








-
In the last two years, two anti-amyloid-β (Aβ) monoclonal antibodies (lecanemab and donanemab, commercially as Lequmbi and Kisunla) have been approved by the FDA for the treatment of Alzheimer's disease (AD), following aducanumab[1,2]. Clinical success soon demonstrated anti-Aβ immunotherapy as one of the most effective treatments to slow AD progression for now. Although the Aβ-tau-neurodegeneration cascade hypothesis still cannot fully explain AD disease pathogenesis[3], the clinical benefits from these antibodies showed that amyloid removal can successfully slow down cognitive decline in patients with AD. These results further highlight the necessity to include Aβ PET imaging in diagnostic criteria to identify appropriate candidates before the treatment[4,5]. To date, progress in this area has involved a large amount of research into what Aβ species are actually the therapeutic targets; early failures to target monomers led to later successes with specific aggregates or plaque-associated Aβ[6,7]. However, it turns out that the cellular and molecular mechanisms behind how anti-Aβ antibodies remove amyloid aggregates are much more complicated than we thought, where multiple biological processes are just revealed in a tangled system (Fig. 1a).
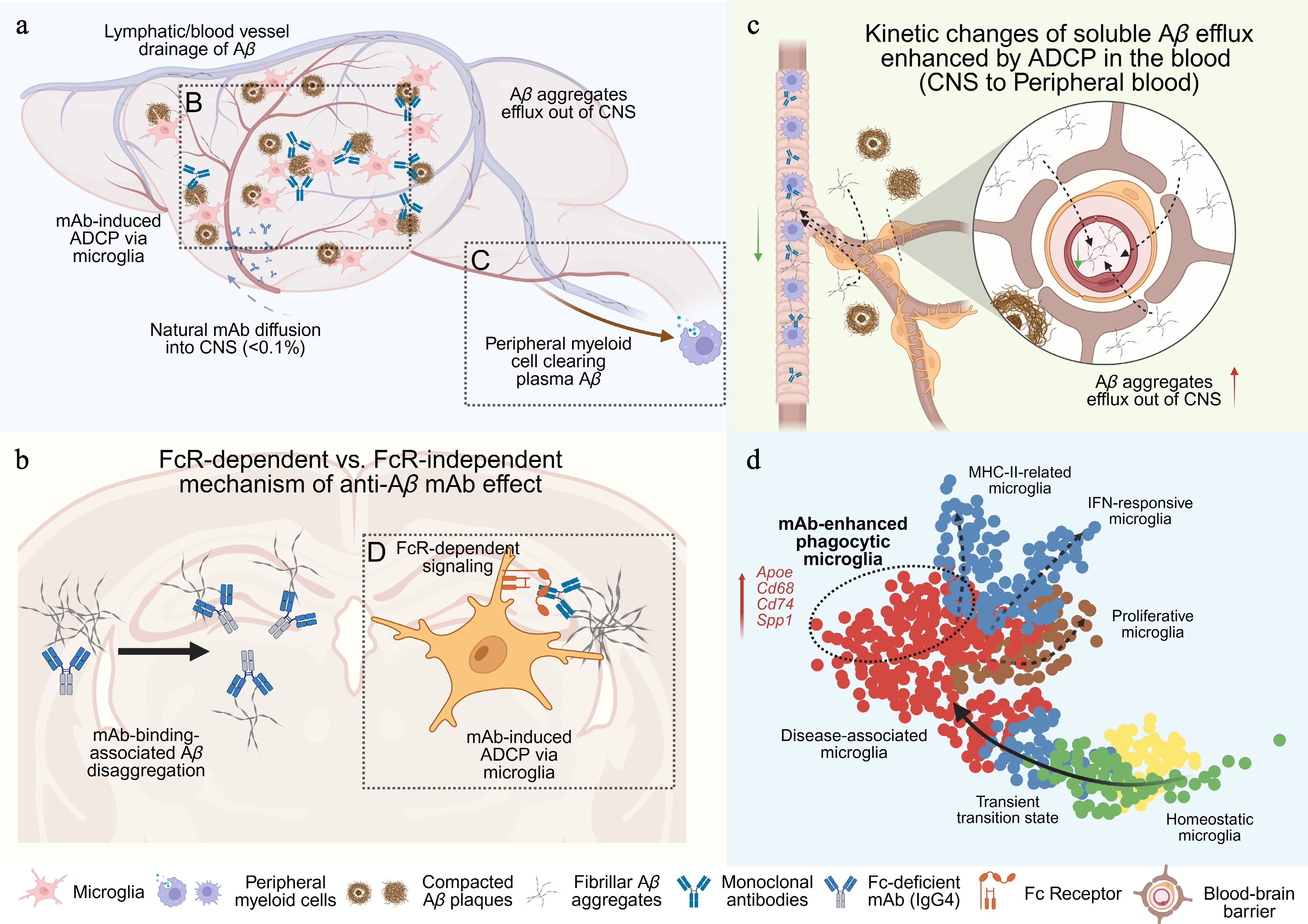
Figure 1.
