










-
The tea plant (Camellia sinensis), a member of the Theaceae family, predominantly prospers in warm and humid environments. Nevertheless, drought has increasingly emerged as a significant limitation to tea cultivation and quality enhancement, influenced by factors such as geographic location and climate change[1]. Under escalating drought stress, tea plantations suffer from diminished yields, elevated mortality rates, and substantial economic detriments[2]. Therefore, understanding the mechanisms underlying drought resistance in tea plants and improving their adaptability to water scarcity constitute essential research priorities in this domain.
The rhizosphere microbiome has been shown to affect plant growth parameters, including overall size[3] and root biomass[4]. In drought conditions, root-associated bacterial communities play a crucial role in sustaining host plant health[5]. Drought stress influences the composition of the rhizosphere microbial community by modifying root exudation profiles[6], selecting for microbial taxa tolerant to stress[7], and altering the physical structure of the soil environment[8]. Furthermore, drought can exert persistent effects on rhizosphere microbiota, including the enrichment of specific taxa that contribute to the host plant's recovery processes[9]. Empirical evidence suggests that microbial communities can alleviate drought-induced stress in plants; for instance, soil microorganisms have been observed to mitigate adverse drought effects in trees by reducing declines in leaf water potential and photosynthetic efficiency[10]. Under conditions of limited water availability, drought consistently selects for stress-resistant bacterial phyla such as Actinobacteria, Firmicutes, and Chloroflexi within the rhizosphere[11,12] as well as in other soil habitats[13]. The rhizosphere also serves as a principal reservoir of endophytic microbes that enhance drought tolerance[14,15]. During drought episodes, several bacterial genera, including Bacillus, Micrococcus, Pseudomonas, and Halomonas, have been demonstrated to promote plant growth[16,17]. The selective recruitment of drought-adapted microbial taxa may represent an evolutionary adaptation, whereby recurrent exposure to drought across generations stabilizes beneficial plant-microbe interactions, thereby enhancing the fitness of both host plants and associated microorganisms.
Empirical evidence indicates that drought-induced alterations in the rhizosphere microbiota can produce legacy effects that enhance drought tolerance in subsequent generations of plants[18]. Ulrich et al.[19] reported that microbial regulation of plant physiological responses under moderate drought conditions influenced plant performance during later periods of water deficit. Additionally, soil legacies resulting from climatic stress have been demonstrated to increase biomass production in ryegrass, while simultaneously improving its resistance and recovery capabilities under repeated drought episodes[20]. Under persistent drought conditions, rapeseed plants exposed to multi-generational drought exhibited greater bacterial abundance and diversity in the rhizosphere compared to control plants[21]. Rhizobacteria contribute to enhanced drought resistance through various mechanisms, including the regulation of phytohormones such as abscisic acid[22], facilitation of nutrient uptake during and following drought, modification of soil hydraulic properties[23], and suppression of pathogenic organisms[24]. Relative to consistently moist or single-drought scenarios, recurrent drought events have been shown to alter microbial community composition, increase community divergence, and improve soil multifunctionality during dry periods. These legacy effects arising from repeated drought stress may strengthen plant resilience to future drought occurrences[25]. Collectively, these findings suggest that plants cultivated in soils with a history of drought are better adapted to subsequent dry conditions than those grown in soils without such a drought history. This adaptive advantage further influences plant-soil feedback mechanisms in response to recurrent drought stress.
This study conducted a drought legacy experiment with three primary objectives: (1) to investigate whether drought legacies impact tea seedlings; (2) to elucidate the effects of these legacies on the structure and diversity of rhizobacterial communities associated with tea seedlings; and (3) to clarify the functional role of the core microbiota in the context of drought legacy effects. Previous research has demonstrated that core members of the microbiota maintain potentially essential functional relationships with their host plants, with their roles often being irreplaceable[26,27]. This characteristic renders them particularly suitable for examining microbiota-mediated plant functions[28,29]. Given that core taxa underpin critical community functions, they may exert cascading influences on both microbial community structure and broader ecosystem processes. To achieve these objectives, physiological assays were employed to evaluate plant responses to drought legacies, and utilized high-throughput sequencing techniques were used to characterize the taxonomic composition and functional potential of rhizosphere microbial communities. This approach enabled the identification of structural patterns, diversity metrics, and putative functions of the root-associated microbiota. Furthermore, integration of microbial data with key plant functional traits facilitated deeper insight into the functional contributions of the core microbiota under drought legacy conditions.
-
The soil utilized in this investigation was sourced from a tea plantation operated by Shandong Taishan Chaxi Valley Agricultural Development Co., Ltd (116°95' E, 36°22' N). The basic physical and chemical properties of soil are as follows: soil texture was sandy loam, soil organic matter was 21.5 g/kg, and pH was 5.9. Three-year-old clonal tea seedlings of the ‘Fuding Dabaicha' cultivar, exhibiting uniform growth vigor, were selected as the plant material. The experimental design comprised two cultivation phases (Period I and Period II) subjected to two distinct soil moisture regimes: constant moisture (WET) and drought stress (DRY). Rhizosphere soil from tea plants in the field was collected and thoroughly mixed with sterilized soil at a mass ratio of 1:9, initiating cultivation Period I. The sterilized soil was obtained by high-temperature sterilization (autoclaved at 121 °C for 30 min). Throughout Period I, the WET treatment was maintained at 70% of the soil's field water capacity for a duration of eight weeks. Conversely, the DRY treatment involved a dynamic soil moisture regime, with moisture levels adjusted weekly as follows: Maintain at 70% field capacity (FC) in the first one to four weeks. At the fifth week, the water content gradually decreased to 35% FC within 3 d, and maintained this level for 4 d. At the sixth week, the water content returned to 70% FC and was maintained for one week. The drought process of the fifth week was repeated again in the seventh week. At the eighth week, it returned to 70% FC. In cultivation Period II, new tea seedlings were transplanted into sterilized soil inoculated with rhizosphere soil derived from either the WET or DRY treatments of Period I. Four treatment groups were established based on the combination of moisture conditions across the two periods: WET-W, DRY-W, WET-D, and DRY-D, where 'W' denotes well-watered conditions and 'D' indicates drought stress. This second phase spanned nine weeks, with the initial eight weeks replicating the irrigation regime of Period I, followed by a ninth week maintained at 35% field water capacity.
Determination of biomass of tea seedlings
-
After the cultivation, the tea seedlings were divided into two parts: aboveground and underground parts. The tea seedlings were heated initially at 105 °C for 30 min to deactivate enzymatic activity and then dried to a constant weight at 80 °C. The dry matter weight of the aboveground and underground parts was measured, and the root-to-shoot ratio was calculated.
Determination of plant endogenous hormones
-
Endogenous hormones were extracted and purified according to the protocol established by Zeng et al.[30]. Fresh tea leaf samples were homogenized in a pre-chilled mortar using an extraction buffer. Following centrifugation, the supernatant was collected and subjected to purification through a C18 solid-phase extraction cartridge. The eluate was then vacuum-dried, reconstituted, and adjusted to a fixed volume for further analysis. Quantification of indole-3-acetic acid (IAA), gibberellic acid (GA3), trans-zeatin riboside (TZR), and abscisic acid (ABA) was performed using enzyme-linked immunosorbent assay (ELISA). Each sample was assayed in triplicate to ensure analytical reliability.
