









-
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system, recognized as a revolutionary genome editing technology, has demonstrated remarkable potential in research into plant functional genomics and genetic improvement of crops[1,2]. The editing efficiency of this system is significantly reliant on the successful delivery and efficient expression of exogenous genes within host cells[3]. In Agrobacterium-mediated plant genetic transformation, the entire process of T-DNA, from the cleavage of the border sequence to its final integration into the plant genome, is crucial for the success of transgenes. Nevertheless, this process is not without flaws. Specifically, the left border (LB) region of T-DNA, because of insufficient protection by the VirD2 protein, is vulnerable to degradation by the host nucleases once it enters the plant cells. This vulnerability leads to deletion of or damage to the adjacent expression elements, thereby severely impacting the stability of transgenes, especially those of precisely designed gene editing components[3−5].
Currently, optimization studies of plant CRISPR/Cas9 systems predominantly concentrate on enhancing the Cas protein's activity, optimizing the design of guide RNA, and developing novel editors[1,6−9]. Nevertheless, several crucial technical bottlenecks at the delivery and expression levels remain to be fully resolved. First, the absence of tools capable of intuitively and efficiently monitoring T-DNA LB deletions hampers the systematic assessment of this prevalent phenomenon. Second, in component design, there is a dearth of convenient and reliable in vivo comparison tools for the selection of expression elements such as promoters and codon preferences. Finally, for the increasingly integrated multigene editing systems (e.g., the single transcriptional unit [STU] and two component transcriptional unit [TCTU] systems), there is a lack of an effective evaluation strategy regarding the influence of internal structures, such as added artificial elements, on the efficiency of expression. The lack of these tools restricts the rational design of plant gene editing vectors to achieve higher efficiency and better control.
To systematically tackle these issues, this study aimed at developing a series of universal tools and optimization strategies to comprehensively boost the expression and editing efficiency of CRISPR/Cas9-based plant genome editing systems. First, we constructed a visual reporter vector, pHNCas9-EGFP, to directly monitor T-DNA LB deletion events. Second, we developed a dual reporter gene system (GUS2AeGFP) and its vector (pHN2G), which provides a reliable platform for the precise comparison of promoters' activity. Moreover, we designed fusion reporter vectors based on the 2A peptide, pHN2AGUS/pHN2AGFP, for intuitive assessment of the translation efficiency of Cas genes with different codon optimizations. Finally, by utilizing the modified vector platform, we optimized and validated the function of elements such as artificial poly(A) in integrated editing systems.
Through the development and application of this series of tools, we not only offer effective monitoring and evaluation methods for the critical steps of plant genetic transformation and gene editing processes but also present new strategies and insights for the rational and efficient design of plant gene editing vectors. This is expected to further promote plant genetic engineering technology in both fundamental research and breeding applications.
-
The tomato (Solanum lycopersicum) variety used in this study was 'Micro-Tom', which is utilized for stable genetic transformation. Nicotiana benthamiana was used for the Agrobacterium-mediated transient transformation experiments. The Arabidopsis thaliana Columbia ecotype (Col-0) was used for floral dip transformation.
Tomato and tobacco materials were cultivated in a controlled climate chamber with the following conditions: a day/night temperature of 25−28/18−20 °C, a 16-h light/8-h dark photoperiod, and a relative humidity of 60%−70%. Arabidopsis thaliana plants were grown at 20−22/16−18 °C under the same long-day conditions.
Vector construction
-
PHNCas9-EGFP: The EGFP expression cassette driven by a constitutive promoter was cloned into the pHNCas9 vector immediately adjacent to the 3' end of the T-DNA LB using enzyme digestion and ligation methods (Kpn I and Sal I).
pHN2G: First, 2AeGFP and GUS were amplified by PCR. The 5' and 3' ends of 2AeGFP were set with EcoR I-Sac I; the 5' and 3' ends of the GUS gene were set with BamH I and Bsa I, respectively. The genes at both ends were digested with Bsa I and then ligated using T4 DNA ligase. The ligated product was used as a template to amplify the full-length GUS2AeGFP gene by PCR, with the Kpn I–Xho I–BamH I, and EcoR I–Sac I digestion sites set at the 5' and 3' ends, respectively. The Hsp250 terminator and the KpnI–XhoI–BamHI–GUS2AeGFP–EcoRI–SacI structure were constructed into a Peasy cloning vector. The laboratory's previously stored PNOS::NPTII expression cassette was cloned into the pHN vector[10]. Finally, the KpnI–XhoI–BamHI–GUS2AeGFP–EcoRI–SacI–THsp–SalI structure was cloned into the 5' end of the PNos::NPTII expression cassette.
pHN2G1pHN2G7: The promoters to be tested (P35SI[11], P35SII[11], P35SII47, PCmpC[12], PC47, P35SIC47, and PC911[13]) were inserted upstream of the GUS2AeGFP gene in the pHN2G vector with the Xho I and BamH I sites, resulting in the series of vectors from pHN2G1 to pHN2G7.
pHN-2AGUS: Using the PHN2G vector as a template, 2AGUS was amplified by PCR, with BamH I–Bsa I and EcoR I–Sac I introduced at the 5' and 3' ends, respectively. The GUS2AeGFP in pHN2G was replaced using enzyme digestion and ligation methods.
pHN-2AeGFP: Using the PHN2G vector as a template, 2AeGFP was amplified by PCR, with BamH I–Bsa I and EcoR I–Sac I introduced at the 5' and 3' ends, respectively. The GUS2AeGFP in pHN2G was replaced using enzyme digestion and ligation methods.
Three codon-optimized Cas9 genes (SlCas9[3], AtCas9[14], and hCas9[15]) without stop codons were fused with either 2AGUS or 2AeGFP via the BamH I/Bsa I sites, constructing expression vectors for testing translation efficiency.
pHN2G6-PTST50/100/150: The nucleotide sequences for PTST50, PTST100, and PTST150 were synthesized by Beijing Tsingke Biotech Co. Ltd. and subsequently inserted into the EcoR I and Sac I sites of the pHN2G6 vector.