The conceptual schemes of biological processes involved in the anti-Aβ monoclonal antibody treatments. This figure was created by
biorender.com . (a) Scheme of how anti-Aβ monoclonal antibody therapy interplays with pathological processes regarding the distribution and species of Aβ aggregates. Five crucial pathways for improving Aβ removal were highlighted with text. The blue dash arrow indicates the mAb diffusion into the CNS system. The brown solid arrow shows the efflux of Aβ aggregates from CNS to the peripheral system. Two dashed boxes highlight the antibody-involved biological processes presented in (b) and (c). (b) Scheme of two independent mechanisms to reduce Aβ aggregation in the parenchyma. The dashed box shows the microglial ADCP induced by anti-Aβ antibody, which is associated with the transcriptomic changes discussed in (d). (c) Scheme of how peripheral anti-Aβ mAb induced ADCP alters the passive efflux of Aβ aggregates from CNS to the peripheral blood circulation through the blood-brain-barrier. The green arrows indicate the decreased concentration gradient of Aβ aggregates in the blood stream. (d) The conceptual UMAP of lecanemab-induced microglial responses. Solid arrows indicate the transcriptomic trajectory in response to pathology of neurodegenerative diseases defined by previous literature. Dashed arrows show presumable trajectory among different terminal microglial signatures. The dashed oval highlights the theoretical population of phagocytic microglia. Representative genes upregulated in the lecanemab-induced microglial signature are highlighted in red. -
In parallel with efforts to define the optimal antigenic species, many studies have focused on the role of Fc effector functions[8−10] and the potential modulations of distinct phagocytes in antibody-mediated aggregate clearance[11−14]. It may appear counterintuitive that the field debated whether anti-amyloid antibodies require Fc effector functions to remove amyloid aggregates in the brain for more than two decades[15,16]. Unlike antibodies targeting antigens on living cells, antibodies targeting extracellular proteins or aggregates does not exclusively function through ADCP (Fig. 1b). Antibody binding to Aβ aggregates can directly destabilize and disassemble pre-formed aggregates or fibrils[17]. Fc-effector-deficient antibodies could also disrupt Aβ aggregation without engaging with Fc receptors, and trigger detrimental proinflammatory signaling[18]. For example, crenezumab (an hIgG4 antibody) was specifically designed to neutralize toxic Aβ aggregates through direct binding, largely independent from Fc receptor–mediated responses due to its hIgG4 backbone[19]. However, unfortunately, crenezumab ultimately failed in clinical trials because of insufficient amyloid removal in patients[20]. This negative result, in turn, indicates the importance of Fc effector functions for high efficacy of anti-Aβ monoclonal antibody therapies.