Rhizosphere soil sampling, DNA extraction, and 16S rRNA gene amplicon sequencing
-
Following the completion of the drought induction phase, whole seedlings with intact root systems were meticulously excavated. Carefully dig out the entire root system of the tea seedlings, gently shake off the non-tightly attached soil. Subsequently, rhizosphere soil samples were obtained by gently removing soil particles adhering closely to the root surface (1−3 mm) using a sterile brush. These samples were promptly stored at −80 °C until subsequent analyses. Microbial DNA was extracted from the soil samples employing the HiPure Soil DNA Kit (Magen, Guangzhou, China). The V3-V4 hypervariable region of the bacterial 16S rRNA gene was amplified utilizing the primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). The resulting amplicons were subjected to paired-end sequencing on an Illumina HiSeq 2500 platform following equimolar pooling. Raw sequencing reads were merged using FLASH (version 1.2.11), and high-quality clean tags were generated through quality filtering implemented in QIIME (version 1.9.1). Subsequently, sequences were clustered into operational taxonomic units (OTUs) at a 97% similarity threshold using Uparse software (version 8.1.1861).
Statistical analysis
-
Statistical analyses were conducted utilizing one-way analysis of variance (ANOVA) implemented in IBM SPSS (version 27). A threshold of p < 0.05 was established to determine statistical significance. Data visualization was accomplished using GraphPad Prism (version 10.1.2).
Microbial alpha-diversity indices were calculated utilizing the 'vegan' package in R (version 4.4.3)[31], whereas beta-diversity was evaluated employing the 'phyloseq' package. The influence of inoculum source and moisture treatment on the rhizobacterial community composition was examined through PERMANOVA analyses based on Bray-Curtis dissimilarity metrics. Taxon-specific responses were determined via differential abundance analysis of raw operational taxonomic unit (OTU) data using the 'DESeq2' package[32].
To assess the complexity of the soil microbial network, a co-occurrence network was constructed based on the Spearman correlation algorithm and characterized its topological properties using the 'igraph' package[33]. For correlation analysis, a significance level of p < 0.05 was applied. The correlation coefficient (R) threshold was determined through an iterative approach guided by the network topology, without multiple-testing correction of p-values[34]. Network visualization was conducted using Gephi software. Cohesion, a metric reflecting microbial community connectivity, was employed to quantify the stability and complexity of the networks. In biological interaction network analysis, for a given network, the cohesion index was calculated as the sum of significant positive or negative correlations between species weighted by their abundances:
$ \mathrm{C}_{j}^{pos}=\sum \limits_{i=1}^{n}{\mathrm{a}}_{i}{\overline{r}}_{i,r \gt 0} ({\rm{Positive}}\; {\rm{Cohesion}}) $ $ \mathrm{C}_{j}^{neg}=\sum \limits_{i=1}^{n}{\mathrm{a}}_{i}{\overline{r}}_{i,r \lt 0} ({\rm{Negative}}\; {\rm{Cohesion}}) $ ai represented the abundance of species i in sample j; Cjpos and Cjneg denoted positive and negative cohesion, respectively[35,36]. The positive connectivity of species i (
$\overline r_{i,r>0} $ $\overline r_{i,r<0} $ To identify potential keystone taxa within the microbial network, an integrative analytical strategy was employed, combining DESeq2, LEfSe, Indicator Species Analysis, and the specificity-occupancy (SPEC-OCCU) framework. DESeq2 was utilized to detect operational taxonomic units (OTUs) exhibiting differential relative abundance between drought and well-watered conditions, with taxa considered responsive if they demonstrated statistically significant changes (p < 0.05) and an absolute log2 fold change of at least two (|log2FC| ≥ 2)[32]. LEfSe identified discriminative OTUs demonstrating significant differences between groups (LDA score > 2, p < 0.05), effectively detecting species with subtle abundance changes yet distinct distribution patterns[37]. Consequently, DESeq2 and LEfSe served complementary roles in microbial differential analysis by jointly confirming taxa that exhibited either pronounced abundance differences or unique distribution profiles despite minimal abundance variation[38]. Indicator Species Analysis was employed to select ecologically relevant taxa associated with specific habitats or treatments by combining measures of specificity and fidelity (IndVal > 0.5, p < 0.05)[39]. Additionally, SPEC-OCCU plots facilitated the identification of potential specialist species within the community[40]. In this study, taxa satisfying dual criteria of specificity and occupancy values equal to or exceeding 0.7 were classified as specialists, reflecting distinct habitat specificity alongside consistent presence across replicates within the target environment[39,40]. Collectively, these four methodologies constitute a systematically structured, multi-tiered screening framework. This integrated approach comprehensively addresses variations in abundance, inter-group discriminative capacity, ecological stability, and habitat specificity, thereby minimizing false positives and enabling a more robust and thorough identification of potential core species within microbial networks.
The putative metabolic functional pathways of the tea rhizosphere microbial community were predicted using PICRUSt2 and annotated against the KEGG database (
www.genome.jp/kegg ). -
At the conclusion of cultivation period II, seedlings subjected to drought stress demonstrated a marked decrease in both shoot and root dry biomass relative to their well-watered counterparts, irrespective of inoculation with rhizosphere soil derived from either the previously well-watered (WET) or drought-exposed (DRY) microbial communities established during period I (Fig. 1a, b). The root-to-shoot ratio exhibited a significant decline of 20.83% in the WET-D compared to WET-W. Conversely, this ratio increased by 3.5% in the DRY-D relative to DRY-W. Importantly, the root-to-shoot ratio in the DRY-D group was 55.26% greater than that observed in WET-D (Fig. 1c). These findings suggest that inoculation with a microbial consortium conditioned by prior drought exposure (DRY) imparts a legacy effect that preferentially enhances root development in tea seedlings under subsequent drought conditions.
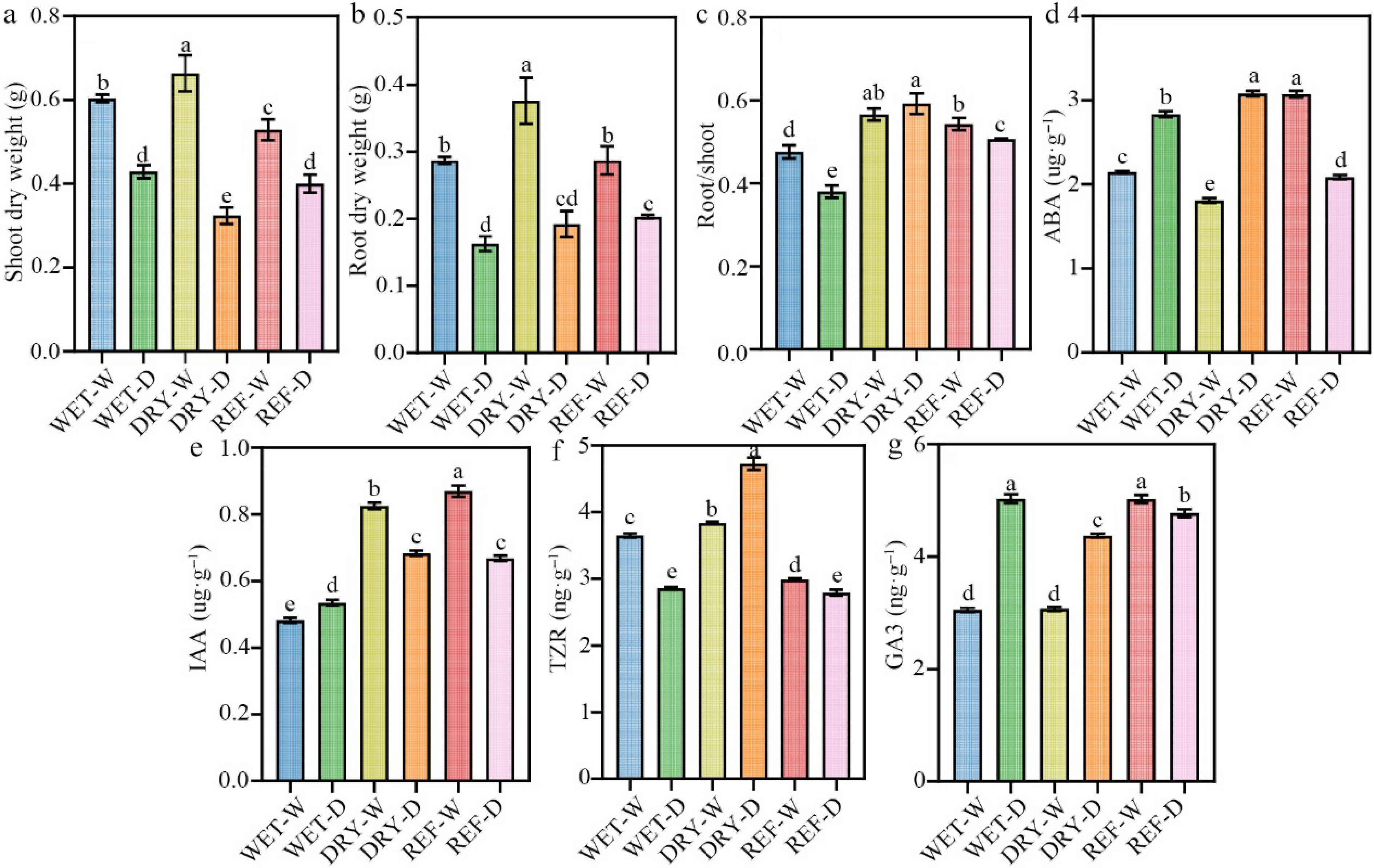
Figure 1.
