










-
Maize is one of the most important staple crops, contributing to approximately 40% of the global cereal food supply[1]. High-vigor seeds, characterized by high germination rate and uniform germination across diverse field conditions, are essential for maximizing yield production[2−4]. Studies have demonstrated that early harvesting can achieve the highest seed vigor in maize and rice[5,6]. However, these early-harvested seeds invariably possess a high moisture content (MC). Seeds with high MC are more susceptible to damage from mechanical harvesting and elevated drying temperatures, which generally lead to a decline in seed vigor during seed processes[7−9]. Furthermore, high MC seeds incur greater harvesting and drying costs, and they require immediate drying to prevent damage from spontaneous heating caused by increased respiratory activity[10]. Therefore, reducing seed MC at harvest is particularly critical for producing high-vigor seeds.
Previous works have extensively explored the physiological and genetic characteristics of seed dehydration, a critical trait directly linked to seed MC at harvest in maize. Results suggest that the entire dehydration process can be divided into two distinct stages: pre-physiological maturity (PM) dehydration and post-PM dehydration[11,12]. Pre-PM dehydration is a physiological process regulated by the sink-source relationship and primarily controlled by genetic factors; conversely, post-PM dehydration is a physical process mainly influenced by environmental factors such as temperature and humidity[13−16]. Since the final seed harvest MC is regulated by both pre- and post-PM dehydration, breeding varieties that exhibit rapid dehydration is an efficient strategy for reducing the MC of harvested seeds.
Significant differences in seed MC at harvest have been observed across maize varieties. For example, white-endosperm maize often shows a significantly higher MC than yellow-endosperm varieties[17], and Spanish hybrid varieties averaged 2.1% higher seed MC at harvest maturity than American varieties[18]. Because almost all cultivated maize varieties are hybrids produced by crossing inbred lines, researchers have focused on the relationship between parental inbreds and their derived hybrids, suggesting a close genetic relationship for seed MC traits. For instance, the inbred line B14 was reported to have a significantly higher seed MC at harvest than Oh45, and its hybrid offspring displayed the same difference[19]. Moreover, data from 25 parental inbreds and 31 single-cross hybrids across three environments revealed high and significant correlations between inbred and hybrid performance for seed MC at physiological maturity[20]. An investigation using genomic prediction for the seed dehydration rate in diverse inbred lines also confirmed that selecting for low MC in inbreds can contribute effectively to hybrid improvement[21]. These reports collectively confirm the substantial influence of parental inbred lines on the final MC performance of hybrids.
In general, General Combination Ability (GCA) is considered the main effect reflecting additive gene action, while Specific Combination Ability (SCA) is regarded as an interaction effect indicating non-additive gene action, specifically dominance variance, of the parental inbred lines on hybrid performance[22−24]. Despite the frequent use of GCA and SCA analyses for yield traits, their application to the dehydration trait remains largely unexplored.
Genomic imprinting is a key epigenetic mechanism that underpins a specific type of SCA, representing a unique form of non-additive gene action. This phenomenon, which is widely present in hybrid plants, refers to the expression of certain genes that is dependent on the parental origin of the allele[25]. Imprinted genes are categorized as Maternally Expressed Genes (MEGs) or Paternally Expressed Genes (PEGs), based on the principal parental source of expression[25]. In plants, gene imprinting was first discovered in maize, where the expression of an R gene in the endosperm showed maternal allelic specificity[26]. Subsequently, genes such as FIE1, FIE2, NRP1, MEZ1, MEG1, and PEG1 have also been found to be imprinted in maize endosperm. Specifically, FIE1 is maternally expressed and may influence the imprinting status of PEGs in hybrids, with a biological function in regulating nutrient balance between the endosperm and embryo[27]. In contrast, ZmFIE2 exhibits a paternal allele-preferential expression pattern, though its function has not yet been elucidated[28]. NRP1, identified by a genome-wide mRNA profiling study, is an imprinting gene with a putative function in the endosperm[29]. MEZ1 display maternal allele-preferential expression in the endosperm but not in the embryonic tissues[30], while PEG1 is a paternally expressed gene in the endosperm[29]. MEG1 is maternally expressed, showing a maternal parent-of-origin expression pattern during early stages of endosperm development but switching to biallelic expression at later stages[31].
Regarding the seed dehydration trait, no imprinting gene has been identified to date. Although several genes regulating this trait have been cloned from inbred lines[32−34], their performance in hybrid seeds has not yet been tested. Therefore, in this work, a genome wide screening of imprinting genes related to the seed dehydration trait was conducted by performing transcriptome sequencing on a pair of reciprocal hybrids. These hybrids were constructed by crossing two inbred lines with contrasting dehydration rates. We successfully identified 226 MEGs and 112 PEGs, and further verified imprinted expression of two candidate genes in the reciprocal hybrids using qPCR. These results introduce new candidate imprinted genes for dehydration, which hold significant potential for the breeding of low-MC maize hybrids.
-
Four inbred lines were selected from an inbred line panel, and cultivated in Zhuozhou, Hebei Province, China (39°48′ N, 115°97′ E) during the summer of 2019 and 2020. Seeds were sown on April 30, 2019 and May 20, 2020, using a row length of 5 m, a row spacing of 60 cm, and a plant spacing of 20 cm. The inbred lines and their reciprocal crosses were planted using a Randomized Complete Block Design (RCBD), with four replicates. All female ears were covered with paper bags prior to silking. On the day of silking, manual pollination was conducted for self-pollination of parental lines and reciprocal cross-pollination of hybrids. Cobs were manually harvested every 5 d from 30 DAP (Days After Pollination) to 70 DAP. Seeds were manually threshed from the cob after natural indoor drying at a temperature of 20 to 30 °С.
Before sowing, 85 kg·ha−1 P2O5, 90 kg·ha−1 K2O and 100 kg·ha−1 nitrogen (217 kg·ha−1 urea) were applied during field forking. An additional 60 kg·ha−1 nitrogen was applied to fertilize the plants at the shooting stage (six expanded leaves) and the silking stage, along with timely irrigation. Turf machinery and weeding were applied before sowing to ensure proper seedling establishment. Sprinkler irrigation was applied to ensure a sufficient water supply and to eliminate the possibility of water deficits during plant growth.
Seed MC measurement
-
Upon harvest at each time point, a portion of the seeds was randomly selected to immediately record the seed number and fresh weight. Then, seeds were placed in a paper bag for air drying outdoors, transported to the laboratory, and dried to constant weight at 130 °C. The final MC was calculated, allowing for the subsequent calculation of dehydration rate (daily decrease in seed MC), and water loss rate (daily decrease of water in one seed, expressed in mg).