On the other hand, if the primary therapeutic effect of anti-Aβ monoclonal antibodies depends on ADCP, it remains unclear which phagocytic cell plays the major role, including the peripheral myeloid cells, perivascular macrophages, and parenchymal microglia[11–14]. Because anti-Aβ antibodies are administered systemically and Aβ antigens are present from cerebrospinal fluid (CSF) to plasma, some studies suggested that myeloid cells outside the brain may facilitate amyloid clearance during treatment[11,12] (Fig. 1c). Recent discoveries of the roles of the meningeal lymphatic system further highlight the complexity of brain-periphery communication in both spontaneous Aβ clearance and aducanumab-mediated amyloid removal[21]. These findings are particularly significant for the field to consider how antibody-based strategies could apply to other neurodegenerative diseases. Because if the peripheral system contributes largely to the therapeutic effect, it remains uncertain for other pathological protein aggregates such as tau tangles, α-synuclein, and TDP-43, as therapeutic targets, given the differences in their distributions between plasma and CSF during the disease progression.
-
To address the questions above, Albertini et al.[22] published a brief study using a human microglia (iMGL) xenograft mouse model together with a lecanemab human immunoglobulin G1 (IgG1) variant engineered to abolish Fc-mediated effector functions (lecanemab LALA-PG). Using advanced omics techniques including single-cell RNA sequencing (scRNAseq) and spatial transcriptomics, the authors showed that lecanemab induces distinct transcriptomic changes, driving human microglia toward upregulation of lysosomal and phagosomal pathways, as well as MHC-II and interferon(IFN)-associated signatures. Importantly, the comparison with the LALA-PG variant demonstrates that Fc effector function is required for efficient amyloid clearance during lecanemab treatment. Furthermore, by the recently reported FIRE mouse model (Csf1rΔFIRE/ΔFIRE) lacking CNS myeloid cells (both parenchymal microglia and perivascular macrophages), Albertini et al[22]. demonstrated that CNS-resident microglia can play a predominant role in lecanemab-mediated amyloid removal in vivo, with minimal contribution from peripheral mouse myeloid cells in this system. In line with the reduced amyloid pathology, lecanemab treatment also partially protects against amyloid-induced synaptic loss and neuronal dystrophy. Finally, although lecanemab treatment did not alter microglial signatures of individual clusters, it was evident that microglia localized near amyloid plaques constantly upregulate Spp1 in treated animals. Recombinant human osteopontin (OPN, encoded by Spp1) treatment further improved amyloid aggregate clearance by H9-derived microglia in brain slice cultures in vitro.
-
Since the scRNAseq era, pathological microglial responses and disease-associated transcriptomic states have been comprehensively characterized across a range of neurodegenerative disorders[23,24]. Human and mouse microglia signatures are never identical because of both species divergence and technical variability, but shared microglial gene expression patterns occur, which indicates a conserved transcriptional regulation that underlie common activation and response pathways such as disease-associated microglia (DAM) signature[23,25]. However, it remains unresolved whether the DAM response is uniformly protective or varies across different stages of Alzheimer's disease pathology[26]. Even less is known about the physiological and pathological roles of other shared microglial signatures, including IFN- and MHC-II-associated responses. However, the clinical effectiveness of lecanemab and donanemab supports that a distinct, functionally protective microglial transcriptomic state through the FcRγ-dependent phagocytosis signaling exists, which may be partially overlapped with DAM-related features observed across neurodegenerative diseases.
In the original study, the transcriptomic data demonstrates that lecanemab treatment with an intact Fc domain can activate phagosome-related gene programs in microglia around amyloid plaques. Importantly, the differentially expressed genes (DEGs) in the lecanemab treatment are distinct from the ones in the LALA-PG-treated group when comparing plaque-associated microglia to plaque-distant microglia, indicating a treatment-specific transcriptional response. Although the comparative framework in the original analysis may appear somewhat complex and the identified DEG sets may partially reflect intrinsic spatial differences (close to plaque vs. distant from plaque) rather than pure treatment effects (lecanemab vs LALA-PG), this observation nevertheless represents a key insight of the study: anti-Aβ antibody appears to induce a specific phagocytic process rather than simply amplifying all existing, spontaneous reactive states (Fig. 1d).
In line with this interpretation, a recent study examining microglial responses to aducanumab treatment in mice reported a similar activation pattern associated with ADCP[27], including upregulation of MHC-II-related genes (e.g., Cd74, H2-Ab1, H2-Aa) and interferon-responsive genes (e.g., Ifit2, Ifit3, Ifitm3). Notably, aducanumab treatment did not induce a significant increase in Spp1 expression in that study; instead, Spp1 upregulation was primarily associated with age-associated changes in their model. In our own previous work, administration of an inhibitory anti-LILRB4 antibody lacking Fc effector function similarly induced a transcriptional change characterized by increased Cd74 and Apoe expression in highly phagocytic microglia[28].