Effects of different treatments on physiological parameters of tea seedlings in cultivation period II. (a) Shoot dry weight. (b) Root dry weight. (c) Root-to-shoot ratio. (d) Abscisic acid (ABA) content. (e) Indole-3-acetic acid (IAA) content. (f) Trans-zeatin riboside (TZR) content. (g) Gibberellic acid (GA3) content. Different letters represent significant differences between treatments (p < 0.05).
During the second cultivation period under drought stress, concentrations of ABA and GA3 in tea leaves exhibited a significant increase across all treatment groups (Fig. 1d, g). In arid conditions, the IAA content in the WET-D was markedly higher than that in WET-W, whereas the TZR content was significantly lower (Fig. 1e, f). Conversely, seedlings inoculated with drought-preconditioned microbiota (DRY-D compared to DRY-W) demonstrated inverse patterns in IAA and TZR levels under identical drought conditions. Notably, under drought stress, the DRY-D resulted in significantly elevated concentrations of ABA, IAA, and TZR by 8.65%, 27.66%, and 65.17%, respectively, relative to WET-D, while GA3 content decreased by 13.06%. These findings suggest that the residual effects of inoculating the tea rhizosphere bacterial community, conditioned by early drought exposure during the first cultivation period, can modulate hormone distribution and accumulation, thereby enhancing the drought tolerance of tea plants during the subsequent cultivation phase.
Influence of soil drought legacy on rhizobacterial community diversity and composition in tea seedlings
-
The rhizobacterial communities across all four inoculation treatments demonstrated significantly greater richness and evenness compared to those observed in the REF-W and REF-D (Fig. 2a−c). The Shannon, Chao1, and Pielou indices were markedly higher in the DRY-W treatment relative to the WET-W (Fig. 2a−c). Conversely, under drought conditions, the Pielou index was significantly reduced in DRY-D compared to WET-D (Fig. 2c). These findings indicate that reinoculation with drought-preconditioned bacteria followed by rewatering enhances the richness and evenness of the rhizobacterial community, whereas subsequent exposure to drought stress diminishes community evenness. Non-metric multidimensional scaling (NMDS) and principal coordinates analysis (PCoA) revealed significant differences in β diversity among treatments (Fig. 2d), demonstrating distinct bacterial community compositions between groups, while samples within each group exhibited compositional similarity (Fig. 2e, PERMANOVA: R2 = 0.993, p < 0.01). Further PERMANOVA analysis confirmed that both the source of inoculum (R2 = 0.260, p = 0.001) and moisture treatment (R2 = 0.512, p = 0.001) exerted significant effects on the structure of the rhizobacterial communities (Table 1).
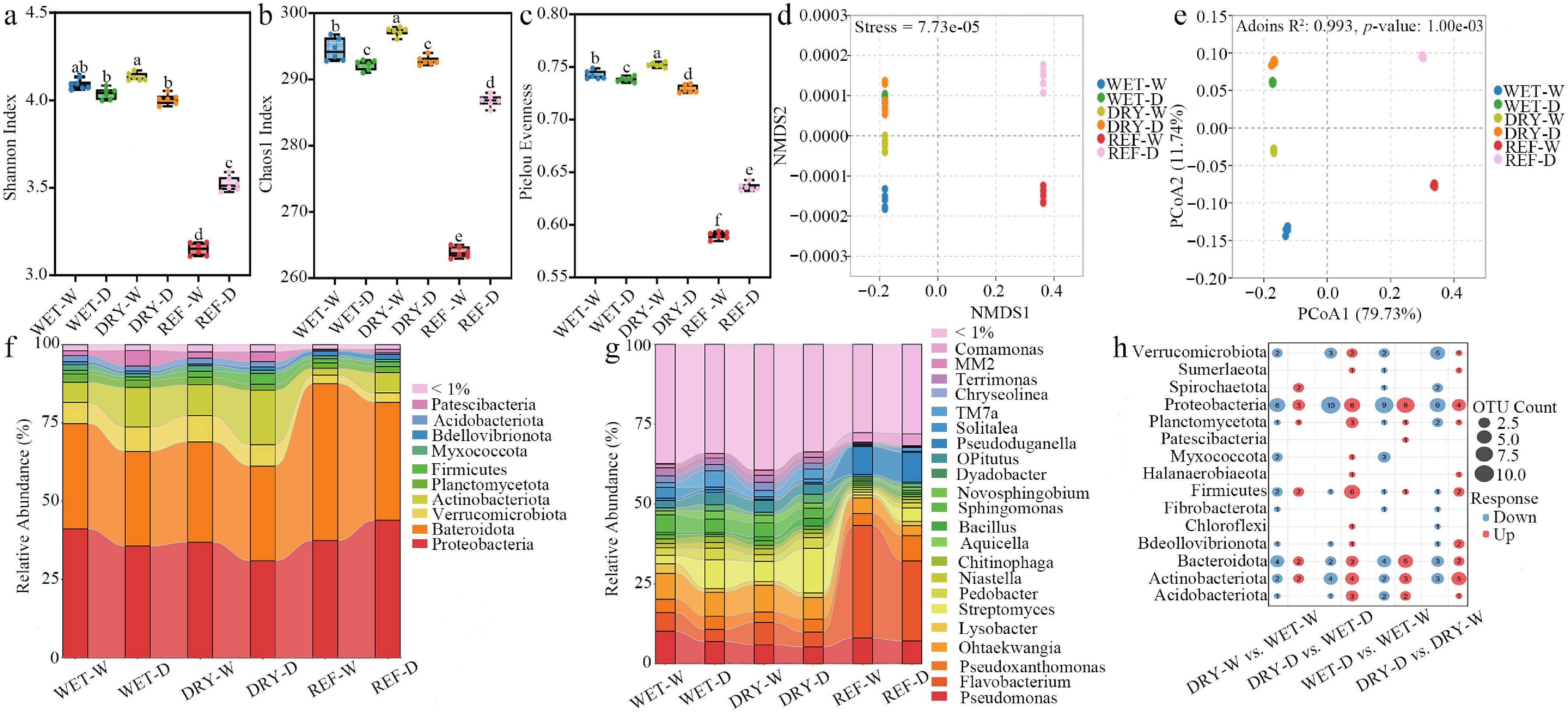
Figure 2.
