic data, this study provides a comprehensive framework for understanding the evolutionary history of Malvaceae s.l. Two major clades corresponding to Byttneriina and Malvadendrina were recovered, and the monophyly of most subfamilies was confirmed. Divergence time estimates and ancestral area reconstructions suggest an origin spanning Africa and South America. Furthermore, investigations into gene tree discordance identify incomplete lineage sorting (ILS) as the predominant driver of phylogenetic incongruence, whereas introgression appears to be restricted to localized events among closely related species. The findings from this study highlight the importance of integrating multiple genomic partitions and analytical approaches to resolve relationships within taxonomically challenging plant groups.
The authors appreciate the constructive comments and suggestions from the editor and reviewers on our initial manuscript. This work was supported by the National Natural Science Foundation of China (Grant No. 32270221), the Hainan Provincial Natural Science Foundation of China (Grant No. 421RC486 and 822QN314), the Hainan Province Science and Technology Special Fund (Grant No. ZDYF2022XDNY190), the Project of Sanya Yazhou Bay Science and Technology City (Grant No. SCKJ-JYRC-2022-83), and the Collaborative Innovation Center for Nanfan and High-Efficiency Tropical Agriculture (Grant No. XTCX2022NYB09). We acknowledge the staff of the Laboratories of Analytical Biology at the National Museum of Natural History, Smithsonian Institution, for technical support and assistance. We also thank the High-Performance Computing Platform of YZBSTCACC for providing computational resources that supported data analyses.
-
The authors confirm their contributions to the paper as follows: study conception and design: Wang HF, Li HY; experimental work and data analysis: Yang WL, Liu PF; draft manuscript preparation and revision: Yang WL, Liu PF, Zuo SY, Jiang DZ, Zhang YH, Chen B, Li HY, Wang HF. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed in the currentstudy are available from the corresponding author on reasonable request.
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
accompanies this paper online at: https://doi.org/10.48130/tp-0026-0001.
-
Received 10 September 2025; Accepted 22 December 2025; Published online 5 February 2026
-
Malvaceae s.l. splits into two primary clades (Byttneriina and Malvadendrina) supported by 353 nuclear loci.
Incomplete lineage sorting is the predominant driver (> 70% of loci) of nuclear gene tree discordance.
Ancestral reconstructions indicate an African origin, with crown diversification at ~119.38 Ma.
Significant topological discordance exists between nuclear and plastid phylogenies across eight subfamilies.
Divergence time estimates suggest an Early Cretaceous origin (~134.31 Ma) and rapid radiation.
- Supplementary Table S1 Average length of intact plastids and nuclear G/C content of each subfamily of Malvaceae s.l.
- Supplementary Table S2 Statistics for the six models considered in the nuclear-based analysis in BioGeoBEARS.
- Supplementary Table S3 Model selection of different ancestral traits.
- Supplementary Table S4 List of species and vouchers used in this study.
- Supplementary Table S5 Length information of each partition in assembled chloroplast genome data.
- Supplementary Fig. S1 Complete time-calibrated phylogeny showing mean divergence times and their 95% highest posterior density intervals for all nodes.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang WL, Chen B, Liu PF, Zuo SY, Jiang DZ, et al. 2026. Phylogenetic framework and evolution of Malvaceae s.l. Tropical Plants 5: e002 doi: 10.48130/tp-0026-0001 

Phylogenetic framework and evolution of Malvaceae s.l.
- Received: 10 September 2025
- Revised: 16 December 2025
- Accepted: 22 December 2025
- Published online: 05 February 2026
Abstract: Understanding the evolutionary relationships and diversification of large, ecologically important plant families, such as Malvaceae s.l., is crucial for understanding angiosperm evolution and biogeographic patterns. Malvaceae s.l., known for its morphological diversity and complex evolutionary history, presents unique challenges in resolving phylogenetic relationships due to factors such as hybridization, introgression, polyploidy, and incomplete lineage sorting (ILS). This study addresses these complexities by reconstructing phylogenetic relationships, estimating divergence times, and inferring ancestral geographic distributions of Malvaceae s.l. using both plastid and nuclear genomic data. The analysis includes 134 species of Malvaceae s.l. and two outgroup species, strongly supports the division of Malvaceae s.l. into two primary clades, Byttneriina and Malvadendrina, while clarifying relationships among the subfamilies Dombeyoideae, Brownlowioideae, Sterculioideae, and Tilioideae. This study reconstructs the evolutionary history of Malvaceae s.l. based on plastid and nuclear genomic data, revealing deep phylogenetic discordance largely driven by incomplete lineage sorting, with additional signals of localized introgression within subfamilies. Divergence time estimates place the origin of Malvaceae s.l. at approximately 134.31 Ma (95% HPD = 123.16–138.33 Ma), representing its initial split from the outgroup lineage. The crown diversification of the family, corresponding to the divergence between its two major clades, Byttneriina and Malvadendrina, occurred around 119.38 Ma (95% HPD = 106.48–130.92 Ma). Ancestral range reconstructions support an African origin, followed by dispersal to tropical regions worldwide. Specifically, the ancestors of the Malvadendrina clade likely dispersed from Africa to South America, while Byttneriina shows strong ties to a North American origin.