EGFP fluorescence analysis: The expression of the EGFP gene in plant materials was detected via fluorescence microscopy. ImageJ software was used to analyze the intensity of EGFP fluorescence. An EGFP fluorescent flashlight (3280RB) produced by Shanghai LUYOR was used to detect EGFP fluorescence in transgenic plants.
Quantification of GUS enzyme activity: Tobacco leaf tissue from the injection area was weighed and ground in liquid nitrogen to extract total protein. Using 4-methylumbelliferyl-β-D-glucuronide (4-MUG) as a substrate, the fluorescence intensity of the reaction product was measured with a fluorescence spectrophotometer to calculate the GUS enzyme activity. For comparing the sampling methods, Method A involved nine independent biological replicates, and Method B involved nine mixed samples (each mixed sample derived from three discs), with a total of three biological replicates.
Genome extraction and genome DNA resequencing and analysis: Tomato genomic DNA was extracted using the Genstar Genome Extraction Kit (D135). Subsequently, genomic DNA samples from 27 transgenic plants were mixed in equal amounts. Library construction, sequencing, and data analysis were carried out by Biomarker Technologies.
-
A key process of Agrobacterium-mediated plant genetic transformation strategies involves the adsorption of Agrobacterium onto plant cells and the subsequent introduction of T-DNA (the T-strand) into plant cells, where it integrates randomly into the plant's genome. However, because of the lack of VirD2 protection at the LB end of the T-strand, it is more prone to degradation by the host's nucleases[4,5]. Regardless of the type of expression element coupled at the LB end, its function is more easily compromised, which is also true for its use in constructing gene editing vectors. Moreover, the degree of LB end loss seems to vary among different plants[5]. Currently, there is a lack of an effective tool to monitor this phenomenon intuitively. To facilitate monitoring of this phenomenon, we have developed a vector named pHNCas9-EGFP (Fig. 1a) for visual detection of LB end loss. An enhanced green fluorescence protein (EGFP) expression cassette initiated by the C47 promoter[3] is coupled with the 3' end of the LB to determine if there is a necessary link between the damage to the LB end and the function of the expression cassette being compromised.
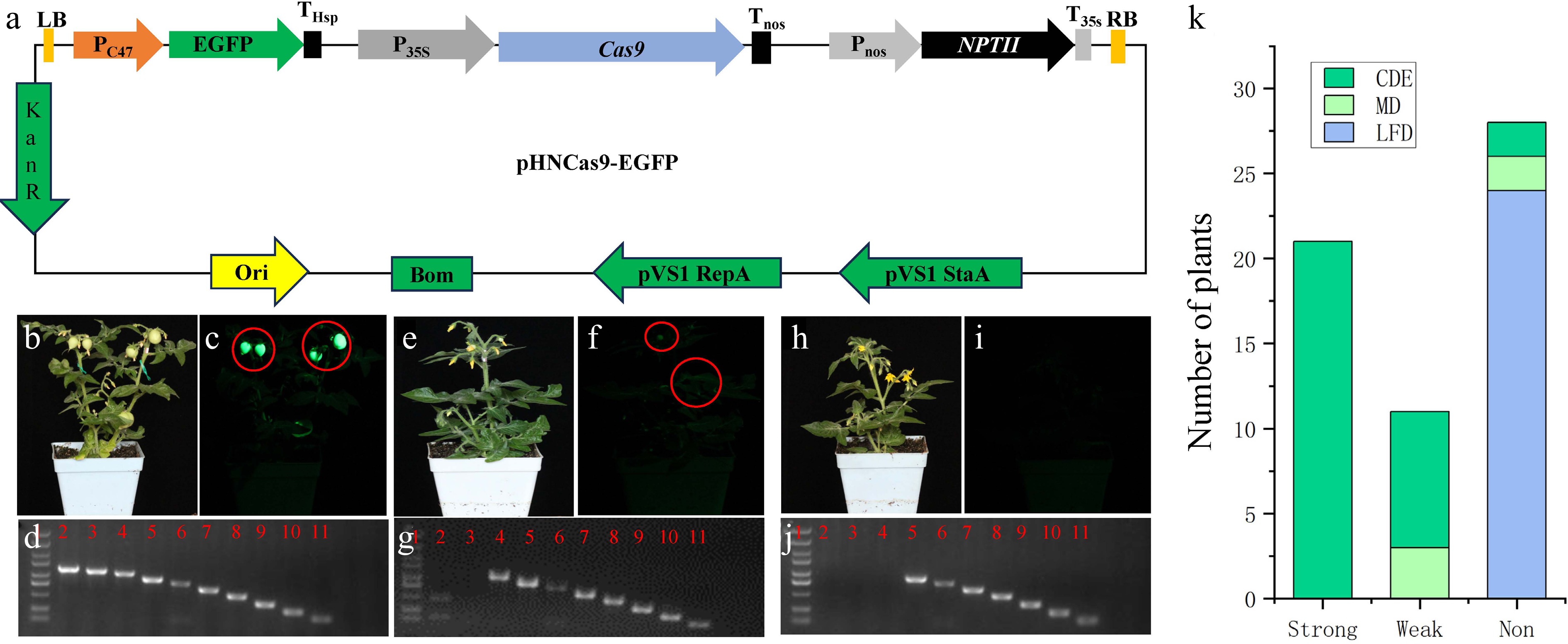
Figure 1.