Effects of different treatments on the diversity and composition of the rhizobacterial community in tea seedlings during cultivation period II. (a) Shannon, (b) Chao1, and (c) Pielou evenness index, respectively. (d) NMDS, and (e) PCoA ordination plots, respectively; Relative abundance of bacterial communities at (f) the phylum, and (g) genus levels, respectively. (h) Bubble plot showing differentially abundant bacterial phyla between treatments, based on DESeq2 analysis (p < 0.05, |log2FC| ≥ 2). Different letters represent significant differences between treatments (p < 0.05).
Table 1. PERMANOVA based on Bray-Curtis distance was used to analyze the effects of inoculum and dry-wet treatment on the composition of bacterial community in the rhizosphere of tea seedlings at culture stage II.
df Sum sq. Mean sq. F model R2 p Inoculant (WET vs. DRY) 1 0.095 0.095 7.733 0.260 0.001*** Treatment (C vs. D) 1 0.188 0.188 23.045 0.512 0.001*** ***, p < 0.001. At the phylum level, the rhizobacterial communities in WET-W, WET-D, DRY-W, and DRY-D were predominantly composed of Proteobacteria, Bacteroidota, Actinobacteriota, and Verrucomicrobiota (Fig. 2f, Supplementary Table S1). In addition, there were differences in the relative abundance of rhizosphere bacteria among treatments at the genus level (Fig. 2g, Supplementary Table S2).
To evaluate the legacy effects of drought on specific bacterial lineages and to identify the operational taxonomic units (OTUs) contributing to community differences, a differential abundance analysis utilizing DESeq2 was conducted. The comparison between DRY-D and WET-D revealed 54 OTUs with altered relative abundance, of which 32 were up-regulated, and 22 were down-regulated. In contrast, the smallest compositional shift was observed between DRY-W and WET-W, involving only 34 differentially abundant OTUs (Fig. 2h). Among the affected phyla, Proteobacteria demonstrated the most substantial alterations, characterized by a higher number of down-regulated OTUs relative to up-regulated ones. Bacteroidota displayed a greater number of down-regulated OTUs in the DRY-W vs. WET-W, whereas more OTUs were up-regulated in the WET-D vs. WET-W. In the DRY-D vs. WET-D, significant changes were observed in Actinobacteriota and Firmicutes, with Firmicutes exhibiting an increase in OTU abundance. Additionally, the comparison between DRY-D and DRY-W revealed a predominant enrichment of Actinobacteriota and a marked down-regulation of Verrucomicrobiota (Fig. 2h).
Influence of soil drought legacy on rhizobacterial network properties in tea seedlings
-
Bacterial co-occurrence networks were constructed to assess the effects of various treatments on network complexity within the rhizosphere during the second cultivation period. All six networks demonstrated high clustering coefficients (ranging from 0.691–0.881), short average path lengths (0.539–2.487), and elevated modularity values (0.831–0.903) (Supplementary Table S3), indicative of 'small-world' characteristics and modular organization. Comparative analysis between DRY-W and WET-W, as well as DRY-D and WET-D, revealed that networks under DRY-W and DRY-D exhibited reductions in node counts, edge numbers, average degrees, modularity, and average clustering coefficients (Fig. 3g−k). These findings suggest that inoculation with drought-induced recombinant bacteria results in a smaller rhizosphere bacterial network characterized by sparser connections, diminished intra-module connectivity, increased inter-module interactions, and less distinct modular boundaries. Furthermore, comparisons between WET-D and WET-W, and between DRY-D and DRY-W, indicated that drought-stressed groups (WET-D and DRY-D) possessed lower node counts, modularity, and average path lengths (Fig. 3g, j, l). Collectively, these patterns imply that drought stress reduces network size and modularity while concurrently shortening average path length, which may enhance the functional efficiency of microbial interactions.
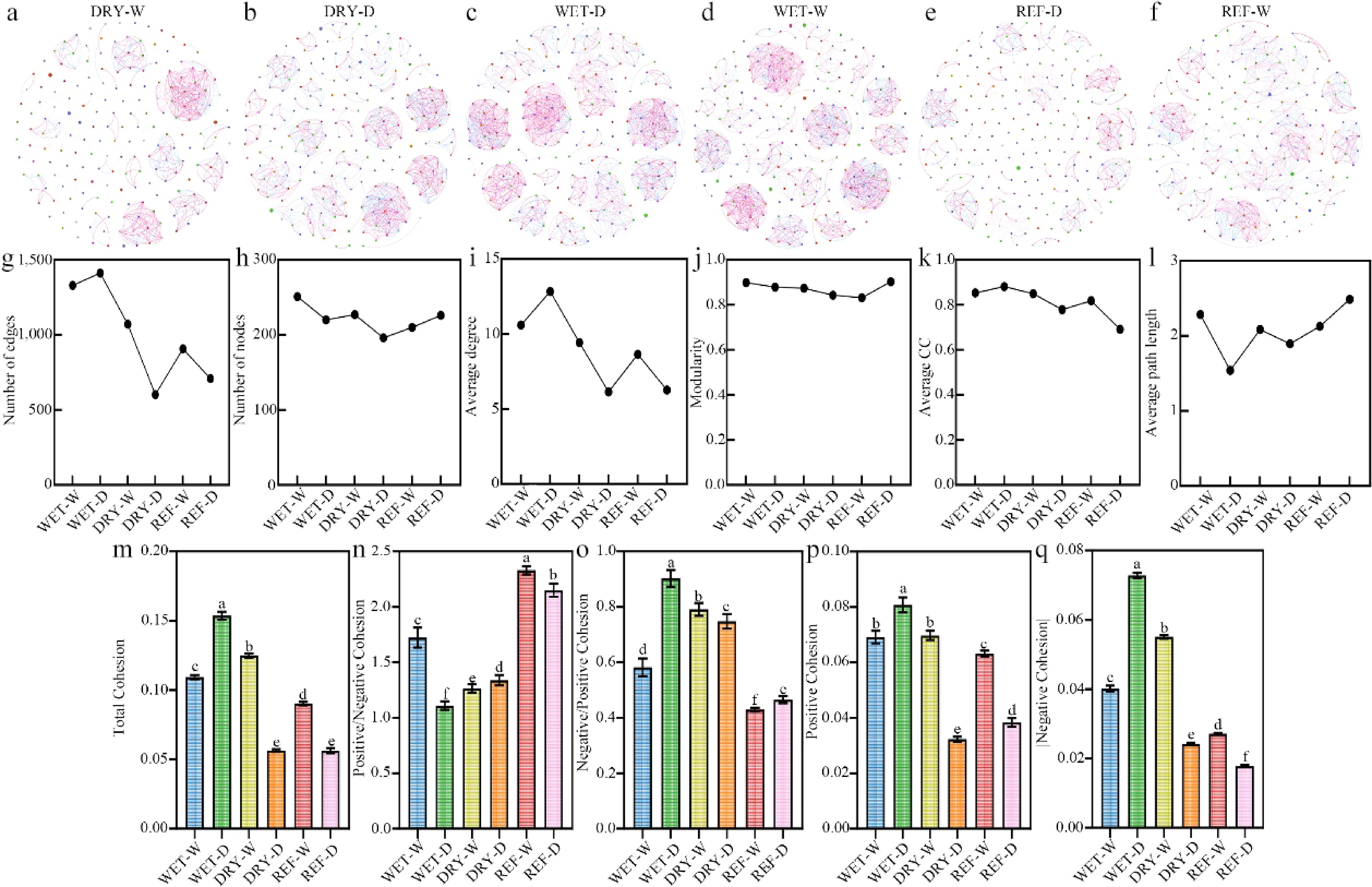
Figure 3.