Together, these studies suggest a microglial activation state that could be favorable for amyloid clearance and highly expressed genes such as Apoe, Cd68, Cd74, and MHC-II genes. Of note, these transcriptomic shifts may not be exclusively driven by Fc domain engagement. Instead, they reflect broader changes in microglial activation induced by the treatments. Lecanemab, however, may trigger additional responses, such as Spp1 upregulation, beyond the shared MHC-II and IFN-related responses. Having said that, the overall transcriptomic differences between lecanemab-treated and control groups remain subtle, and antibody treatment does not induce large-scale shifts of microglial states. Whether this apparent subtlety reflects a biological phenomenon or technical limitations of current single-cell-based techniques remains an open question for future studies.
Moreover, it is particularly interesting that current anti-Aβ monoclonal antibody therapies demonstrate greater efficacy in early-stage patients[2]. One hypothesis is that late-stage pathology with extensive amyloid deposition and tauopathy already induces microglial overactivation or functional exhaustion prior to antibody administration[29,30]. This idea is also supported by findings from multiple anti-TREM2 studies, including the lesson from the Alector clinical trial, in which additional TREM2 stimulation appeared ineffective, likely because amyloid-driven, TREM2-dependent signaling was already maximally engaged[31−33]. An alternative hypothesis is that additional inflammatory, immune, and degenerative stimuli (such as lymphocyte infiltration, tau aggregates, and myelin debris) at late stages of AD will overwhelm the microglial signaling. At this stage, these signals, together with modifiers such as APOE genotype, may dominate microglial state transitions and overshadow ADCP-associated responses, thereby diminishing the therapeutic impact of amyloid removal[34−36]. A third hypothesis is that amyloid clearance could only slow downstream pathological cascades without reversing pre-occurred neuronal damage, such as irreversible tau-mediated neurodegeneration. Following this concept, therapeutic developments should aim at promoting neuronal regeneration, rather than further enhancing amyloid removal, which may offer greater benefit in advanced AD stages[37,38]. Investigating these aspects will require a systematic, stage-dependent characterization of microglial activation states across treatment timepoints and pathological contexts, which are crucial for advancing our understanding of microglial biology and optimizing therapeutic strategies for neurodegenerative diseases.
-
Several limitations exist in the current study, which warrant consideration. First, the lecanemab treatment merely led to suboptimal amyloid clearance in this study, and the effect was variable across experiments, as illustrated by the representative images. This raises concerns about the consistency of antibody effectiveness within their experimental system. Given the well-documented lecanemab studies in other rodent models and human patients, one may raise concerns regarding the intrinsic limitations of the chimeric transplantation approach: xenografted human microglia may adopt atypical transcriptional states due to the interaction between human-intrinsic programs and the murine brain environment, which cannot be translated into either human or mouse microglial responses[39].
Second, although Spp1 upregulation was consistently observed across multiple lecanemab treatment experiments and supported by in vitro data in this study, it remains unclear whether Spp1 is involved in functional phagocytosis in vivo. The spatial association between Spp1 expression and lecanemab-FcR engagement alone is insufficient to demonstrate its function. The inclusion of additional genetic approaches, such as OPN (Spp1) loss-of-function or overexpression models, would have strengthened the causal link between Spp1 and improvement of ADCP of amyloid aggregates.
Moreover, FcR-dependent immune activation should not be conflated with FcR-dependent phagocytosis[8]. Downstream signaling of FcR engagement can broadly reshape immune transcriptional programs. Within the scope of the original study, many of the DEGs may be indirectly regulated by FcR signaling rather than directly involved in amyloid clearance, such as driving parallel immune responses like IFN-associated responses.
