









-
With the rapid advancement of electric vehicles and grid-scale energy storage systems, there is an ever-growing demand for high-energy-density rechargeable batteries[1,2]. In this context, lithium–oxygen (Li–O2) batteries (LOBs) emerge as one of the most promising next-generation energy storage technologies, primarily due to their exceptional theoretical specific energy (~3,500 Wh kg−1, active mass based on Li2O2, O2 + 2Li+ + 2e− ↔ Li2O2, E0 = 2.96 V vs Li/Li+)[3−5]. However, their practical application is severely hindered by the persistently high overpotentials, poor high-rate performance, low round-trip efficiency, and poor cycling stability[6,7]. These limitations fundamentally originate from the insulating and insoluble nature of the discharge product (Li2O2), which causes inevitable surface passivation or even pore blockage, resulting in notoriously sluggish charge transfer and mass transport kinetics during the discharge and charge process[8−10]. To address these challenges, considerable research has focused on the modulation of cathode porous property and binding energy of intermediates to catalytic sites to regulate the properties of discharge products[11−15].
Typically, the morphology and structure of discharge product were correlated to the competition of two discharge routes, the solution route and the surface route, facilitating a three-dimensional (3D) growth in the solution and a space-confined growth on the cathode surface, respectively[16−18]. Studies have shown that toroidal Li2O2, formed via a solution-mediated pathway, facilitates a high specific capacity due to its substantial volume but suffers from poor electronic contact with the electrode, leading to severe polarization and sluggish decomposition kinetics during charge. Conversely, flower-like and film-like Li2O2, grown via a hybrid pathway, exhibit better contact and lower charge overpotentials but typically show a lower capacity due to rapid surface passivation[19,20]. Therefore, the two typical Li2O2 morphologies have led to an inherent trade-off between high capacity (favored by toroidal growth) and high efficiency (favored by film-like/flower-like growth).
To this end, substantial research has been dedicated to manipulating Li2O2 growth through catalyst and electrolyte design to break the trade-off. Yang et al. revealed that catalysts exhibiting LiO2 adsorption energies near the top of the volcano plot, such as Pd-modified graphene and planar noble-metal surfaces, facilitate the growth of worm-like amorphous Li2O2 and achieve optimal electrochemical performance with minimal discharge/charge overpotentials[21,22]. Zhang et al. employed computed tomography and geometric modeling to reveal that near-free Co sites dynamically evolve and match the Li2O2 (100) facet, promoting oriented sheet-like Li2O2 growth with enhanced electron conductivity and weakened *LiO2 binding, thus improving oxygen-evolution kinetics[23]. Chen et al. demonstrated via ab initio simulations that low-DN (Donor Number) electrolytes paired with high-adsorption catalysts promote surface-mediated Li2O2 growth, while high-DN electrolytes with weak-adsorption catalysts favor solution-mediated deposition, highlighting the coupled electrolyte–catalyst control over Li2O2 growth pathways. In summary, most current strategies focus on the sole effect of charge transfer modulation on discharge pathways. This controlled approach has revealed critical mechanisms, such as LiO2 adsorption optimization and electrolyte–catalyst synergy, yet leaves the question of how to actively orchestrate pore-level transport and active sites within 3D porous electrodes.
Numerous studies have also focused on designing porous structures and employing computational simulations and experiments to investigate the morphology of discharge products and battery performance, with a primary emphasis on mass transport optimization. Confining Li2O2 within mesoporous carbon channels has been found to enhance intermediate diffusion and improve electrode–product contact, thereby reducing charging overpotential[24]. Tunable two-dimensional (2D) ordered channels can not only provide efficient pathways for Li+ and O2 transport but also promote reversible Li2O2 deposition/decomposition, leading to simultaneous improvements in capacity and cycling stability[25]. Combined computational and experimental studies further indicate that lower mass loading, reduced current density, and higher oxygen pressure collectively enhance discharge capacity by alleviating transport limitations[26]. Modeling work also reveals that the balance between charge transfer and oxygen transport governs the LiO2(s)/Li2O2 ratio during discharge, directly affecting specific capacity[27]. While progress has been made, further experimental research is needed to systematically examine how the coupling between porous structure and catalytic site activity shapes discharge product morphology and battery performance, particularly regarding capacity and charge polarization.