Effects of different treatments on rhizobacterial network properties in tea seedlings during cultivation period II. (a)−(f) Visualizations of co-occurrence networks under each treatment. (g)−(l) Topological properties of the networks, including node count, edge number, average degree, modularity, average clustering coefficient, and average path length. (m)−(q) Cohesion analysis of the rhizobacterial communities, showing total cohesion, positive/negative cohesion ratio, negative/positive cohesion ratio, and absolute values of positive and negative cohesion. Different letters represent significant differences between treatments (p < 0.05).
Network cohesion metrics were utilized to assess potential species interactions within rhizobacterial communities across different treatment conditions. The total cohesion values ranked as follows: WET-D > DRY-W > WET-W > REF-W > DRY-D ≈ REF-D (Fig. 3m), indicating that the WET-D exhibited the highest community complexity, whereas DRY-D demonstrated relatively low complexity. Both positive and negative cohesion values exceeded one across all treatments (Fig. 3n); however, the ratios of negative to positive cohesion remained below 1 (Fig. 3o), suggesting a predominance of positive interactions and overall maintenance of community stability. The highest positive-to-negative cohesion ratio (1.723) was observed in the WET-W, indicative of a community primarily driven by mutualistic interactions. Although WET-D showed the greatest absolute values for both positive and negative cohesion, it exhibited the lowest positive-to-negative ratio alongside the highest negative-to-positive ratio (Fig. 3n−q). This pattern suggests an increase in synchronous negative interactions within the WET-D community, reflecting intensified competition that diminished the relative influence of positive interactions. In the DRY-W, positive and negative cohesion values were second only to those in WET-D, yet the positive-to-negative ratio was higher, indicating a more balanced interaction dynamic. Conversely, the DRY-D displayed the lowest absolute cohesion values but maintained a relatively high positive-to-negative ratio, implying an overall reduction in species interactions with a relative predominance of positive relationships.
Identification and analysis of core bacterial taxa
-
To further investigate the bacterial community with the aim of identifying potential core species within the network and assessing their influence on network characteristics and community cohesion, a combined analytical approach integrating DESeq2, LEfSe, Indicator Species Analysis, and SPEC-OCCU was employed to screen for core microorganisms (Fig. 4a−i, Supplementary Table S4). This comprehensive screening revealed 13, five, 14, and four core operational taxonomic units (OTUs) corresponding to the WET-W, WET-D, DRY-W, and DRY-D, respectively (Fig. 4j, k, Supplementary Table S5). Notable differences were observed in the dominant bacterial phyla constituting the core OTUs across the different treatments. Specifically, the WET-W was predominantly characterized by core phyla including Verrucomicrobiota, Proteobacteria, Fibrobacterota, and Myxococcota. In contrast, the WET-D featured core phyla such as Actinobacteriota, Bacteroidota, Proteobacteria, and Acidobacteriota. The DRY-W treatment's core phyla were mainly Actinobacteriota, Proteobacteria, Acidobacteriota, and Bacteroidota, whereas the DRY-D was primarily associated with Verrucomicrobiota, Firmicutes, Planctomycetota, and Halanaerobiaeota (Supplementary Table S5).
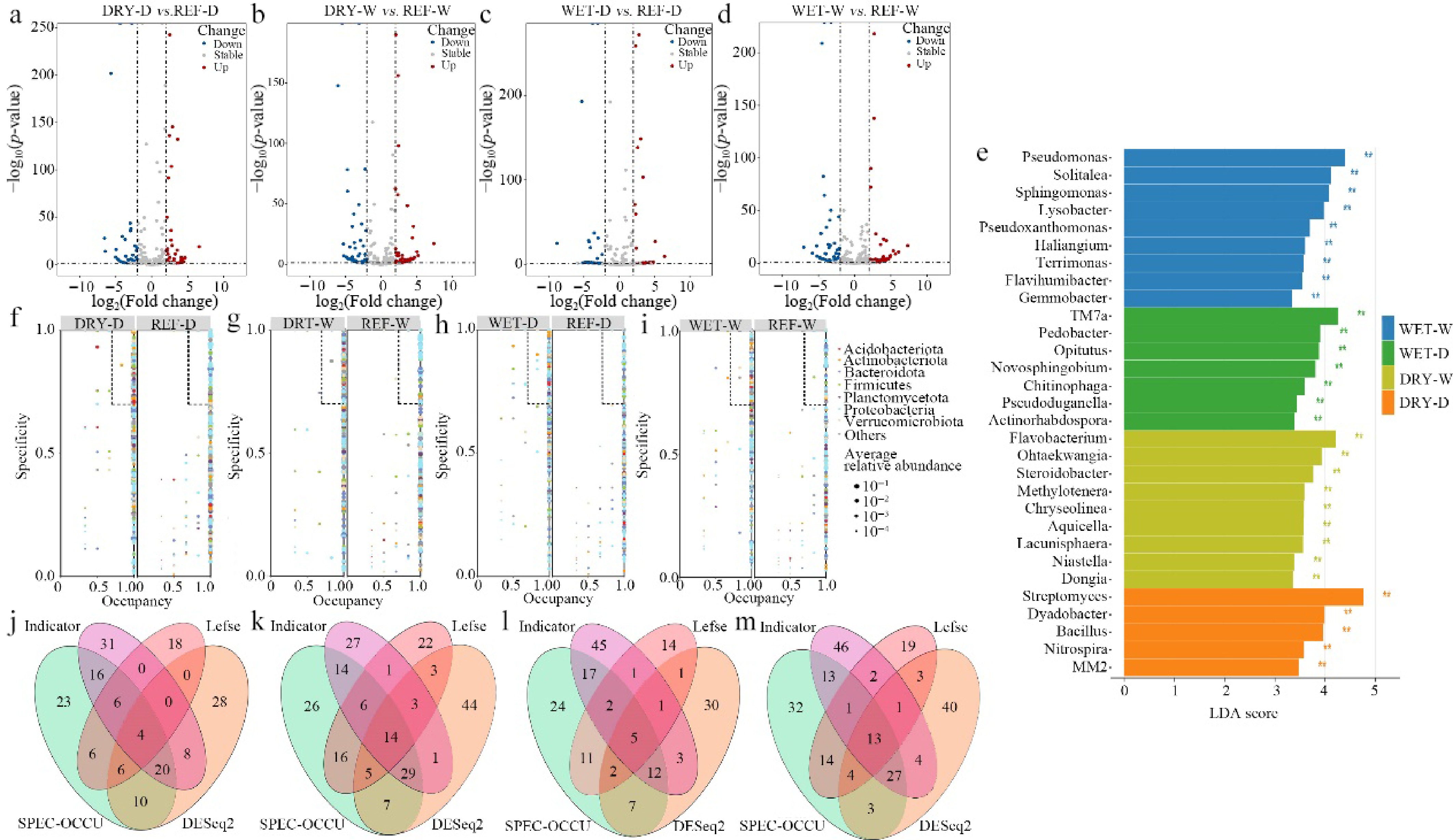
Figure 4.
Identification of core bacterial taxa in the rhizosphere. (a)−(d) Volcano plots showing differentially abundant OTUs in each treatment group (DESeq2). (e) LEfSe analysis illustrating taxa with significant abundance differences among treatments (LDA score > 2, p < 0.05). (f)−(i) Occupancy-specificity plots for each treatment; OTUs with specificity and occupancy values ≥ 0.7 were classified as specialist species. (j)–(m) Venn diagrams representing core bacterial OTUs identified by the combined DESeq2-LEfSe-Indicator Species Analysis-SPEC-OCCU approach.