Finally, an limitation of the human–mouse chimeric model is its requirement for inhibiting the adaptive immune system to enable the transplantation of human iMGL. Given increasing evidence for T cell infiltration and adaptive immune involvement in neurodegenerative diseases[36,40], this constraint prevents assessment of the complex interplay between ADCP and antibody-dependent cellular cytotoxicity that may occur simultaneously in patients receiving lecanemab. Using new models to study these processes will be an important direction for future studies.
-
The demonstration on the Fc effector function of anti-Aβ monoclonal antibody therapies gives important directions for the next generation of antibody engineering. Different from the work on infectious diseases that usually focuses on Fc modifications to expand plasma half-life[41], the major challenge in monoclonal antibody development for CNS diseases stands on limited blood-brain-barrier permeability[42]. Pharmaceutical companies like Roche and Denali Therapeutics made pioneering progress introducing new brain delivery methods by fusing the Fc domain with transferrin domain to induce transferrin receptor-mediated transcytosis[43,44], while more research groups are actively exploring alternative transcytosis pathways, such as CD98hc[45,46]. However, whether such Fc modifications would change Fc effector functions remains uninvestigated and will require future studies. Moreover, the brain microenvironment in AD and other neurodegenerative diseases differs fundamentally from that of the peripheral immune system, particularly with the restricted repertoire of Fc receptors expressed by CNS-resident cells[47,48]. In addition, the expression and functions of neuronal Fc receptors remain a puzzle and should not be overlooked when designing Fc-engineering strategies for enhancing ADCP of amyloid aggregates[49,50]. Collectively, these highlight critical aspects for future research in optimizing antibody-based therapies for neurodegenerative diseases.
On the other hand, Spp1, widely recognized as a marker of extracellular matrix remodeling, was first identified in 2017 as a core DAM marker in plaque-associated microglia regulated by TREM2[25]. Since then, Spp1+ microglia have been discovered across multiple neurodegenerative diseases[51−53], as well as in developmental stages when activated microglia facilitate synaptic pruning[54,55]. Beyond the CNS microglia, Spp1 upregulation is also associated with peripheral inflammation, fibrosis, and cancer suppression[56−58], where it can indirectly influence myeloid cell activation and adhesion to extracellular matrices. Although direct administration or overexpression of Spp1 may yield limited effects to improve microglia phagocytosis, the findings in the lecanemab study suggests that combination strategies, such as cocktail therapies or fusion proteins with anti-amyloid antibodies, could potentially further boost amyloid clearance. Given the shared upregulation of Spp1 across different neurodegenerative diseases, targeting this pathway may offer broader therapeutic applicability than antigen-specific antibodies alone. At the same time, other microglial gene signatures revealed by those antibody treatments warrant further consideration. Small-molecules or alternative approaches to induce similarly high phagocytic microglial states may be a promising direction for developing therapies that require efficient removal of extracellular junk, including AD, multiple sclerosis, and traumatic brain injury.
-
Not applicable.
-
The author confirms sole responsibility for the following: conception and design, data collection, analysis and interpretation of results, and manuscript preparation.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current commentary.
-
The author declares that there is no conflict of interest.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen Y. 2026. Anti-amyloid antibody therapies for Alzheimer's disease: how much do we really understand? Targetome 2(1): e004 doi: 10.48130/targetome-0026-0004 

Anti-amyloid antibody therapies for Alzheimer's disease: how much do we really understand?
- Received: 07 January 2026
- Revised: 20 January 2026
- Accepted: 23 January 2026
- Published online: 09 February 2026
Abstract: A new study published inNature Neurosciencedemonstrated that Fc effector function is essential for amyloid aggregate clearance during lecanemab treatment, a recently FDA-approved therapy for Alzheimer's disease. These findings underscore the requirement for intact Fc-mediated activity to achieve therapeutic efficacy. In addition, single-cell RNA sequencing identified microglia transcriptomic changes that are unique to lecanemab treatment and extend beyond previously characterized microglial activation signatures. Together with a retrospective review of anti-amyloid antibody development and related mechanisms, this commentary provides important insights into new directions for the design of next-generation therapies for Alzheimer's disease.
-
Key words:
- Lecanemab /
- Alzheimer's disease /
- Amyloid-β /
- Therapeutic target /
- Microglia