Detection tools and strategies for T-DNA LB terminal deletion. (a) Schematic diagram of the pHNCas9-EGFP vector's structure. (b) Photograph of a strong fluorescent transgenic tomato plant under white light. (c) Photograph of a strongly fluorescent transgenic tomato plant under excitation with 485-nm blue light. (d) Example of a PCR electrophoresis image of a plant with an intact EGFP expression cassette. Lane 1 is the DNA marker, and Lanes 2 to 11 are the electrophoresis bands of the PCR products from the 10 pairs of primers used to assess the integrity of the EGFP expression cassette. (e) Photograph of a weakly fluorescent transgenic tomato plant under white light. (f) Photograph of a weakly fluorescent transgenic tomato plant under excitation with 485 nm blue light. (g) Example of a PCR electrophoresis image of a plant with a slightly damaged EGFP expression cassette. Lane 1 is the DNA marker, and Lanes 2 to 11 are the electrophoresis bands of the PCR products from the 10 pairs of primers used to assess the integrity of the EGFP expression cassette. (h) Photograph of a nonfluorescent transgenic tomato plant under white light. (i) Photograph of a nonfluorescent transgenic tomato plant under excitation with 485 nm blue light. (j) Example of a PCR electrophoresis image of a plant with a severely damaged EGFP expression cassette. Lane 1 is the DNA marker, and Lanes 2 to 11 are the electrophoresis bands of the PCR products from the 10 pairs of primers used to assess the integrity of the EGFP expression cassette. (k) Statistics of various types of transgenic tomato plants. The photos in (b), (e), and (h) were taken under white light; those in (c), (f), and (i) were taken under EGFP fluorescence. The shutter speed for white light was 1/80 s, and that for fluorescence was 1/10 s. The ISO was set at 3,200, and the aperture value was f/6.3 for both conditions. The EGFP fluorescence photos were taken using an EGFP fluorescent flashlight and a LUV-520 filter (LUYOR). Strong, strong fluorescence; weak, weak fluorescence; non, nonfluorescent; CDE, complete DNA ends; MD, minorly damaged DNA end; LFD, large-fragment DNA damage at the end.
To test whether the pHNCas9-EGFP vector can effectively detect the impact of LB end loss on the function of nearby expression cassette elements, we used this vector for the genetic transformation of tomato (Solanum lycopersicum), yielding 60 transgenic plants. We used a fluorescent flashlight to detect EGFP fluorescence in transgenic tomatoes to determine if the function of the EGFP expression cassette was compromised. Additionally, we designed a reverse primer and 10 forward primers on the EGFP expression cassette to detect the intactness of the T-DNA LB end sequence using a simple polymerase chain reaction (PCR) technique[3]. According to the number of agarose gel electrophoresis bands of the PCR products, we can roughly categorize the results into three types: CDE, which refers to complete T-DNA ends and includes all 10 DNA electrophoresis bands; MD, meaning minor T-DNA LB end deletions, with no more than two missing DNA electrophoresis bands; and LFD, indicating large fragment deletions at the T-DNA LB end, more than two missing DNA electrophoresis bands. The results showed that 28 transgenic lines did not exhibit visible EGFP fluorescence (Fig. 1h, i, k), and the PCR results indicated that at least 24 of them (Fig. 1k) exhibited large fragments of DNA LB end loss (an example of an electrophoresis image of this is shown in Fig. 1j), two had minor damage to the T-DNA left borders (an example of electrophoresis image of this is shown in Fig. 1g), and two plants had intact T-DNA LBs (an example of an electrophoresis image of this is shown in Fig. 1d). Thus, the visibility of EGFP fluorescence had a detection efficiency of 85.71% (24/28) (Fig. 1k) for large-fragment deletions at the LB end. Moreover, 11 plants displayed weak fluorescence (Fig. 1e, f, k), and the PCR results showed that at least 3 had minorly damaged DNA at the LB end (Fig. 1k) in the PC47 promoter[3] region (an example of an electrophoresis image of this is shown in Fig. 1g); furthermore, all the EGFP expression cassettes in the 21 strong EGFP fluorescent lines (Fig. 1b, c, k) were complete (an example of an electrophoresis image of this is shown in Fig. 1d). We equally mixed the genomic DNA of these 27 transgenic plants, which experienced T-DNA loss at the LB end, for Illumina sequencing. In total, 25 types of T-DNA LB end loss events were identified, starting from the 3' end of the LB, with the smallest loss length being 175 bp and the largest 2,285 bp[3]. This further confirmed that pHNCas9-EGFP can serve as a visual tool for detecting T-DNA LB end loss.
Screening strategies and tools for efficient expression promoters
-
Promoters are the most important elements used to regulate the intensity of expression in plant genetic engineering. To better identify the strength of expression of different promoters, we first developed a dual reporter gene named GUS2AeGFP (Fig. 2a). The GUS2AeGFP sequence is provided in Supplementary Fig. S1. This gene comprises β-glucuronidase (GUS) gene, a 2A self-cleaving peptide gene, and an EGFP gene, and is expected to simultaneously exhibit GUS enzyme activity and green fluorescence upon expression. Subsequently, the Nos::NPTII expression cassette[10], the Hsp terminator[16], and GUS2AeGFP were sequentially connected on the pHN vector backbone[10], with Xho I and BamH I restriction sites set at the 5' end of the GUS2AeGFP gene, ultimately obtaining an expression vector for detecting promoters' activity named pHN2G (Fig. 2a).