To evaluate how core microbial taxa influence network architecture, the OTUs identified in each treatment group were selectively removed using an integrated approach combining DESeq2, LEfSe, indicator species analysis, and the SPEC-OCCU framework. The number of OTUs removed was 13 in WET-W, 5 in WET-D, 14 in DRY-W, and 4 in DRY-D (Supplementary Table S5). The influence of core bacterial taxa on the structural configuration of rhizosphere networks was assessed by analyzing topological metrics before and after their simulated removal. Across all experimental conditions, the exclusion of core taxa consistently resulted in reductions in the number of nodes, edges, and average degree (Supplementary Table S6), thereby corroborating their role as connectivity hubs whose absence generally diminishes network size and connectivity. In the WET-W, the removal of core taxa significantly decreased the average path length without affecting modularity, underscoring their critical function in preserving global network connectivity. Comparable directional changes in path length and clustering coefficient were observed in the WET-D, albeit with lesser magnitude, indicating a reduced dependence of network stability on core taxa under these conditions. In the DRY-W, core taxa removal led to an increase in clustering coefficient alongside a decrease in path length, suggesting a more pronounced regulatory influence of core taxa on local clustering relative to WET-W. Finally, in the DRY-D, the elimination of core taxa resulted in elevated modularity and average path length coupled with a reduction in clustering coefficient, highlighting their vital role in maintaining overall network connectivity and interaction strength (Supplementary Table S6).
Spearman correlation analysis identified significant relationships between core bacterial taxa and key physiological parameters in tea seedlings, with distinct correlation patterns observed across different treatment conditions. Under the WET-W, the genera Nannocystis, Puia, Candidatus_Udaeobacter, and Subgroup_10 demonstrated positive correlations with root mass, while exhibiting negative correlations with the levels of IAA, ABA, and TZR. Conversely, Pajaroellobacter and Hymenobacter displayed opposite correlation trends. Additionally, possible_genus_04 and Terrabacter were positively correlated with GA3 (Fig. 5a). In the WET-D, ABA and TZR levels were positively correlated with Acidothermus and negatively correlated with Actinorhabdospora (Fig. 5b). Within the DRY-W, Rhodopseudomonas showed positive correlations with IAA and ABA, and negative correlations with TZR and GA3, whereas Rhodoplanes exhibited the inverse pattern. Root mass was positively associated with Ellin516 and Phycisphaera, but negatively associated with Rhodoplanes (Fig. 5c). In the DRY-D, both Pedosphaera and Halocella were positively correlated with root mass and ABA, and negatively correlated with IAA (Fig. 5d).
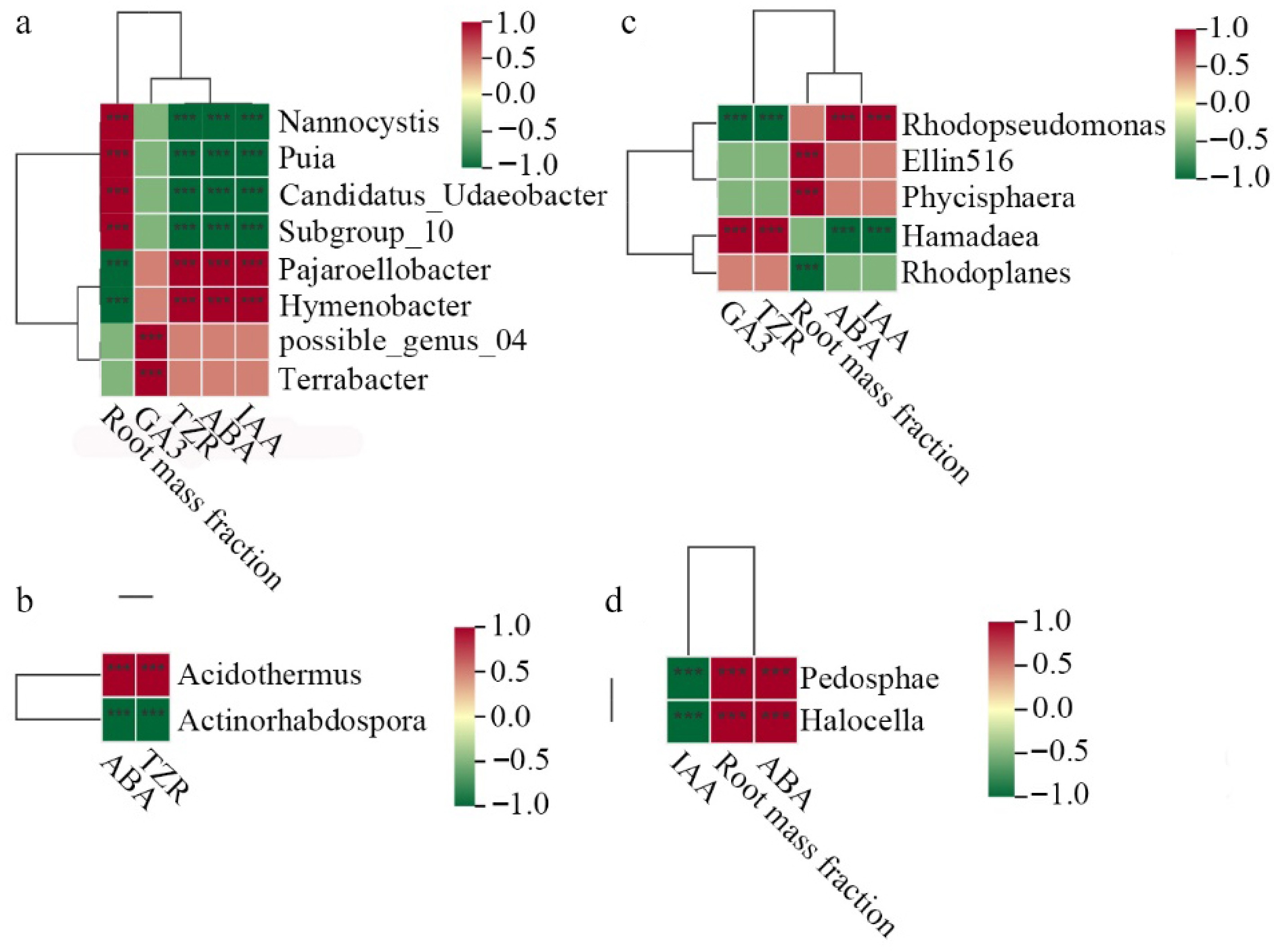
Figure 5.
Spearman correlation analysis between core bacterial and physiological parameters of tea seedlings. (a) WET-W. (b) WET-D. (c) DRY-W. (d) DRY-D. *** p < 0.001.
Functional annotation of the core bacterial communities, conducted using PICRUSt2 in conjunction with the KEGG database, identified microbial metabolism as the predominant pathway across all four treatment groups. Subsequent enrichment analysis demonstrated significant variations in metabolic and pathway activities among the different treatments. Among the top 20 enriched functional categories, pathways associated with Carbon metabolism, Biosynthesis of amino acid, and Biosynthesis of cofactors were notably prominent across all treatments (Fig. 6). Furthermore, the WET-W exhibited significant enrichment in environmental information processing pathways, specifically Bacterial chemotaxis, as well as in Cellular processes including Flagellar assembly and the Caulobacter cell cycle (Fig. 6a). Both the WET-D and DRY-W demonstrated enrichment in environmental information processing, particularly the Bacterial secretion system, alongside cellular processes such as Flagellar assembly (Fig. 6b, c). Additionally, the DRY-W showed notable enrichment in the Caulobacter cell cycle (Fig. 6c). In the DRY-D, cellular processes related to Peptidoglycan biosynthesis and Flagellar assembly were markedly prominent (Fig. 6d).