The seed MC was determined according to our previous work[6]. Briefly, 150 seeds were randomly collected from the middle sections of fresh-harvested ears (five ears, 30 seeds per ear). Seeds were immediately weighed and dried in an air-drying oven at 105 °C for 5 h. The water loss, expressed as a percentage, was denoted as S1. Then, all seeds were ground into powder, and a 5 g subsample was dried again at 130 °C for 1 h. The water loss was expressed as S2. The seed WC was calculated using the following formula:
$ \mathrm{WC=S1+S2-S1\times S2/100} $ Transcriptome sequencing
-
Seeds without seed coat were harvested and used for transcriptome analysis, with two replicates. Ten pooled seeds per replicate were ground in liquid nitrogen for total RNA extraction using the RNAprep pure Plant Kit (Tiangen Biotech, Beijing, China). Sequencing libraries were constructed from 1.5 µg RNA using the NEBNext UltraTM RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA). Libraries were sequenced on an Illumina platform to generate 150 bp paired-end reads. FastQC (version 0.11.9) was used for data quality detection, and Trimmomatic (version 0.39) was used to filter the data, removing adaptors and low-quality reads[35]. Subsequently, clean reads were aligned to the B73 reference genome (Zm-B73-REFERENCE-GRAMENE-4.0) using Hisat2[36]. Uniquely mapped reads were extracted for gene-expression quantification using Stringtie (version 2.2.1). The DESeq2 R package was used to calculate normalized FPKM (fragments per kilo-base of transcript per million reads of base pairs mapped) values. Genes with a p < 0.05 threshold were identified as Differentially Expressed Genes (DEGs) between comparable groups. Heatmap figures were plotted using the pheatmap R package (v1.0.12). Gene Ontology (GO) enrichment analyses were performed using the web-based software AgriGOv2 at
http://systemsbiology.cau.edu.cn/agriGOv2 . Significant GO categories (FDR < 0.05) were identified for biological processes, molecular function, and subcellular locations. Gene annotations were sourced fromwww.maizegdb.org .SNP calling and imprinting gene identification
-
STAR (version 2.7.6a) was used to align the RNA-seq data. The aligned data were processed using Picard (version 1.119), and GATK (version 4.1.9.0). Variation between the parental lines Z1 and P was performed following the GATK document (
https://gatk.broadinstitute.org/hc/en-us/articles/360036194592-Getting-started-with-GATK4 ). In brief, the Select Variants tool was used to screen for biallelic SNPs, with the parameters of --restrict-alleles-to BIALLELIC -select 'vc.getHetCount()==0 && vc.getHomVarCount() >= 1' --select-type-to-include SNP; then the norm --rm-dup exact parameter of bcftools (version 1.3.1) was used to remove the duplicate variation site information. Then, the ASEReadCounter tool of GATK was used to obtain read counts from the Z1 and P sources covering each SNP site in the reciprocal hybrids. Only sites with a total read count ≥ 10 were selected, and the expression ratio of maternal and paternal alleles at each SNP locus was calculated. SNPs were screened based on the chi-square test (χ2) to determine whether the observed ratio of maternal/paternal to total alleles significantly deviated (p < 0.05) from the expected ratios. For the imprinted SNP identified in both crosses, a 401 bp sequence (200 bp on each side) from the maternal genome was input into the BLAST tool atwww.maizegdb.org website to find the target genes in B73 (identity > 90%, E-value < 1e-10).qRT-PCR validation of RNAseq data
-
qRT-PCR was performed using the TransStart Green qPCR SuperMix kit (Tiangen), on the 7300 Real Time PCRSystem (Applied Biosystems, Waltham, MA, USA). The maize ACTIN gene (Zm00001d010159) served as the endogenous control. Primers are listed in Supplementary Table S8. Three biological replicates were conducted, with each being technically repeated three times. Gene transcript relative abundance was calculated using the 2−ΔΔCᴛ method.
-
Four inbred lines, Zheng58 (Z1), PH4CV (P), Zheng30 (Z2), and Dan360 (D) were cultivated in 2019 and 2020, and three pairs of reciprocal hybrids were generated: Z1P (Z1 maternal × P paternal) and PZ1, Z1Z2 and Z2Z1, and DP and PD. Seed MC was measured from 30 DAP to 60 DAP at 5- or 10-d intervals. As seed development progressed, the MC of all inbreds and hybrids uniformly decreased. At every time point, significant MC differences were observed between the parental lines of each cross. Lines Z1 and D consistently showed higher MC values compared to their respective paired parents, P and Z2, in both 2019 and 2020 (Supplementary Fig. S1). Consequently, P and Z2 were classified as faster dehydration rate (DR) lines relative to Z1 and D.
Concerning the reciprocal hybrids, the MC of Z1P was consistently higher than that of PZ1 at all investigated time-points in both years, indicating that PZ1 dehydrated faster (Supplementary Fig. S1a, S1b). Similarly, Z1Z2 and DP showed higher MC than Z2Z1 and PD, respectively (Supplementary Fig. S1c−S1f). These results collectively demonstrate that hybrids created using the higher MC inbred as the maternal parent maintained a higher MC than their reciprocal crosses. This finding strongly suggests that a maternal effect significantly influences the dehydration rate of hybrid seeds.
The average daily dehydration rate from 30 DAP to 60 DAP was calculated (Fig. 1a–c). In both years, P had the fastest rate (1.17% per day in 2019, and 1.11% per day in 2020), while Z1 had the slowest (0.85% per day in 2019, and 0.77% per day in 2020). Crucially, the hybrids PZ1, Z2Z1, and PD (faster line as female parent) exhibited significantly faster dehydration rates than their reciprocal hybrids (Z1P, Z1Z2, and DP) in both years.
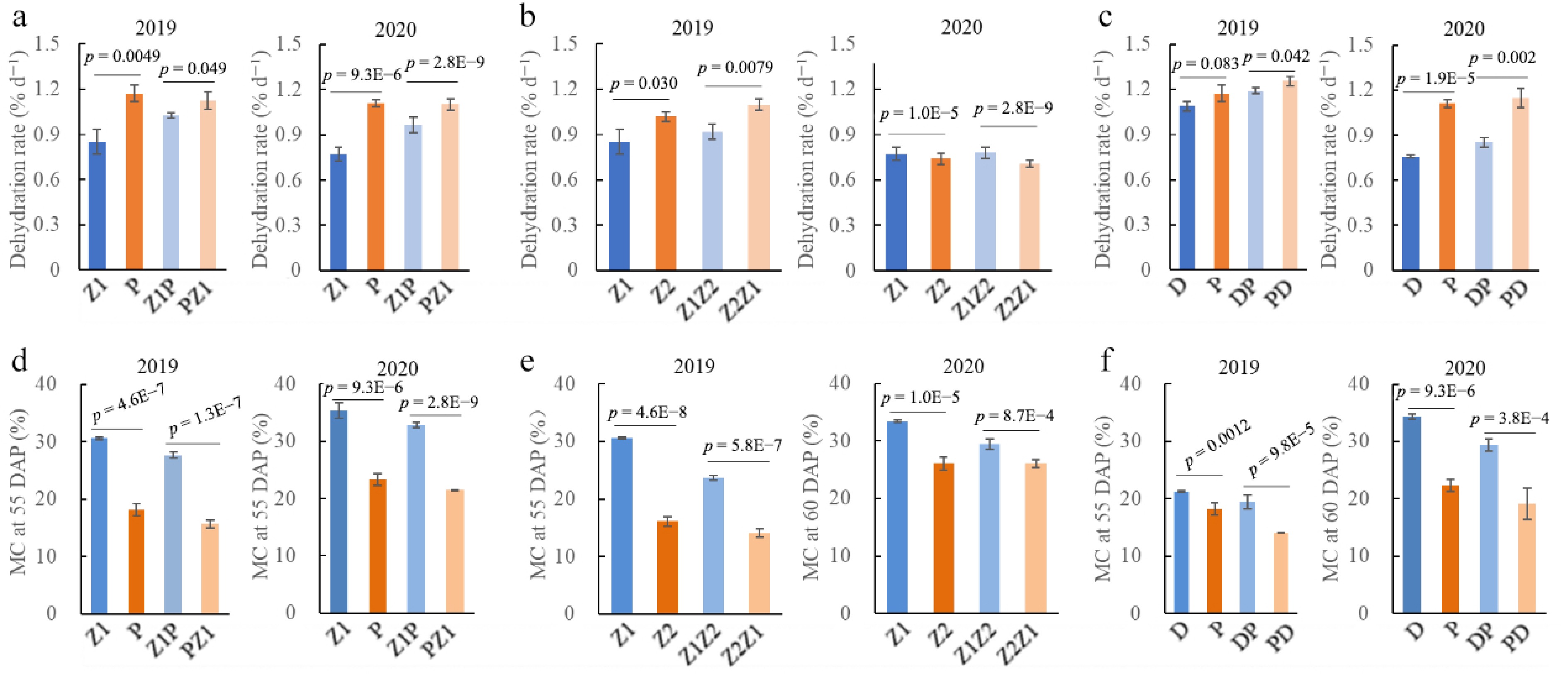
Figure 1.