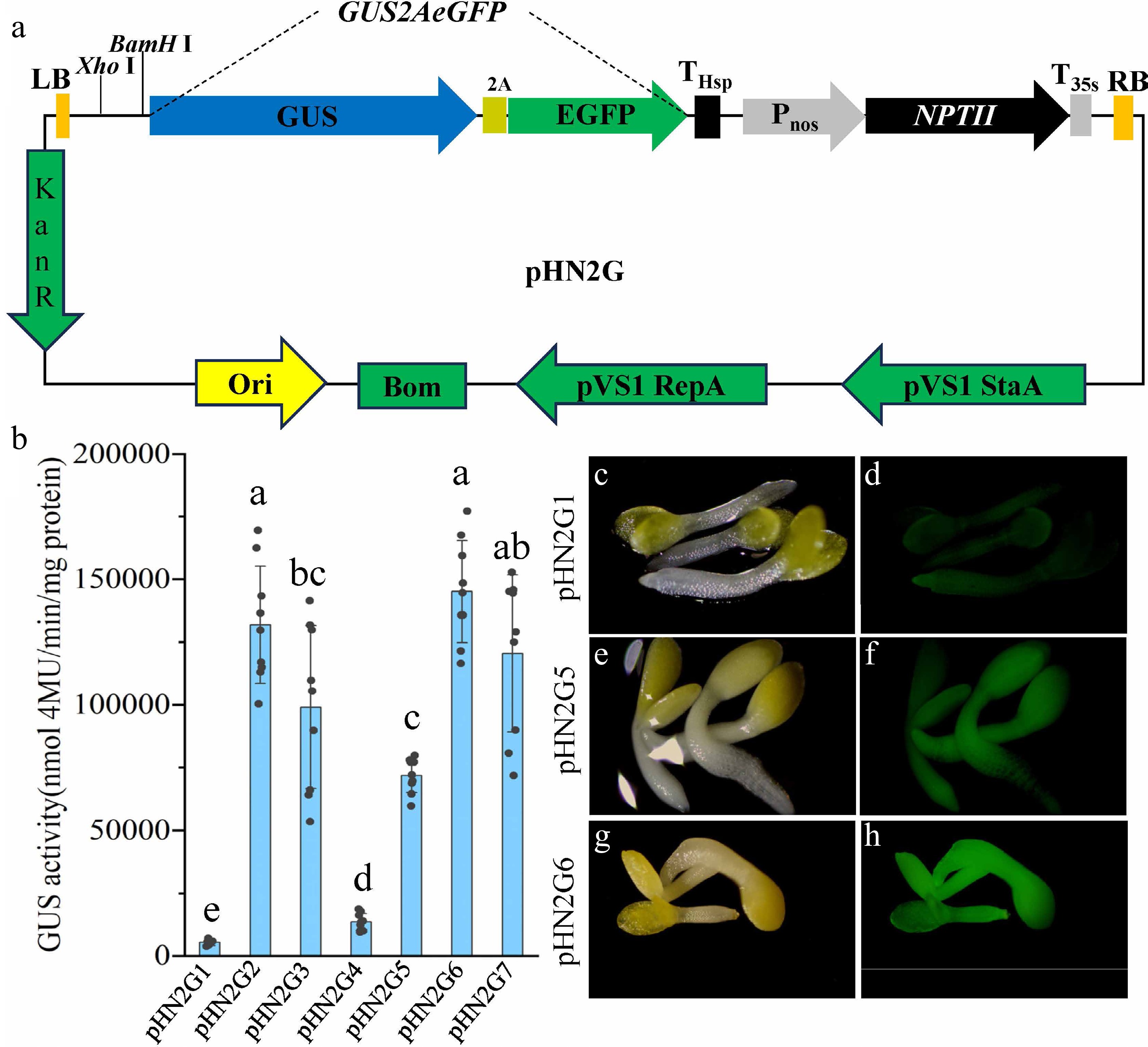
Figure 2.
Tools and strategies for efficient promoter screening. (a) Schematic diagram of the pHN2G vector's structure. (b) Quantitative analysis of GUS enzyme activity resulting from transient expression in tobacco (Nicotiana benthamiana) leaves using pHN2G1 to pHN2G7. Data are the mean ± standard deviation (SD). Different lowercase letters indicate significant differences between groups. (c) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G1 under white light. (d) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G1 under excitation with 485 nm blue light. (e) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G5 under white light. (f) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G5 under excitation with 485 nm blue light. (g) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G6 under white light. (h) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G6 under excitation with 485 nm blue light. The photos in (c), (e), and (g) were taken under white light (exposure 30 ms); those in (d), (f), and (h) were taken under EGFP fluorescence (exposure 80 ms) using a fluorescence microscope, respectively.
To verify whether this gene can achieve the detection of two signals and the impact of different sampling methods on the detection results, we first constructed a P35SI promoter (derived from the Pcambia2301 vector, it contains only one 35S enhancer) between Xho I and BamH I, named pHN2G1, and transformed it into Agrobacterium. This vector was further transiently expressed in tobacco (Nicotiana benthamiana) leaves. Using a fluorescence microscope, we observed bright EGFP fluorescence in the leaf cells, indicating the successful expression of EGFP. Further, we set up two different sampling modes, Method A and Method B. In the experimental plan of Method A, three biological replicates were conducted, with each biological replicate involving the injection of one healthy leaf. Three samples with a diameter of approximately 0.8 cm were taken around the injection hole from each tobacco leaf, and each sample was individually ground to extract protein, resulting in a single data point for GUS enzyme activity. In the experimental plan of Method B, three biological replicates were also conducted, with each biological replicate involving the injection of three healthy and uniformly growing leaves. From each tobacco leaf, three samples with a diameter of approximately 0.8 cm were taken as one mixed sample. The mixed sample was ground to extract protein, resulting in one outcome of GUS enzyme activity. The analysis showed that the mean enzyme activity obtained using both methods was comparable, but the standard deviation of the data obtained using Method A was twice that of Method B (Supplementary Fig. S2), suggesting that the data obtained using Method B may be more stable and reliable.