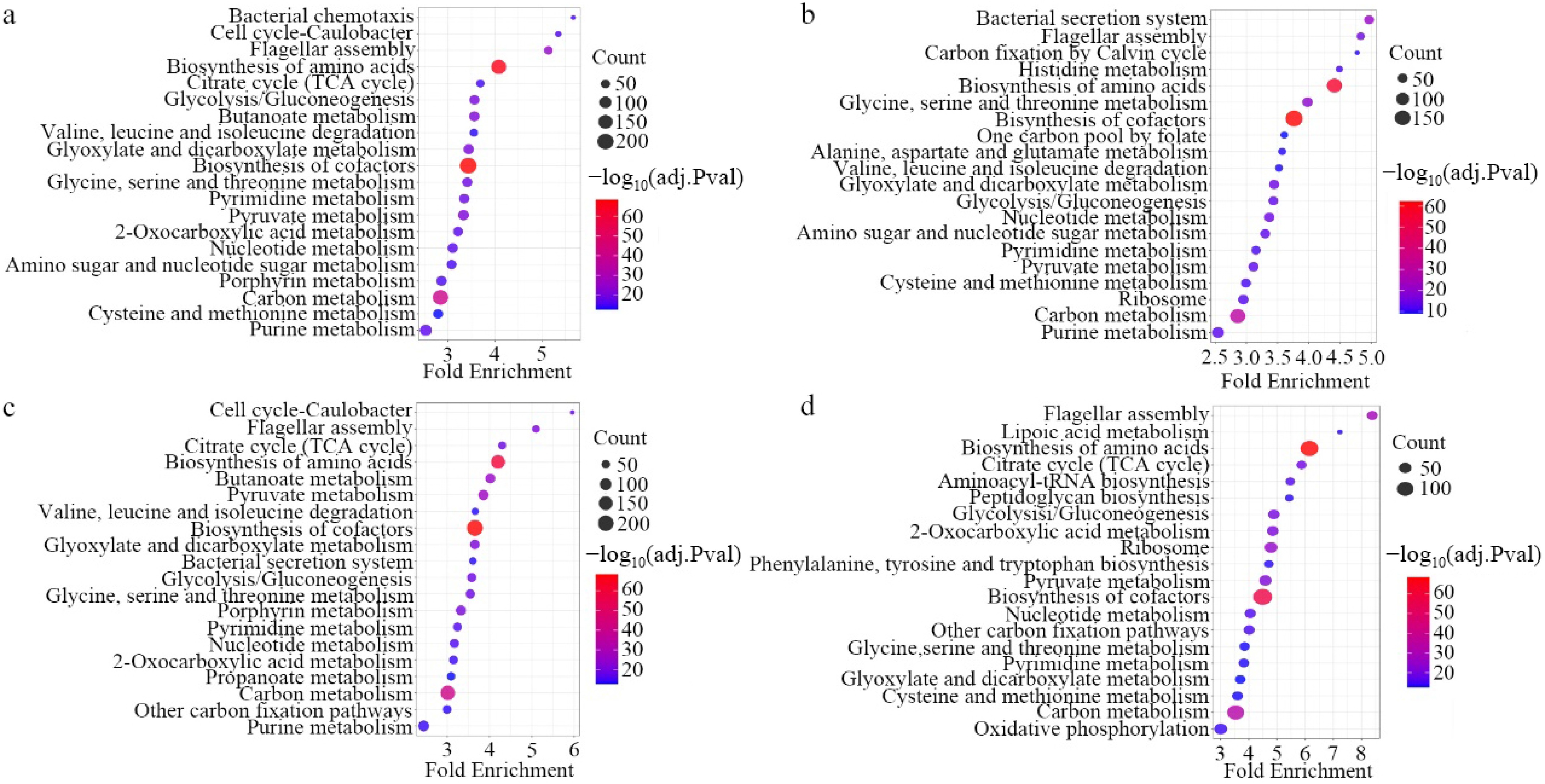
Figure 6.
Functional prediction of core rhizobacteria in different treatments. (a) WET-W. (b) WET-D. (c) DRY-W. (d) DRY-D.
-
In this study, drought legacy effects were demonstrated to be not merely attributable to the enrichment of drought-tolerant microorganisms. Rather, it involved the formation of a core rhizosphere microbial community characterized by a streamlined structure and specialized functional capabilities. This community systematically enhanced the plant's drought resistance through the modulation of its hormonal signaling network.
Drought conditions could significantly influence plant growth and development, producing legacy effects that profoundly impact plant species composition, biodiversity, community structure, and ecosystem functions, thereby affecting subsequent plant growth. These effects were likely mediated by soil microorganisms[41], which might function as 'bioregulators' by modulating hormonal balance to enhance drought resistance in tea plants. This study demonstrated that the drought legacy driven by a reconstructed rhizosphere core microbiome might significantly enhance the drought resistance of tea plants by establishing a more optimized hormonal homeostasis (Fig. 1). These results revealed a complex microbially mediated regulatory mechanism that fine-tunes plant physiological responses to bolster drought resilience. A particularly noteworthy physiological finding was the distinctive hormonal profile observed in tea seedlings subjected to both a drought-history microbial inoculum and subsequent drought stress (DRY-D). Unlike the conventional drought response seen in WET-D plants, characterized by increased IAA and decreased TZR, DRY-D plants exhibited a synergistic upregulation of ABA, IAA, and TZR (Fig. 1). This unique hormonal pattern suggested a targeted microbial influence rather than a random effect. ABA was widely recognized as a principal stress hormone that induced stomatal closure and enhanced water conservation[42]. The concurrent elevation of IAA, a key regulator of root growth and architecture, indicated a strategy that not only conserves water but also actively promotes water acquisition under stress conditions[43]. This observation aligns with recent studies demonstrating that bacteria such as Bacillus and Enterobacter could produce IAA to modify root morphology in wheat, thereby increasing root surface area and improving water uptake[41]. Furthermore, DRY-D plants accumulated higher levels of TZR, a cytokinin known to antagonize ABA signaling and promote growth[44]. The increased TZR under drought stress might help alleviate stress-induced senescence, thus maintaining metabolic activity and enhancing recovery potential. Phenotypically, this optimized hormonal balance favored resource allocation to the roots, as evidenced by a significantly higher root-to-shoot ratio in DRY-D plants compared to WET-D counterparts.
Spearman correlation analysis revealed significant associations between specific core microbial taxa and the observed hormonal profiles, offering clues to their potential ecological roles. In the DRY-D treatment, the core bacterial genera Pedosphaera and Halocella showed significant positive correlations with ABA levels and root biomass (Fig. 5d). Although their precise mechanistic contributions to tea plant drought tolerance remain to be experimentally confirmed, it has been previously linked to drought-adapted soil environments[45], and these strong correlations position them as high-priority candidate taxa. More notably, members of the genus Halocella had been reported to interact with multiple groups within the soil microbial network[46] and to participate in the metabolism of sugars, lipids, and proteins under osmotic stress conditions[47], which could potentially contribute to rhizosphere microenvironment modification or interspecific signaling. It is plausible that these core taxa, possibly through direct or indirect interactions, are associated with the modulated hormonal landscape observed in DRY-D plants. The observed negative correlation between these core taxa and IAA (Fig. 5d) further suggests a complex, fine-tuned regulatory scenario rather than a simple, uniform upregulation of plant growth-promoting hormones. This nuanced pattern contrasts with the more conventional stress response observed in WET-D plants, which lacked a microbiome conditioned by prior drought.