Seed dehydration rate and moisture content (MC) of maize inbred line and their reciprocal hybrids in 2019 and 2020 years. Seed dehydration rate from 30 to 70 days after pollination (DAP) for maize inbred line Z1, P, Z2, and D, and their reciprocal hybrid pairs (a) Z1P and PZ1, (b) Z1Z2 and Z2Z1, and (c) DP and PD. Seed MC at 55 DAP for inbred line Z1, P, Z2, and D, and their reciprocal hybrid pairs (d) Z1P and PZ1, (e) Z1Z2 and Z2Z1, and (f) DP and PD. Z1 (Zheng58), P (PH4CV), Z2 (Zheng30), and D (Dan360) are parental inbred lines. Z1P and PZ1, Z1Z2 and Z2Z1, and DP and PD represent reciprocal hybrids between Z1 and P, Z1 and Z2, and D and P, respectively. In the hybrid nomenclature, the first listed inbred was used as the female parent, and the second as the male parent.
Identification of differently expressed genes
-
Since the biggest MC difference across all three reciprocal crosses was observed around 55–60 DAP in both years (Fig. 1d–f), 55 DAP seeds were selected (with seed coats removed) from Z1, P, and their reciprocal hybrids Z1P and PZ1 for transcriptome sequencing. The sequencing generated 21,599,657–23,370,500 clean reads for each sample, with 87.73%–94.93% (mean 92.30%) reads being successfully mapped to the maize B73 reference genome, and 83.37%–90.90% (mean 87.48%) mapped reads being single-site mapping (uniquely mapped reads). The uniquely mapped reads were used to estimate the normalized gene expression values by an indicator of FPKM. After defining an expressed gene as FPKM value ≥ 1, 28,959–32,189 (mean 30,589) expressed genes were identified across the samples (Supplementary Table S1). High Pearson correlation coefficients (> 0.98) between replicates indicated the repeatability of the RNA-seq data (Supplementary Fig. S2),
Comparing the two parents (Z1 and P) identified 9,899 parent DEGs, under a threshold of Log2(FoldChange) > 1 and adjusted p < 0.05. Of these, 4,161 were up-regulated, and 5,738 down-regulated in the faster dehydration line P relative to the slower one Z1 (Fig. 2a; Supplementary Table S2). Similarly, comparing the two reciprocal hybrids (PZ1 vs Z1P) identified 1,158 hybrid DEGs, with 545 up- and 613 down-regulated in the faster line PZ1 relative to the slower one Z1P (Fig. 2b; Supplementary Table S3).
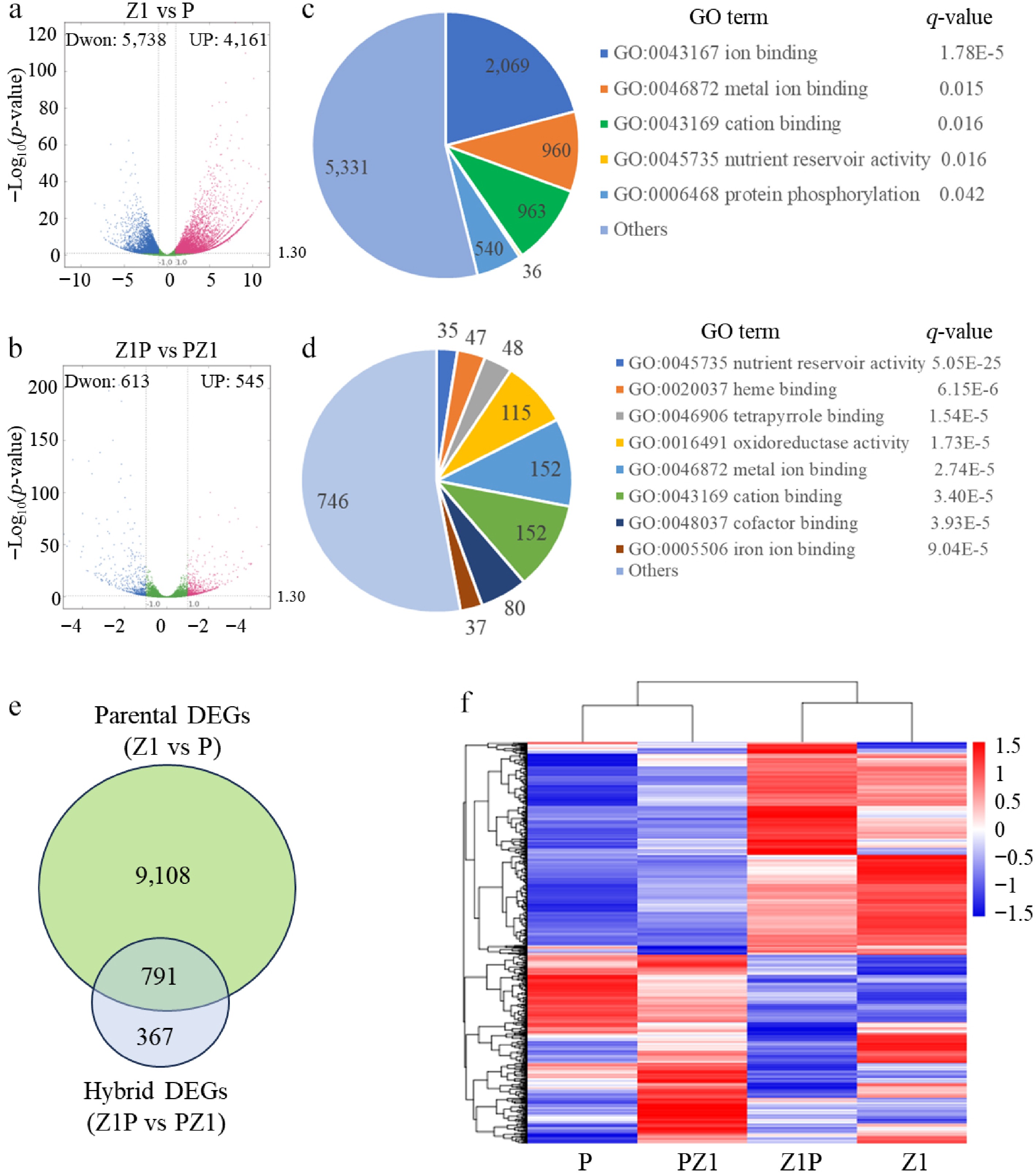
Figure 2.
Identification of different expression genes (DEGs) between parents Z1 and P (parent DEGs), and between reciprocal hybrids Z1P and PZ1 (hybrid DEGs). Volcano plots representing the (a) parent DEGs and the (b) hybrid DEGs. Blue and red points represent significantly down-regulated and up-regulated genes, respectively. The most significantly enriched GO terms for the (c) parent DEGs and the (d) hybrid DEGs. For each GO term, the number of genes is displayed within the chart. The q-value, calculated using a hypergeometric distribution test followed by the Benjamini-Hochberg correction, is listed to the right of the term. (e) Venn diagram showing the overlap between parent DEGs and hybrid DEGs. (f) Heatmap showing the expressions profiles of the DEGs overlapping between parent DEGs and hybrid datasets.
GO analysis was conducted to find that the parent DEGs were primarily enriched in ion binding (GO:0043167), metal ion binding (GO:0046872), cation binding (GO:0043169), nutrient reservoir activity (GO:0045735), and protein phosphorylation (GO:0006468) (Fig. 2c). Meanwhile, the hybrid DEGs were significantly enriched in nutrient reservoir activity (GO:0045735), heme binding (GO:0020037), tetrapyrrole binding (GO:0046906), and oxidoreductase activity (GO:0016491) (Fig. 2d). Then, KEGG clustering analyses was conducted to show that the most enriched functions of the parent DEGs were in stibenoid, diarylheptanoid and gingerol biosynthesis, alpha-linolenic acid metabolism, and flavonoid biosynthesis (Supplementary Fig. S3a), and the most enriched functions of the hybrid DEGs were in stibenoid, diarylheptanoid, and gingerol biosynthesis, phenylalanine metabolism, and flavonoid biosynthesis (Supplementary Fig. S3b). Thus, the functional enrichments showed some level of similarity between the parent DEGs and the hybrid DEGs in secondary metabolism pathways such as stibenoid, diarylheptanoid, and gingerol biosynthesis, and flavonoid biosynthesis.