To test the vector's ability to screen promoters with different expression strengths, six other promoters were cloned into pHN2G. These promoters are P35SII (derived from Pcambia2301, containing two tandem E35S and one m35S)[11], P35SII47 (couples Cor47[17] to the 3' end of P35sII with the 5' untranslated region [UTR] removed)[3], PCmpC (original CmpC promoter)[12], PC47 (couples Cor47[17] to the 3' end of PCmpC)[3], P35SIC47 (couples one E35S, the PCmpC[12], and Cor47[17]), and PC911 (contains the original CmYLCV9.11 promoter)[13]. The constructs were named pHN2G2−pHN2G7. Agrobacterium containing these vectors was used for transient transformation in tobacco leaves, and a quantitative analysis of GUS enzyme activity was performed using the Method B. The results indicated that the expression activity of the PCmpC promoter was higher than that of the P35SI promoter[11], consistent with previous research results[12] (Fig. 2b). The differences in GUS enzyme activity among different promoters were significant, with the GUS enzyme activity driven by the P35SIC47 promoter being 27 times that of the P35SI promoter, whereas the activity driven by the PC47 promoter was 13 times that of the P35SI promoter (Fig. 2b). To test whether these differences were real, the pHN2G1, pHN2G5, and pHN2G6 vectors were transformed into Arabidopsis thaliana using the floral dipping method. The EGFP fluorescence of the T2 generation Arabidopsis seedlings was observed using a fluorescence microscope. The results showed that Arabidopsis transformants of pHN2G1, pHN2G5, and pHN2G6 exhibited significant fluorescence gradients (Fig. 2c−h). These results indicate that the pHN2G vector can serve as a powerful tool for optimizing or screening promoters.
Strategies and tools for evaluating genome editing protein expression capabilities
-
Genome editing systems rely on the activity of Cas enzymes, which, in turn, depend on translation of the Cas enzyme's mRNA into more Cas proteins. However, Cas genes are often large, making codon optimization particularly necessary[14]. Currently, there is a lack of tools that can intuitively compare Cas genes with different codon optimizations. Therefore, we synthesized two reporter genes, 2AGUS (Fig. 3a) and 2AeGFP (Fig. 3b). The sequences of 2AGUS and 2AeGFP are provided in Supplementary Figs S3 and S4, respectively. 2AGUS is composed of a 2A self-cleaving peptide gene and the GUS gene, whereas 2AeGFP is composed of a 2A self-cleaving peptide gene and an EGFP gene. Using these, we constructed two vectors, named pHN2AGUS (Fig. 3a) and pHN2AGFP (Fig. 3b), respectively. The pHN2AGUS vector was obtained by coupling PNos::NPTII and P35SIC47::2AGUS on the pHN vector[10]. The pHN2AGFP vector was obtained by coupling PNos::NPTII and P35SIC47::2AeGFP on the pHN vector[10]. The BamH I and Bsa I sites were reserved before 2AGUS and 2AeGFP for gene cloning.
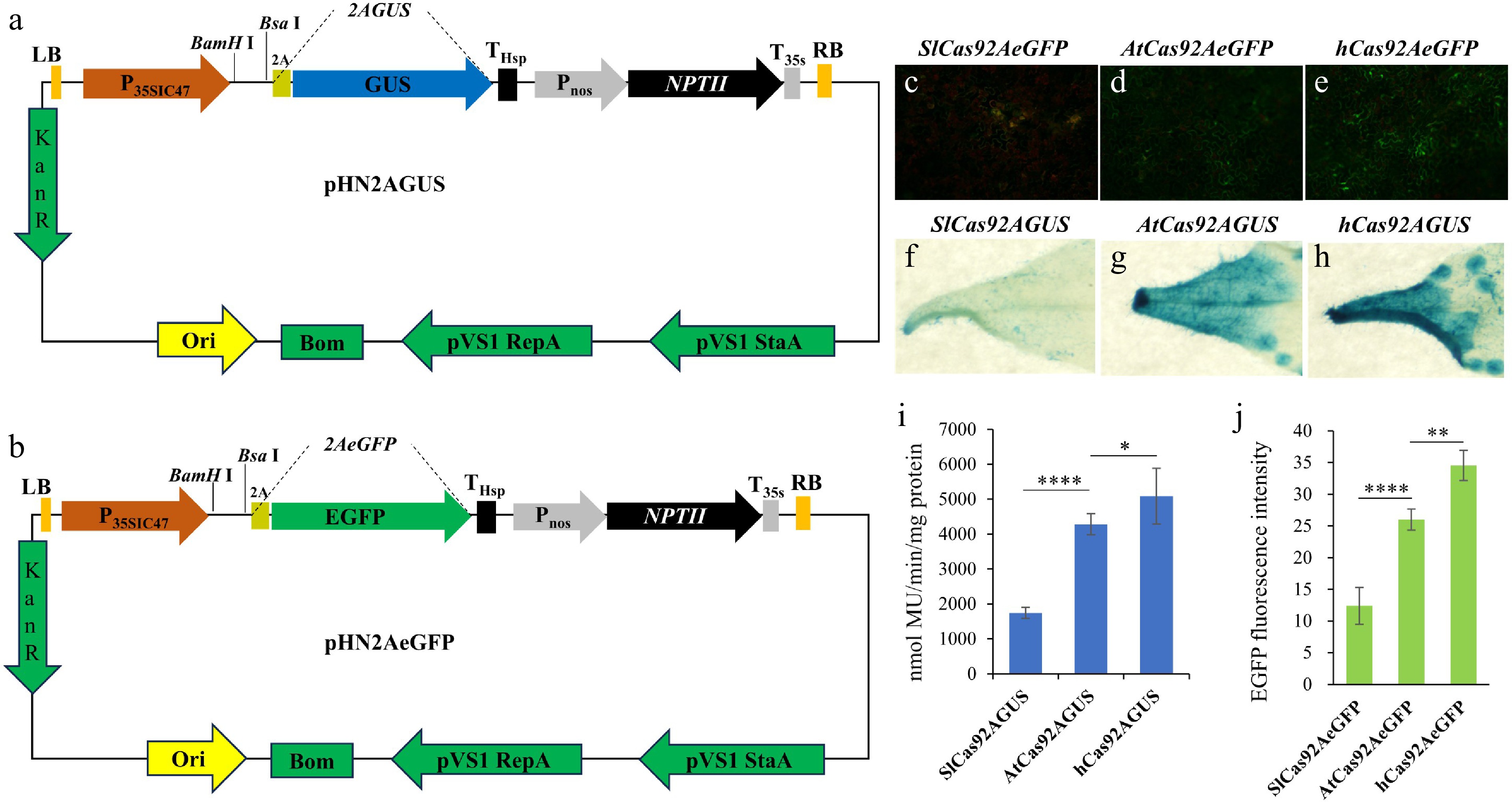
Figure 3.