The ecological stability of the rhizosphere ecosystem was fundamental to plant health[48,49]. Microbial co-occurrence networks served as valuable tools for evaluating the complexity and stability of microbial communities[36]. Findings indicated that drought stress led to a reduction in the number of network nodes and the average path length, accompanied by a decline in modular structure, collectively driving the simplification and homogenization of the microbial co-occurrence network (Fig. 3h–j). The simplification of the co-occurrence network could be considered a double-edged sword. On the one hand, the intensity of reciprocal and competitive interactions among species diminishes (Fig. 3p, q), suggesting that drought stress compromised the stability and resilience of the rhizosphere microbial community[50]. On the other hand, this phenomenon might signify the removal of redundant species and the reinforcement of critical interactions, consequently enhancing the efficiency of energy and material transfer[51]. When comparing WET-D and DRY-D, it was observed that following inoculation with drought-induced recombinant bacteria, although the network exhibited a reduced number of nodes and edges, positive cohesion remained predominant. This indicated that mutualistic symbiosis continued to dominate within the bacterial communities. This phenomenon might be related to the 'survival of the fittest' of microbial communities under drought conditions. Microorganisms in arid soils had undergone an ecological screening, and the groups with strong adaptability and important functions were retained, while the redundant or sensitive groups were eliminated, thus forming a more streamlined but functionally specialized network structure[52]. Considering the improvement of plant drought resistance, the remodeling of this network structure was considered more beneficial than harmful. This reorganization could be understood as an adaptive and efficient strategy employed by the microbial community in response to recurrent drought conditions.
The core microbiota played a pivotal role in sustaining the stability of the microbial network. After removing them, the size of each treatment network diminished, and the connectivity became sparse (Supplementary Table S6); however, the extent of this impact varied across treatments. In the WET-W, they played an important role as a 'connection hub', significantly contributing to global network connectivity. Conversely, in the DRY-D, they primarily served as 'structure maintainers', suggesting that under drought legacy conditions, these core bacteria were essential for preserving the overall architecture and functional integration of the microbial network. These findings corroborated the perspective that core microorganisms acted as ecological drivers, facilitating community responses to environmental perturbations[53]. Furthermore, the legacy of drought influenced the interaction patterns and functional outputs of the entire network by selectively enriching core groups possessing specific functional potentials, exemplified by the Firmicutes in the DRY-D.
The functional prediction of the core microbiota using PICRUSt2 offers genomic insights into their potential mechanisms of action. Notably, there was a significant enrichment of pathways associated with peptidoglycan biosynthesis, flagellar assembly, and bacterial secretion systems within the core taxa derived from drought-legacy treatments (e.g., DRY-W, DRY-D). These features underpinned root colonization and facilitated the establishment of intimate plant-microorganism interactions[54,55]. Efficient bacterial secretion systems constituted a molecular foundation enabling the delivery of effector proteins or metabolites that directly modulated plant hormonal pathways. For example, bacteria might enhance plant growth through the secretion of indole acetic acid and siderophores[56], or modulate endogenous hormone metabolism by releasing 1-aminocyclopropane-1-carboxylate deaminase, thereby influencing root architecture, stomatal behavior, and stress signaling via the reduction of ethylene synthesis[57]. The observed enrichment of these functional traits within the core taxa supported the hypothesis that these microorganisms actively engage in cross-kingdom signaling with the tea plant host.
This study demonstrated that the drought legacy effects enhance the drought resistance of tea plants through the regulation of rhizosphere soil microorganisms; however, several limitations were identified. Although a significant correlation was observed among the bacterial community composition, hormone concentrations, and plant biomass, a direct causal relationship could not be established due to the inability to construct and inoculate a synthetic microbial community (SynCom). This limitation arises from the difficulty in cultivating many soil microorganisms under laboratory conditions, which impeded the isolation and enrichment of all target strains[58]. Additionally, the functional roles of the core microbiome were primarily inferred through bioinformatic predictions using PICRUSt2 analysis[59]. The principal aim of this study was to elucidate how drought legacy effects reshape the structure of the core rhizosphere microflora in tea plants and to explore the association between the core microbiome and plant drought resistance phenotypes. Within this framework, PICRUSt2 analysis served predominantly as a tool for hypothesis generation rather than mechanistic validation. Consequently, the proposed molecular mechanisms remain hypothetical and require further empirical verification. Consequently, the proposed molecular mechanisms remain hypothetical and require further empirical verification. In the future, the work will focus on the screening of core bacteria in order to obtain core bacteria and perform related functional verification.
-
In conclusion, this study demonstrated that drought legacy effects were associated with the recruitment of a distinct core rhizosphere microbiota. This restructured microbial community was linked to an optimized hormonal profile in tea plants, characterized by coordinated increases in ABA, IAA, and TZR under drought stress, which in turn correlates with enhanced root investment and drought tolerance. These strong correlations suggest that the core microbiota might act as a ‘biological regulator'. To establish causality, future research should focus on isolating these core bacteria and conducting re-inoculation experiments to confirm their direct role in enhancing drought tolerance in tea plants.
-
The authors confirm their contributions to the paper as follows: study conception and design: Han X, Xie X; data collection: Xie T, Zhao X, Han X; analysis and interpretation of results: Zhao X, Ma W, Chang Z; draft manuscript preparation: Xie T, Zhao X; manuscript review, funding acquisition, supervision: Han X, Xie X. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
This work was supported by the National Natural Science Foundation of China (Grant No. 32573088; Grant No. 32302437), and the Natural Science Foundation of Shandong Province (Grant No. ZR2019BC062).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Tongtong Xie, Xiaoxia Zhao
- Supplementary Table S1 The composition and relative abundance of bacterial communities in the rhizosphere of tea seedlings under different treatments in culture period II (phylum level).
- Supplementary Table S2 The composition and relative abundance of bacterial communities in the rhizosphere of tea seedlings under different treatments in culture period II (genus level).
- Supplementary Table S3 Topological parameters of bacterial network in rhizosphere of tea seedlings under different treatments in culture period II.
- Supplementary Table S4 Indicator species analysis of rhizosphere bacteria in different treatments.
- Supplementary Table S5 The relative abundance and classification annotation of rhizosphere core bacteria in different treatments.
- Supplementary Table S6 The characteristics, cohesion and variation range of bacterial network in tea rhizosphere before and after core bacteria removal.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Xie T, Zhao X, Han X, Ma W, Chang Z, et al. 2026. Drought legacy effects enhance tea plant drought tolerance by modulating the rhizosphere core microbiota. Beverage Plant Research 5: e013 doi: 10.48130/bpr-0025-0045 

Drought legacy effects enhance tea plant drought tolerance by modulating the rhizosphere core microbiota
- Received: 01 November 2025
- Revised: 05 December 2025
- Accepted: 19 December 2025
- Published online: 14 April 2026
Abstract: The drought legacy effects significantly influence the drought resistance of plants; however, research examining the drought legacy effects of drought on tea plants remains limited. This study employed a two-stage pot experiment to simulate drought residual effects and investigate their influence on tea plants. Results indicated that the root-to-shoot ratio in the drought-drought treatment (DRY-D) was 55.26% higher than in the wetted-drought treatment (WET-D), accompanied by significant increases in the levels of abscisic acid, indole-3-acetic acid, and trans-zeatin riboside. Furthermore, drought legacy effects prompted a restructuring of the rhizosphere microbial communities. Notably, significant differences were observed in the relative abundance of 54 operational taxonomic units (OTUs) between DRY-D and WET-D. The drought legacy also altered the rhizosphere bacterial network of tea seedlings, which exhibited characteristics of a 'small-world' network with modular architecture. However, variations in community cohesion were evident across treatments. Specifically, the bacterial community in WET-W predominantly displayed mutualistic interactions, whereas competitive interactions intensified in WET-D. In drought-wetted treatment (DRY-W), positive and negative bacterial interactions were balanced, while positive interactions predominated in DRY-D. Core bacterial taxa were identified within each treatment group, with 13, five, 14, and four core bacteria detected in wetted-wetted treatment (WET-W), WET-D, DRY-W, and DRY-D, respectively. The core bacterial community in DRY-D played a pivotal role in maintaining the integrity and stability of the microbial network. Overall, the drought legacy enhanced the drought resistance of tea plants by restructuring the rhizosphere core microbial community, thereby providing a foundation for improving tea plant drought tolerance through microbial inoculation strategies.
-
Key words:
- Camellia sinensis /
- Core microbiota /
- Drought legacy effects