Then, 791 common DEGs that overlapped between the parent DEG and hybrid DEG were identified (Fig. 2e), meaning that most hybrid DEGs (68.3%) overlapped with the parent differences. Heatmap clustering of these common DEGs segregated the samples into two groups (P/PZ1 and Z1/Z1P) (Fig. 2f). This pattern suggests that the gene expression profile of a hybrid was more similar to its maternal parent than its paternal parent.
GO and KEGG analyses were also performed on the remaining 367 non-overlapping hybrid DEGs (hybrid-specific DEGs), and it was found that they were primarily enriched in GO terms of transmembrane activity, including obsolete intrinsic components of the plasma membrane, transmembrane transporter activity, carbohydrate transmembrane transporter activity, and so on (Supplementary Fig. S4a). The most enriched KEGG pathways for these hybrid specific DEGs also included transporters (Supplementary Fig. S4b). This indicates that the hybrid-specific expression differences mainly involve the transport of nutrients (starch, sucrose, amino acids) from the maternal plant to the developing seed.
Identification of imprinting SNP for maize seed dehydration
-
To identify imprinted genes, the genetic variation between the two parent lines, Z1 and P were first filtered, obtaining 594,862 high-quality SNPs. At these SNP sites, the sequences were compared to the B73 reference genome. Sites identical to Z1 were retained; for sites that differed, the B73 genotype were replaced with the corresponding SNP genotype from Z1. This process generated an improved reference genome, with all SNP sites made consistent with Z1, which we designated as the Z1 reference genome. The transcriptome data from the reciprocal hybrids were aligned to this Z1 reference genome. This alignment yielded 140,727 SNPs that could be aligned across all four hybrid samples (two replicates per reciprocal cross), of which 117,590 sites were covered by at least 10 reads in every sample (Supplementary Table S4).
The maternal allele proportion (MAP) was defined as the ratio of maternal reads to the total reads (maternal and paternal) at each SNP in a hybrid, and the MAP calculated for the 117,590 SNPs across the four samples (Supplementary Table S4). The MAP for individual SNPs ranged from 0 to 1 within a sample. The average MAP across all SNP was 0.512 in Z1P replicate 1 (Z1P_R1), 0.511 in Z1P_R2, 0.536 in PZ1_R1, and 0.537 in PZ1_R2 (Fig. 3a), resulting in an overall mean MAP of 0.524 across all samples. In a diploid embryo, the theoretical MAP is 0.5, whereas in a triploid endosperm, it is 0.67. The observed overall average MAP of 0.524 in the whole seed is closer to the embryo's theoretical value (0.5) than to the endosperm's (0.67), indicating that embryonic tissue contributes more to the transcriptome abundance in the hybrid seed than endospermic tissues do.
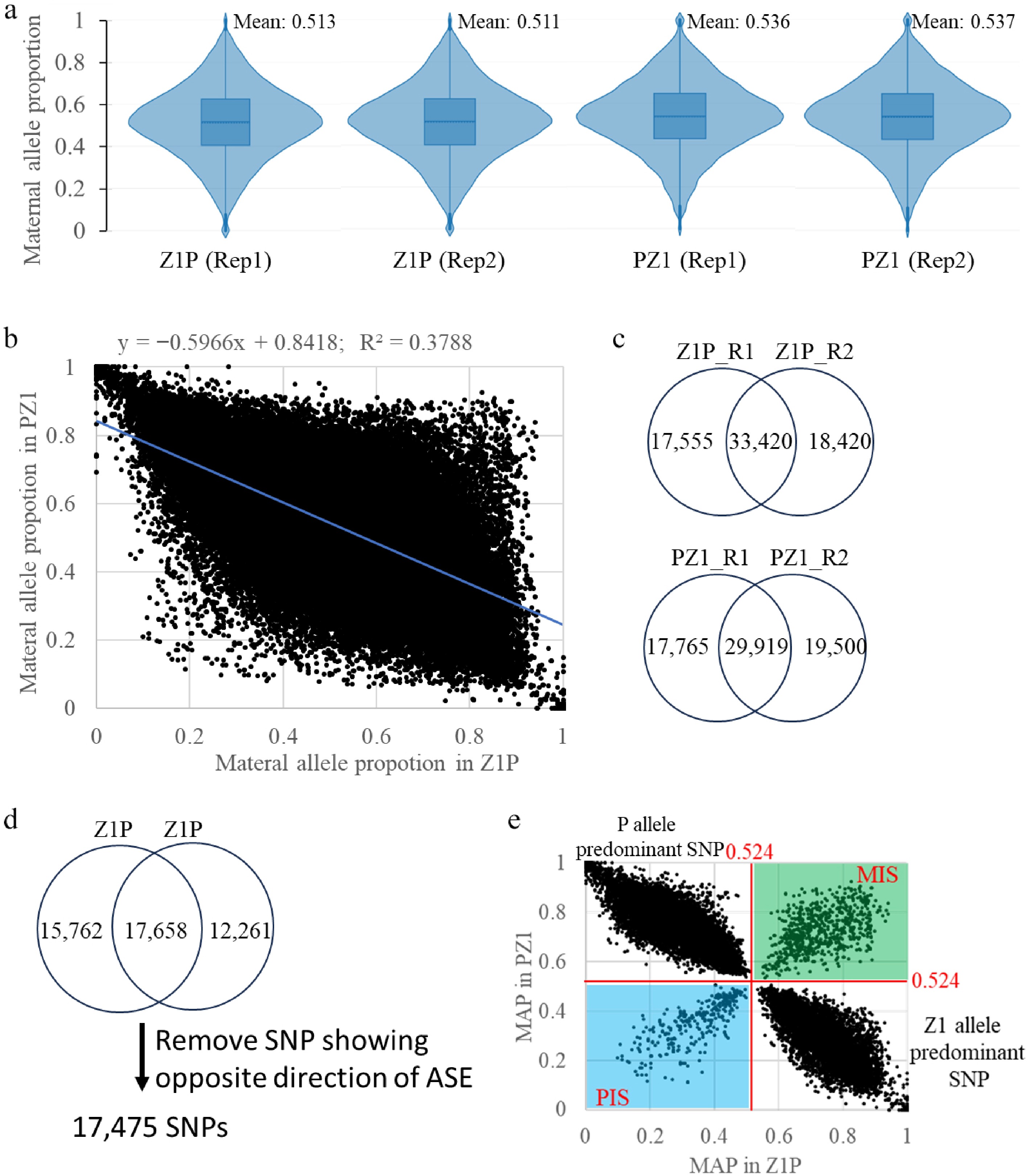
Figure 3.
Identification of maternally expressed SNP (MES) and paternally expressed SNP (PES). (a) Distributions of maternal allele proportions (MAP) in four samples derived from two reciprocal hybrids (Z1P and PZ1) with two replicates (Rep1 and Rep2). (b) Correlation of MAP values between the reciprocal crosses (Z1P and PZ1). (c) Venn diagram depicting imprinted SNPs identified from the two replicates (R1 and R2) within each hybrid cross (Z1P and PZ1). (d) Venn diagram showing the overlap of imprinted SNPs identified from the two reciprocal hybrids (Z1P and PZ1). (e) Scatter plot of allele-specific expression for identified SNPs. The green and blue areas represent SNPs exhibiting consistent maternally or paternally expression bias, respectively, in both reciprocal crosses. The other two areas correspond to SNPs showing expression bias specific to one of the parental lines (P or Z1).