Tools and strategies for detecting the capacity for protein expression. (a) Schematic diagram of the pHN2AGUS vector structure. (b) Schematic diagram of the pHN2AeGFP vector's structure. (c) Photograph of tobacco leaves transiently expressing the pHN-SlCas92AeGFP vector under excitation with 485 nm blue light. (d) Photograph of tobacco leaves transiently expressing the pHNAtCas92AeGFP vector under excitation with 485 nm blue light. (e) Photograph of tobacco leaves transiently expressing the pHN-hCas92AeGFP vector under excitation with 485 nm blue light. (f) GUS staining image of tobacco leaves transiently expressing the pHN-SlCas92AGUS vector. (g) GUS staining image of tobacco leaves transiently expressing the pHN-AtCas92AGUS vector. (h) GUS staining image of tobacco leaves transiently expressing the pHN-hCas92AGUS vector. (i) Quantitative analysis of GUS enzyme activity in tobacco leaves transiently expressing the pHN-SlCas92AGUS, pHN-AtCas92AGUS, and pHN-hCas92AGUS vectors. (j) Quantitative analysis of EGFP fluorescence intensity in tobacco leaves transiently expressing the pHN-SlCas92eGFP, pHN-AtCas92AeGFP, and pHN-hCas92eGFP vectors. Photos in (c), (d), and (e) were taken under EGFP fluorescence with a fluorescence microscope. The exposure time for SlCas92AEGFP was 1/3s. The exposure time for AtCas92AEGFP and hCas92AEGFP was 1/5s. Note: Data are the mean ± SD. *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
To verify whether these two vectors can effectively differentiate the translation efficiency of genes optimized with different codons, we tested three codon-optimized Cas9 genes: SlCas9 (codon optimized for tomato)[3], AtCas9 (codon optimized for Arabidopsis)[14], and hCas9 (codon optimized for humans)[15]. The stop codons were removed from these three genes, and they were inserted at the 5' end of 2AGUS and 2AeGFP, resulting in six expression vectors. These six vectors were transiently expressed in tobacco leaves, and the expression efficiency of the different genes was assessed by fluorescence microscopy (Fig. 3c, d), GUS staining (Fig. 3f, h), and quantitative analysis of GUS enzyme activity (Fig. 3i). The EGFP fluorescence results showed that the fluorescence intensity of hCas92AeGFP was higher than that of AtCas92AeGFP and SlCas92AeGFP (Fig. 3c, e, j); GUS staining and quantitative analysis of GUS enzyme activity displayed a consistent trend (Fig. 3i). These results indicate that pHN2AGUS and pHN2AGFP can be used as tools for detecting the efficiency of protein translation.
Structural optimization strategies for genome editing systems
-
The structure of plant gene editing vectors significantly impacts the expression efficiency of Cas genes and guide RNAs. Currently used plant genome editing system structures include the multi-component transcriptional unit (MCTU) system[6, 10], the TCTU system[18], and the STU system[19], all of which can be used for multigene or multisite editing. The MCTU system was developed first, where Cas9 and each sgRNA are driven by different promoters. The subsequently developed TCTU system drives Cas9 expression by one promoter, whereas the sgRNA array is driven by another promoter and releases different sgRNAs through RNA processing elements (RPEs) such as transfer RNA (tRNA). In the STU system, Cas9 and multiple sgRNAs are integrated into a single expression cassette with multiple RPEs, and adding a poly(A) structure between the Cas9 and sgRNA array can further enhance the editing efficiency of the STU system[19]. This shows that the Cas9/CRISPR system structure is moving towards integration and needs to incorporate some necessary expression elements to improve the translation efficiency of genes at the 5' end. However, there is currently no tool to evaluate the effect of additional elements on improving translation efficiency. We found that the previously constructed pHN2G6 vector has EcoR I and Sac I sites at the 3' end of GUS2AeGFP (Fig. 4a), allowing some expression elements to be cloned. Therefore, pHN2G6 has the potential to become this tool.