Using the 117,590 SNPs, correlation analysis revealed a high reproducibility of MAP between the two biological replicates within each hybrid (Pearson correlation coefficient > 0.93) (Supplementary Fig. S5). In addition, the correlation of MAP between the two reciprocal hybrids (Z1P and PZ1) was negative (−0.38) (Fig. 3b), indicating that allele-specific expression (ASE), an imbalance in parental allele expression, is prevalent in both reciprocal hybrids and tends to be dependent on the inbred-line origin, irrespective of whether the SNP is inherited maternally or the paternally.
Since the overall average MAP in the whole seed is 0.524 (Fig. 3a), an imprinted SNP was defined as when its MAP deviated significantly from 0.524 by chi-square test (p < 0.05; χ2 > 3.84). This analysis identified 33,420 SNPs with significant deviation in both replicates of Z1P, and 29,919 in both replicates of PZ1 (Fig. 3c; Supplementary Table S4). Among these, 17,658 SNPs showed significant deviation in both reciprocal hybrids (Fig. 3d).
An SNP was defined as a maternally expressed SNP (MES)/paternally imprinted SNP (PIS) if its MAP > 0.524; and a paternally expressed SNP (PES)/maternally imprinted SNP (MIS) if its MAP < 0.524. Among the 17,658 overlapping SNPs, 70 showed opposite allelic predominance between the two Z1P replicates (i.e., MES in one replicate but PES in the other), and 120 showed such discordance between the PZ1 replicates, with seven SNPs overlapping between these two sets. After removing these inconsistent SNPs, 17,475 reliable imprinted SNPs were retained that showed a consistent and significant deviation from 0.524 across all four samples (Fig. 3d; Supplementary Fig. S6).
Analysis of these reliable SNPs in the reciprocal hybrids showed that the majority showed ASE. Specifically, 10,094 SNPs displayed Z1-specific predominance (Z1 allele proportion > 0.524 in both Z1P and PZ1), while 6,334 SNPs displayed P-specific predominance (P allele proportion > 0.524 in both Z1P and PZ1) (Fig. 3e). Additionally, 727 MES were identified, characterized by a MAP > 0.524 in both reciprocal hybrids (indicating Z1 allele predominance in Z1P and P allele predominance in PZ1) (Supplementary Table S5). Conversely, 318 PES were identified, with MAP < 0.524 in both hybrids (indicating P allele predominance in Z1P and Z1 allele predominance in PZ1) (Fig. 3e; Supplementary Table S6).
Identification of imprinting genes for maize seed dehydration
-
To identify the genes associated with the identified imprinted sites, a 401 bp sequence (comprising 200 bp upstream and 200 bp downstream) flanking each MES and PES in the Z1 reference genome was used to perform a BLAST search against the maize genome database (
www.maizegdb.org ). The 727 MES mapped to 259 chromosome regions. Of these, 226 regions harbored coding genes (226 MEGs), and 33 were located in non-coding regions (Supplementary Table S5). Among the MEGs, seven were targeted by more than 10 SNPs, 38 by 5–10 SNPs, and 106 by only a single SNP. Similarly, the 320 PES corresponded to 132 chromosome regions. These regions targeted 112 coding genes (PEGs) and 20 non-coding regions (Supplementary Table S6). Among the PEGs, one gene (Zm00001d038791) was targeted by 11 SNPs, 10 by 5–10 SNPs, and 45 by only a single SNP.The expression patterns of these MEGs and PEGs were first examined in the parental lines (Z1 and P), and their reciprocal hybrids (Z1P and PZ1). Heatmap analyses showed that most MEGs and PEGs had similar expression levels across all four genetic materials, suggesting that identification of imprinting (ASE) does not necessarily correlate with a significant difference in total gene expression (material- or paternal-specific expression) (Fig. 4a, b). Furthermore, the identified MEGs and PEGs were compared with previously identified DEGs from the parental lines and the hybrids, and revealed that only 17 MEGs and six PEGs overlapped with the hybrid DEGs (Fig. 4c). Additionally, only 16 MEGs and three PEGs overlapped with the 691 common DEGs identified across both parental and hybrid comparisons (Fig. 4c). These results collectively suggested that the imprinted genes (MEGs and PEGs) are largely distinct from genes showing differential expression between the two parental lines or between the reciprocal hybrids.
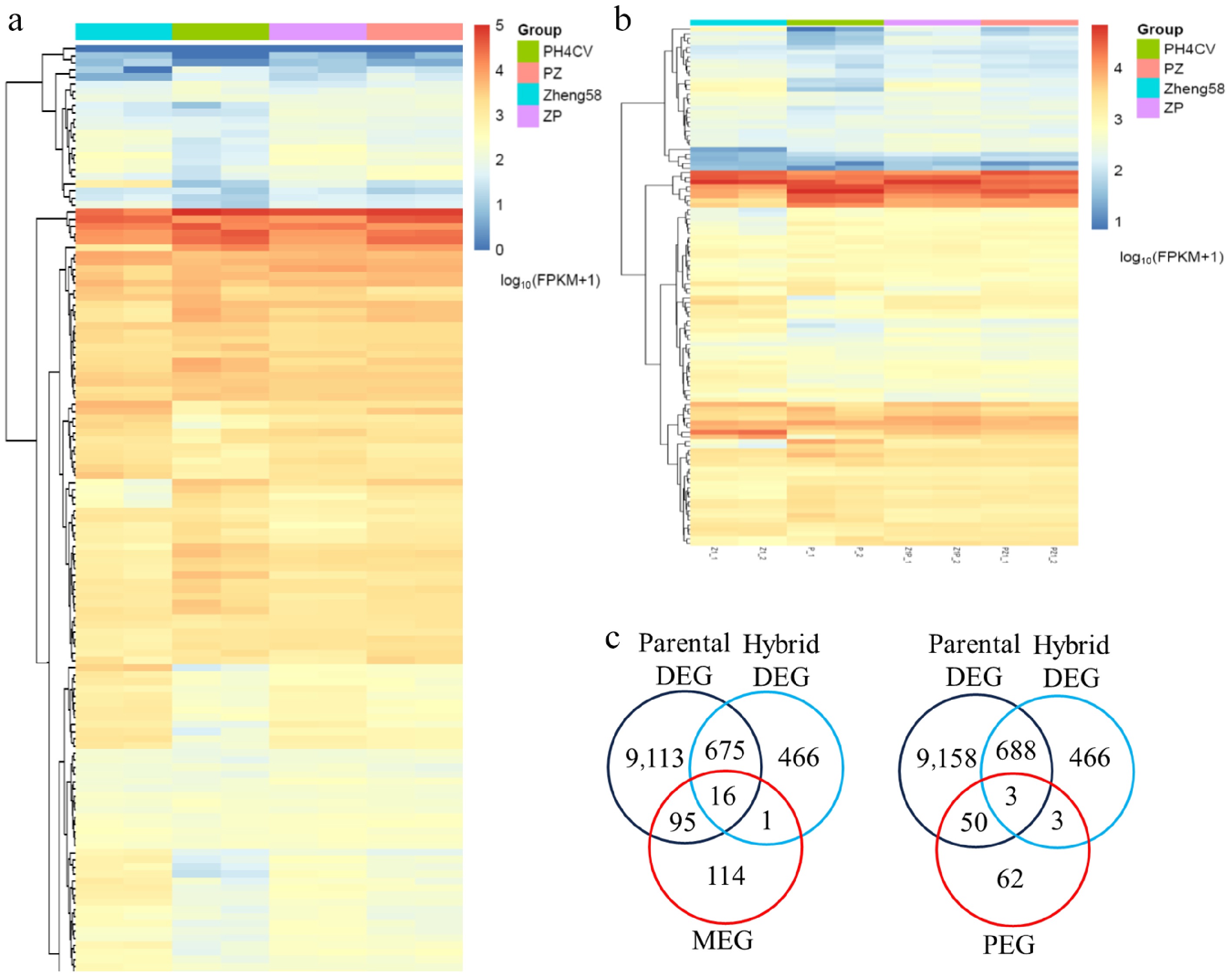
Figure 4.
Characteristics of the maternally expressed genes (MEGs) and paternal expressed genes (PEGs). Heatmap illustrating the total expression levels of the identified (a) MEGs and (b) PEGs across the parental lines (Z1, P), and the reciprocal hybrids (Z1P, PZ1). (c) Venn diagram showing the limited overlap of the MEGs and PEGs with the parental DEGs and the hybrid DEGs.
Next, the tissue-specific expression of the identified imprinted genes were investigated by referring to a published seed/kernel transcriptome dataset from the B73 inbred line[37]. Expression data was successfully obtained for 215 out of 226 MEGs, and 110 out of 112 PEGs. Cluster analysis of the 215 data-extracted MEGs showed that 95 (44.2%) were embryo-specific, 73 (34.0%) were co-expressed in both embryo and endosperm, and only 47 (21.8%) were endosperm-specific (Supplementary Fig. S7). Of the 110 PEGs, 52 (47.3%) were embryo-specific, 43 (39.1%) were co-expressed in both embryo and endosperm, and only 15 (13.6%) were endosperm-specific (Supplementary Fig. S8). These results indicate that a greater number of identified MEGs and PEGs are preferentially expressed in the embryo compared to the endosperm.
Finally, functional enrichment analysis was conducted using GO analysis to find that the 226 MEGs were significantly associated with molecular functions including carbon–carbon lyase activity (GO:0016830), response to oxygen-containing compound (GO:1901700), stomatal movement (GO:0010118), response to chemical (GO:0042221), oxidoreductase activity (GO:0016491), and alkyl/aryl transferase activity (GO:0016765) (Fig. 5a). The 112 PEGs were significantly enriched in microbody (GO:0042579), peroxisome (GO:0005777), obsolete vacuolar part (GO:0044437), vacuolar membrane (GO:0005774), and vacuole (GO:0005773) (Fig. 5b). Then, KEGG analyses was conducted to find that MEGs were primarily enriched in glyoxylate and dicarboxylate metabolism, glycolysis/gluconeogenesis, metabolism of other amino acids, and pyruvate metabolism (Fig. 5c). In contrast, PEGs were enriched in pathways related to chaperones and folding catalysis, and protein processing in the endoplasmic reticulum (Fig. 5d). These results suggested that MEGs are predominantly involved in carbon metabolism, whereas PEGs are more associated with cellular organelle and protein folding functions.
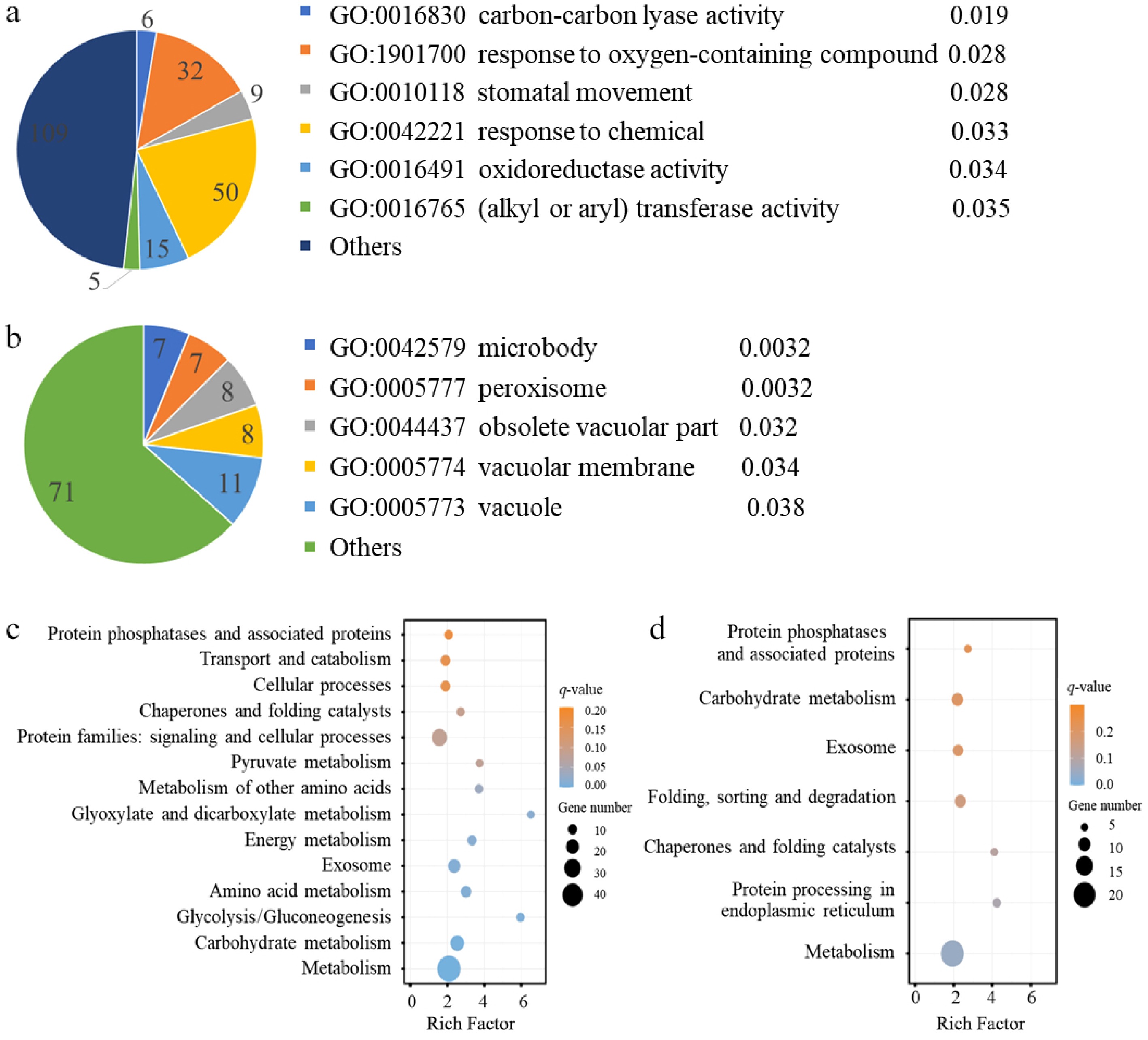
Figure 5.
GO and KEGG analyses of the identified maternally (MEGs) and paternally expressed genes (PEGs). The most significantly enriched GO terms for the (a) MEGs, and the (b) PEGs. For each GO term, the number of genes is displayed in the chart, and the q-value (calculated using the hypergeometric distribution test followed by the Benjamini-Hochberg method) is listed on the right side of the term. KEGG enrichment analysis for the (c) maternally expressed genes and (d) paternally expressed genes.