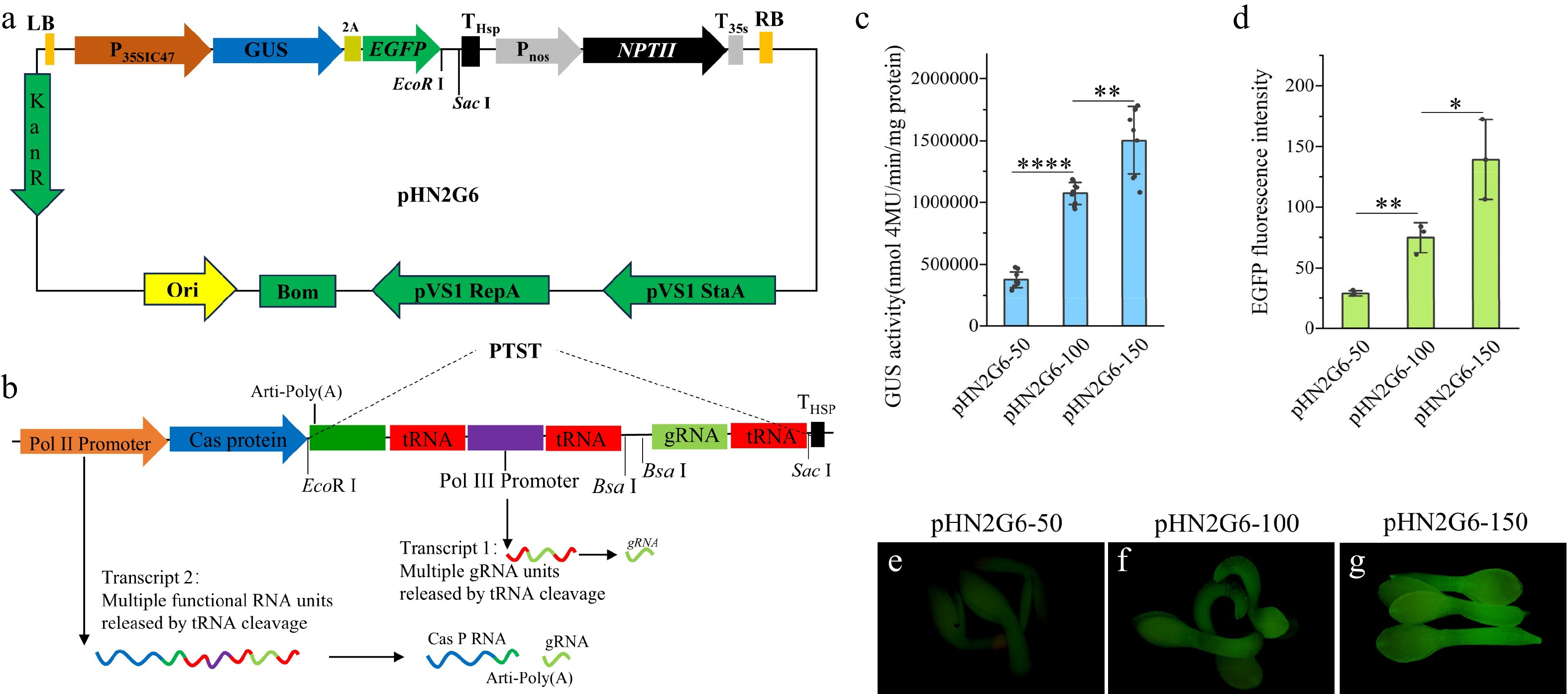
Figure 4.
Tools and strategies for optimizing the structure of a CRISPR/Cas9-based gene editing system (a) Schematic diagram of the pHN2G6 vector's structure. (b) Schematic diagram of the PTST sequence's structure. (c) Quantitative analysis of GUS enzyme activity in tobacco leaves transiently expressing the pHN2G6-50, pHN2G6-100, and pHN2G6-150 vectors. (d) Analysis of the optical density of EGFP fluorescence in T2 seedlings of Arabidopsis transformed with the pHN2G6-50, pHN2G6-100, and pHN2G6-150 vectors. (e) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G6-50 under excitation with 485 nm blue light. (f) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G6-100 under excitation with 485 nm blue light. (g) Photograph of T2 seedlings of Arabidopsis transformed with pHN2G6-150 under excitation with 485 nm blue light. The photos in (e), (f), and (g) were taken under EGFP fluorescence with a fluorescence microscope. The exposure time was 100 ms. Note: Data are the mean ± SD. * p < 0.05; ** p < 0.01; **** p < 0.0001.
To verify this hypothesis, we designed a composite expression element that can be co-expressed with Cas9, named the PTST sequence (Fig. 4b), which consists of artificial poly(A) (arti-poly[A]), tRNA, the Pol III type promoter, sgRNA, and two BsaI sites coupled in the order shown in Fig. 4b. The length of arti-poly(A) may affect the translation efficiency of the 5' end genes. Therefore, we synthesized PTST structures containing arti-poly(A) that were 50, 100, and ~150 bp in length[3]. These three PTST structures were cloned into the 3' end of GUS2AeGFP in the pHN2G6 vector using EcoR I and Sac I restriction sites, named pHN2G6-50, pHN2G6-100, and pHN2G6-150, respectively, and transformed into Agrobacterium, followed by transient expression in tobacco leaves. Quantitative detection of GUS activity revealed that the GUS enzyme activity of pHN2G6-100 and pHN2G6-150 reached 2.37 times and 3.74 times that of pHN2G6-50, respectively (Fig. 4c). The vectors pHN2G6-50, pHN2G6-100, and pHN2G6-150 were transformed into Arabidopsis using the floral dip method. In the T2 generation, the fluorescence of transgenic Arabidopsis seedlings was observed and EGFP fluorescence was quantitatively analyzed using ImageJ. The results showed that the fluorescence value of pHN2G6-150 was as high as five times that of pHN2G6-50 (Fig. 4d−g). This indicates that increasing the length of poly(A) significantly enhances the expression of the 5' end genes in this element, and it also demonstrates that pHN2G6 can conveniently and effectively detect the impact of different elements on the expression of 5' end genes.
Ultimately, on the basis of the optimization results presented above, we developed a new gene editing vector (based on CRISPR/Cas9) named pHNRhCas9NG[3]. This vector is kanamycin-resistant in plants and includes an EGFP expression cassette for visual screening. This vector was used to conduct single-target, multitarget, single-gene, and multigene editing tests on 10 genes in tomatoes. In total, 67 transgenic lines were obtained, and sequencing analysis of the PCR products at the target sites in all 67 lines showed mutations, with a mutation rate of 100%; among them, 62 lines showed significant green fluorescence using an EGFP fluorescent flashlight, with a visualization rate as high as 92.5%[3].
-
In this study, we developed a series of toolkits with broad application prospects and proposed the corresponding optimization strategies to address several key bottlenecks in the optimization of plant CRISPR/Cas9 systems mediated by Agrobacterium. Our work systematically explored the entire chain from the stability of T-DNA delivery and the strength of expression elements to the final output efficiency of the editing protein, providing new insights and practical solutions for the rational design of efficient plant gene editing vectors.