Verification of imprinted gene expressions
-
To identify the most reliable candidate imprinting genes potentially involved in seed dehydration, a stringent threshold was applied to the observed parental allele expression ratio: MAP > 0.82 (0.52 + 0.30) or < 0.22 (0.52–0.30) in both reciprocal hybrids. This approach identified 45 MES from the 727 MES, and 19 PES from the 320 PES (Supplementary Tables S5, S6). After BLAST at
www.maizegdb.org using the 401 bp sequence surrounding these SNPs, we obtained 25 MEGs and 12 PEGs (Supplementary Table S7). Among these, two genes were targeted by four SNPs, six genes by three SNPs, and eight genes by two SNPs; the remaining 21 were targeted by a single SNP.Using the RNA-seq data, sequence alignment was performed on these 37 extremely maternally/paternally expressed genes to identify InDel (Insertion/deletion) molecular markers between the Z1 and P based on the 401 surrounding sequences of the MES/PES. InDels with a length of at least 3 bp for the gene Zm00001d040697 (Fig. 6a), and Zm00001d052744 (Fig. 6b) were identified; however, no suitable InDel was found for the remaining 35 genes. Based on these variations, allele-specific primers were designed for Zm00001d040697 and Zm00001d052744, and their allelic expressions investigated in reciprocal hybrids via qPCR. Initial tests on parent line Z1 and P demonstrated that the primers amplified the target allele significantly in one parent while showing negligible amplification in the other, confirming the high specificity of the primers (Fig. 6c–f). Then, in 55 DAP seeds, the expression levels of the two alleles were amplified separately for the Z1P and PZ1 hybrids. For Zm00001d040697, the expression of the Z1 allele in Z1P was 2.04 times higher than in PZ1, while the P allele expression in PZ1 was 4.55 times higher than in Z1P (Fig. 6c, d). For Zm00001d052744, the Z1 allele expression in Z1P was 2.76 times higher than in PZ1, while the P allele expression in PZ1 was 5.26 times higher than in Z1P (Fig. 6e, f). These results confirmed the maternally expressed characteristics of both Zm00001d040697 and Zm00001d052744 genes.
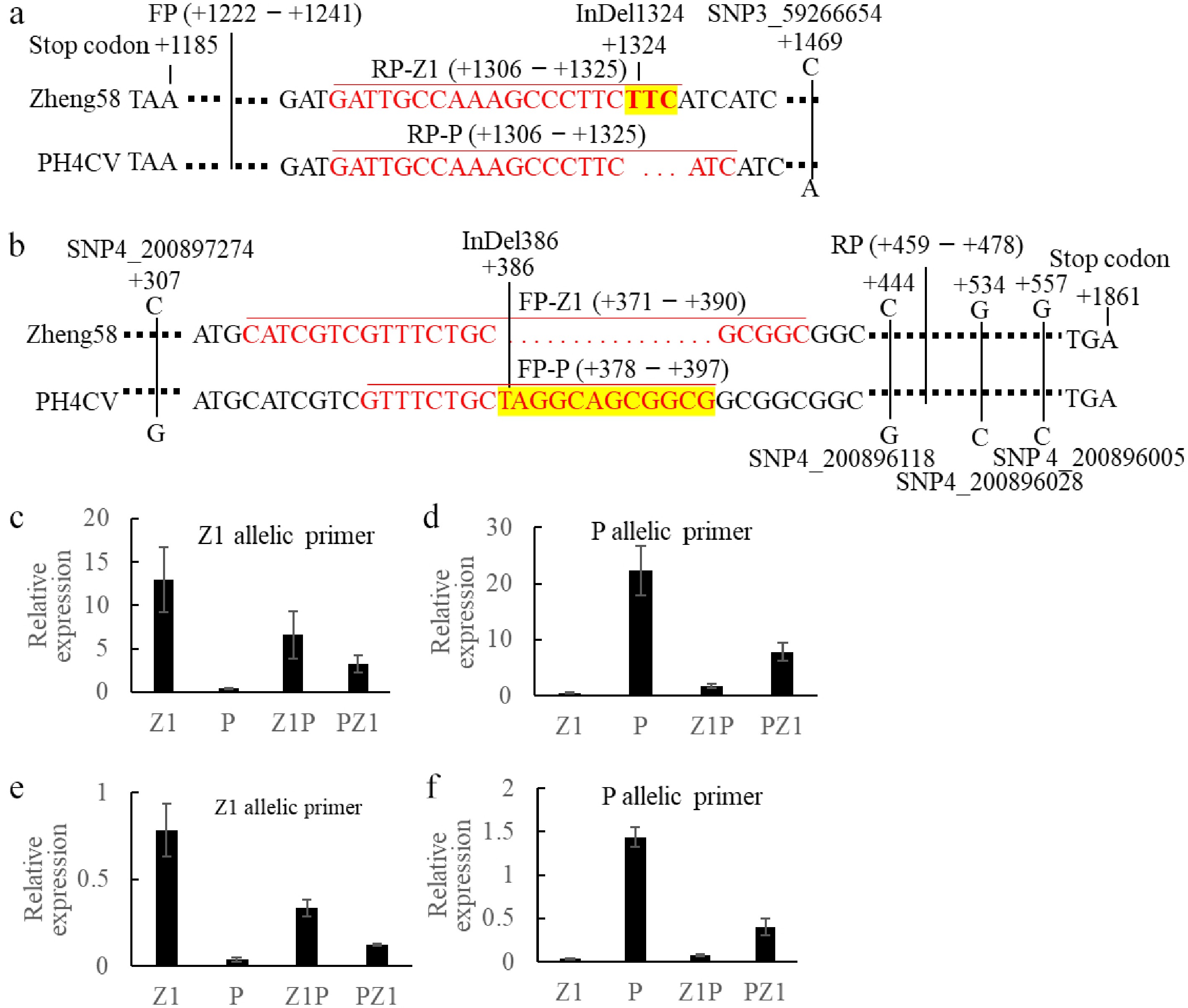
Figure 6.
Verification of imprinted expression for candidate genes in seeds of Z1P and PZ1 reciprocal crosses. (a) Haplotype-specific primer design strategy for Zm00001d040697 gene; (b) Haplotype-specific primer design strategy for Zm00001d052744 gene. Relative expression levels of Zm00001d040697 in the four genotypes (Z1, P, Z1P, and PZ1) using (c) Z1-specific and (d) P-specific primers. Relative expression levels of Zm00001d052744 in the four genotypes (Z1, P, Z1P, and PZ1) using (e) Z1-specific, and (f) P-specific primers.
-
In recent decades, considerable advances have been made in understanding the regulatory mechanisms of kernel dehydration in maize. On the one hand, several genes controlling this trait have been discovered through quantitative trait locus (QTL) mapping and genome-wide association study (GWAS). For example, recent work identified a kernel dehydration rate (KDR) QTL (qKDR1) that functions as a non-coding sequence. It regulates the expression of microRPG1 (a micropeptide encoding gene), which precisely modulates the expression of ZmETHYLENE-INSENSITIVE3-like 1 and 3 within the ethylene signaling pathway[34]. Furthermore, a GWAS identified 71 kernel MC QTLs in maize, and highlighted GRMZM5G805627 and GRMZM2G137211 as candidate genes underlying major QTLs by integrating genetic population analysis, transcriptomic profiling, and gene editing[32]. Using time-series kernel MC data from seven DAP to 70 DAP, another GWAS revealed 377 associated loci, including a gene encoding heat shock 70 kDa protein 5 (HSPa5; Zm00001d047799), which was functionally verified by mutant experiments[38]. On the other hand, many genes have been suggested to associate with maize kernel dehydration through transcriptomes, proteomics, and metabolomics. These studies elucidated that the expressions of genes encoding late embryogenesis abundant (LEA) proteins, heat shock proteins (HSP), serpins, stress/defense responsive proteins, and nutrition accumulation proteins were critical regulatory factors affecting the kernel dehydration process[33,39−42]. In this work, transcriptome analysis was performed on the embryos of two inbred lines with contrasting dehydration rates. The present results revealed that dehydration-related DEGs were mainly enriched in nutrient reservoir activity, involving pathways such as stibenoid, diarylheptanoid, and gingerol biosynthesis, alpha-linolenic acid metabolism, and flavonoid biosynthesis (Fig. 2). Furthermore, the transcriptomes of embryos of two reciprocal hybrids with contrasting kernel dehydration rate were analyzed. It was found that most DEGs between the reciprocal hybrids were epigenetically inherited from the different expression patterns of their parents, and were similarly enriched in nutrient reservoir activity (Fig. 2). These results highlight the important function of nutrient reservoir activity in kernel dehydration, and uncover the genetic mechanisms governing dehydration in hybrid seeds.