First, the pHNCas9-EGFP vector we developed converted the 'invisible' sequence deletion event at the LB end of the T-DNA into a visible EGFP fluorescent signal. Our results provided direct evidence supporting the long-standing hypothesis that the LB end is more prone to damage[4] and systematically revealed the prevalence and diversity of this deletion event in tomatoes. Up to 85.71% of the fluorescence loss was associated with large-segment deletions at the LB end, indicating that the loss of function of LB-adjacent elements (such as specific sgRNA or regulatory elements) is an important factor that must be considered when evaluating gene editing results. It should be noted that in our results, we also found a few plants with complete or slightly damaged EGFP expression cassettes, yet no fluorescence could be detected. This may be because the T-DNA was inserted into the nonactive regions of chromatin. Our tool provides a convenient means to monitor the integrity of T-DNA in different plant species and transformation methods, and warns vector designers to avoid placing core functional elements near the LB.
Second, the pHN2G dual-reporter system we developed provides a reliable and versatile platform for the functional identification of promoters and other expression elements through the combination of a quantitative enzyme activity analysis of GUS and fluorescent visualization of EGFP. Among them, Method B significantly reduces data variation, establishing a more reliable experimental standard for transient expression analysis in plants. Using this system, we not only verified the activity of the well-known strong promoter PCmpC but, more importantly, identified the super-strong promoter P35SIC47, whose activity reached 27 times that of the widely used P35SI promoter. This discovery provides a powerful tool for gene editing applications requiring high-level expression of Cas proteins. The fluorescence gradient observed in the stable transformation of Arabidopsis further proves the system's effectiveness in stable genetic material, making it a bridge connecting transient screening and stable expression validation. Additionally, this system can also be used to verify the activity of hormone-responsive promoters, which has significant implications for the study of hormone signal transduction pathways.
At the protein translation level, our 2A peptide fusion reporter system (pHN2AGUS/pHN2AGFP) successfully visualized and quantified the differences in translation efficiency of Cas9 genes with different codon optimization versions. hCas9 exhibited higher protein yield than SlCas9 and AtCas9, clearly indicating that factors beyond a simple codon adaptation index (CAI), such as mRNA's secondary structure (affecting mRNA's stability, ribosome recognition and binding) and potential inhibitory sequences (nuclease-sensitive sites can cause mRNA to degrade rapidly), collectively determine the final translation efficiency of genes in plants. Our tool provides an indispensable in vivo evaluation platform for more precise codon optimization and gene design in the future.
Finally, by utilizing the modified pHN2G6 vector, we successfully analyzed the impact of the internal structure of integrated editing systems (such as STU systems) on the efficiency of expression. Our experiments are the first to directly demonstrate in plants that increasing the length of arti-poly(A) between Cas9 and the sgRNA array can significantly enhance the translation efficiency of the upstream Cas9 gene. This finding not only provides clear guidance for optimizing existing multigene editing systems—by introducing a longer poly(A) tail as an effective optimization strategy—but, more importantly, it also showcases the tremendous potential of our tool platform in evaluating and optimizing complex genetic circuits. In the future, other potential enhancement elements, such as introns and translation enhancers, can be rapidly and quantitatively screened for function using this platform.
-
The authors confirm their contributions to the paper as follows: research plan and design: Hu N; experiments: Hu N, Dai D; data analysis: Hu N, Dai D; manuscript writing: Dai D, Chen K, Liu H; manuscript revising: Hu N, Dai D, Li J. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
This work was supported by the National Natural Science Foundation of China (No. 32560731, No. 32002029), China Postdoctoral Science Foundation (No. 2021M690909), the Tarim University President's Fund − Poplar Outstanding Youth Science and Technology Talent Support Project (TDZKJC202502), Tarim University President's Fund − Poplar Talent − Scientific Research Start-up Fund for Introducing Talent (Doctoral) Project (TDZKBS202510, TDZKBS202513), and Poplar Talent − Scientific Research Start-up Fund for Introducing Talent (Master's) Project (TDZKSS202504).
-
The authors declare that they have no conflict of interest.
- Supplementary Fig. S1 Sequence of GUS2AeGFP.
- Supplementary Fig. S2 Differences in GUS enzyme activity data measured by method A and method B.
- Supplementary Fig. S3 Sequence of 2AGUS.
- Supplementary Fig. S4 Sequence of 2AeGFP.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Chongqing University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Dai D, Chen K, Liu H, Li J, Hu N. 2026. Strategies and tools for optimizing plant genome editing systems. Plant Hormones 2: e005 doi: 10.48130/ph-0026-0005 

Strategies and tools for optimizing plant genome editing systems
- Received: 03 December 2025
- Revised: 31 January 2026
- Accepted: 19 February 2026
- Published online: 19 March 2026
Abstract: The editing efficiency of the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system in plants is restricted by the delivery of T-DNA and the stability of transgene expression. However, there is a dearth of efficient and uncomplicated evaluation tools in this respect. To tackle this issue, a comprehensive set of tools has been developed. First, we constructed a visual reporter vector, pHNCas9-EGFP, and successfully verified the frequent occurrence of T-DNA left border deletions in transformation of tomato (Solanum lycopersicum), facilitating efficient monitoring of this event. Secondly, we constructed the dual-reporter vector pHN2G, which provides a reliable method for accurately comparing promoters' strength. It also provides potential tools for detecting the response of plant gene promoters to hormones. Moreover, we designed the fusion reporter vectors pHN2AGUS/pHN2AGFP to achieve an intuitive assessment of the translation efficiency of Cas9 genes with different codon optimizations in plants. Finally, by utilizing the pHN2G6 vector, we evaluated key components in the integrated editing system and found that extending the artificial poly(A) to 150 bp can boost the translation efficiency of the upstream gene by 3.74-fold. This study provides a series of original tools and optimization strategies to overcome critical technical bottlenecks in plant genome editing, which is of great significance for promoting the design of efficient genetic transformation systems in plants.
-
Key words:
- CRISPR/Cas9 /
- T-DNA insertion /
- Promoter /
- Genome-editing efficiency /
- Tools /
- GUS /
- EGFP /
- 2A self-cleaving peptide