Since the discovery of the first imprinted gene in 1970, methods for identifying such genes have been applied intensively to traditional traits, such as endosperm development[27−30,42−44]. However, previous studies focused primarily on the identification of single imprinted genes. With the advancement of high-throughput sequencing, genome-level identification has become possible, greatly increasing efficiency. To date, several studies have identified hundreds of imprinted genes in crops like maize, wheat, and castor[45−50]. However, research specifically identifying imprinted genes related to seed dehydration remains scarce. In this study, a genome-wide identification of imprinted genes for seed dehydration was conducted, identifying 226 MEGs and 112 PEGs (Fig. 3). Two of these genes were verified for their imprinted expressions in reciprocal hybrids using qPCR (Fig. 5). These may serve as vital targets for genetic improvement to reduce MC in hybrid maize seed at harvest.
Comparing the present findings with a previous dataset that identified 282, 400, and 307 imprinted genes from 11 DAP maize kernels of BC (B73 × CAU5), MC (Mo17 × CAU5), and BM (B73 × Mo17) reciprocal hybrids, respectively[50], a small number of overlapping imprinted genes were observed. Specifically, only two, four, and three MEGs, and one, three, and one PEGs from the present study overlapped with the BC, MC, and BM datasets, respectively. It was speculated that differences in developmental stages and genetic backgrounds likely account for this limited overlap.
Regarding epigenetic mechanisms, previous works suggested that imprinted gene expression is primarily controlled by DNA methylation, histone modification, or a combined action of both near the target genes[51−55]. Specifically, cytosine methylation and demethylation processes play essential roles in the expression of parental alleles[56,57]. In the future, DNA methylation analysis surrounding the imprinted genes identified in this study will be instrumental in uncovering the complex mechanisms of allele-specific expression in hybrid seeds.
-
In conclusion, most DEGs between the two reciprocal hybrids are inherited from differential parental expressions, with the hybrid expression pattern more closely resembling the maternal parent than the paternal parent. Given that the genotypes of reciprocal hybrids are identical, these results indicate that expression differences are likely epigenetically inherited through maternal or paternal effects. 338 imprinted genes related to seed dehydration (226 MEGs and 112 PEGs) were identified, and two were verified via qPCR. These findings provide both theoretical insights into the molecular mechanism of hybrid seed dehydration, and practical candidates for reducing maize seed MC at harvest.
This work was supported by the Project of Fund for Stable Support to Agricultural Sci-Tech Renovation of Xinjiang (xjnkywdzc-2023001-22), the Tianchi Talents Recruitment Program, the Regional Science Fund Project of the National Natural Science Foundation of China (NSFC: 32360515 & 32372161), and the earmarked fund for China Agriculture Research System-Maize (CARS-02).
-
The authors confirm their contributions to the paper as follows: funding support, study conception and design: Yang J, Gu R; material treatment, data analysis, and manuscript writing: Han D, Li Z; material preparing: Chen R, Du X, Abula A, Lv Y, Qin T, Dong Y; data analysis: Zheng N, Li L, Zhang H, Fu J. All authors reviewed the results and approved the final version of the manuscript.
-
The transcriptome sequencing data are available at the China National Center for Bioinformation with BioProject numbers of PRJCA042265. Other datasets generated and/or analyzed during the current study are available from the corresponding authors upon reasonable request.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Dengxu Han, Zhaoyuan Li
- Supplementary Table S1 Summary of transcriptome sequencing.
- Supplementary Table S2 Different expressed genes (DEGs) in 55 DAP seeds of Zhang58 (Z1) and PH4CV (P).
- Supplementary Table S3 Different expressed genes (DEGs) in 55 DAP seeds of the reciprocal hybrids Zhang58 X PH4CV (Z1P) and PZ1.
- Supplementary Table S4 Identified SNPs between parent Z1 and P with at least 10 reads in the 4 hybrid samples of two genotype (Z1P and PZ1) X two replicates (R1 and R2), and their maternal allele propotion (MAP) in these samples.
- Supplementary Table S5 The identifed maternal imprinted SNP (MIS) and their targeting genes (maternal imprinted genes, MIG).
- Supplementary Table S6 The identifed paternal imprinted SNP (PIS) and their targeting genes (paternal imprinted genes, PIG).
- Supplementary Table S7 The most reliable dehydration related imprinted genes with an extremely bias of parental allele expression of MAP > 0.82 (0.52 + 0.30) or < 0.22 (0.52 − 0.30) in both reciprocal hybrids.
- Supplementary Table S8 Primers used in this work.
- Supplementary Fig. S1 Seed moisture content (MC) of maize inbred lines and reciprocal hybrids during seed development.
- Supplementary Fig. S2 Correlations of gene expression levels between biological replicates. Scatter plots illustrate the correlation of FPKM (fragments per kilobase of transcript per million mapped reads) values between two biological replicates for all expressed genes identified by RNA-seq.
- Supplementary Fig. S3 KEGG pathway enrichment analysis of differentially expressed genes. Bubble plots showing the KEGG enrichment results for parent DEGs.
- Supplementary Fig. S4 Functional enrichment analysis of hybrid-specific differentially expressed genes (DEGs).
- Supplementary Fig. S5 Scatter plots showing the correlations of maternal allele proportion (MAP) calculated from parental SNP between two biological replicates in reciprocal hybrid Z1P (a) and PZ1 (b).
- Supplementary Fig. S6 Scatter plots showing the MAP of the imprinted SNP in two replicates of Z1P (a) and PZ1 (b).
- Supplementary Fig. S7 Heatmap showing the expression patterns for the 215 MEGs in developing embryo (Em) and endosperm (En) from 10 to 38 days after pollination (DAP).
- Supplementary Fig. S8 Heatmap showing the expression patterns for the 112 PEGs in developing embryo (Em) and endosperm (En) from 10 to 38 days after pollination (DAP).
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Han D, Li Z, Chen R, Du X, Zheng N, et al. 2026. Transcriptome-wide identification of imprinting genes at the seed dehydration stage in maize. Seed Biology 5: e005 doi: 10.48130/seedbio-0025-0030 

Transcriptome-wide identification of imprinting genes at the seed dehydration stage in maize
- Received: 22 September 2025
- Revised: 18 December 2025
- Accepted: 25 December 2025
- Published online: 04 March 2026
Abstract: Seeds harvested at lower moisture contents (MC) are advantageous, leading to both high-efficiency drying and high-vigor seed production. While maize is the most successful crop in utilizing heterosis, the regulatory mechanism governing seed MC in hybrids remains largely unknown. Here, three pairs of reciprocal hybrids were constructed using four distinct inbred lines: the fast-dehydration line PH4CV (P) and Zheng30 (Z2), and the slow lines Zheng58 (Z1) and Dan360 (D). Field experiments demonstrated that hybrids crossed using the faster lines as maternal parents (PZ1, Z2Z1, and PD) exhibited a faster dehydration rate during seed development compared to their corresponding reciprocal hybrids (Z1P, Z1Z2, and DP). Seeds were then collected from the PZ1 and Z1P reciprocal hybrids at 55 days after pollination (DAP) for transcriptome analysis. The results showed that 68.3% of the Different Expression Genes (DEGs) between reciprocal hybrids overlapped with the DEGs between parental lines. By comparing the Allele Specific Expression (ASE) of the expressed genes within each hybrid, an average maternal allele proportion of 0.524 was determined in these 55 DAP seeds. Furthermore, 226 Maternally Expressed Genes (MEGs), and 112 Paternally Expressed Genes (PEGs) were identified, and the ASE of two candidate imprinting genes, Zm00001d040697 and Zm00001d052744, were subsequently verified in the reciprocal hybrids by qPCR. Expression analyses indicated that the imprinted genes are likely distinct from the genes differentially expressed between the reciprocal hybrids. Together, these results confirm a maternal effect on seed dehydration rate, and provide a strong set of candidate imprinting genes potentially regulating seed MC.
-
Key words:
- Dehydration /
- Imprinting gene /
- Transcriptomic analysis /
- Reciprocal hybrid /
- Seed /
- Maize