Herein, we report a metal-free ZIF-derived carbon cathode catalyst for Li–O2 batteries constructed through a solvent-modulated synthesis coupled with vacuum-assisted heat treatment (Fig. 1a and Supplementary Fig. S1). By tailoring the precursor environment, this strategy enabled controlled regulation of surface nitrogen functionalities together with the pore architecture. A series of carbon materials with varied nitrogen moieties and pore characteristics was thereby obtained, allowing for an experimental investigation of how these features collectively influence the discharge and charge processes. Combined structural and electrochemical analysis revealed that coordinated control of active-site chemistry and pore topology governed the Li2O2 formation/decomposition kinetics and polarization behavior. These findings are supposed to provide design principles for porous carbon cathodes where catalytic functionality and pore structure can be concurrently optimized.
-
All the chemical reagents used were of analytical grade without further purification. About 470 mg of benzimidazole (bim) and 92 mg of zinc acetate (Zn(CH3COO)2) were added to 20 and 5 mL of N,N-dimethylformamide (DMF), respectively, and stirred to obtain the evenly distributed solutions. Under rapid stirring, the zinc acetate solution was added to the benzimidazole solution and poured into a 100 mL beaker. The solution was heated in a vacuum oven at 80 °C under 0.003 MPa for 36 h. After the complete evaporation of DMF, deionized water was poured into the beaker and subjected to an ultrasonic process at 60 Hz for 10 min. The mixture was filtered and dried to obtain the ZIF-7 crystal synthesized using DMF solvent, which was labeled as ZIF-7-D. The resulting powder was pyrolyzed at 950 °C for 3 h in N2 flow at a heating rate of 5 °C min−1 to obtain ZDC. To prepare ZWC and ZMC, similar procedures were employed, differing only in the solvent (deionized water and methanol, respectively).
Preparation of cathodes and assembly of Li-O2 cells
-
The carbon materials ZDC, ZWC, and ZMC were mixed with Super P and polyvinylidene fluoride (PVDF) binder in a mass ratio of 8:1:1, respectively. The mixtures were then dissolved evenly in N-methylpyrrolidinone (NMP) and coated on circular carbon cloth discs with a diameter of 11 mm, followed by a drying procedure in an oven at 80 °C for 24 h. The active mass of catalyst loaded onto each disc cathode was 0.1 ± 0.02 mg.
The cathode was assembled in a Swagelok-type cell, with a lithium metal anode and a glass fiber separator, and 1 M LiTFSI/TEGDME as electrolyte. The assembly operation was performed in a glovebox, and the cell was sealed into a glass jar, which was subsequently filled with pure O2 (99.999%).
Material characterizations
-
Field emission scanning electron microscopy (FE-SEM; Hitachi S4800) was used to examine the morphology and surface elemental composition of the samples. N2 adsorption–desorption isotherms (Micromeritics ASAP2460) were measured at 77.3 K. The specific surface area and pore size distributions of material samples were determined by Brunauer-Emmett-Teller (BET) and Barrett-Joyner-Halenda (BJH) analyses, respectively. X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, USA) measurements were performed with a monochromatic Al source, and the binding energies of all element data were calibrated using the C 1s peak at 284.8 eV as reference. X-ray diffraction (XRD, Rigaku Ultima IV, Japan) analysis was performed with Cu Kα radiation at 40 kV and 40 mA. Raman spectra (Horiba LabRAM HR Evolution, Japan) were measured using a 514.5 nm laser. Cathode samples were obtained by disassembling the Li–O2 cells in a glovebox under an inert atmosphere and subsequently stored in a vacuum tank to avoid side effects due to air exposure.
Electrochemical measurements
-
All electrochemical measurements were performed in a two-electrode system at ambient temperature. Cyclic voltammetry (CV) tests were performed using a CHI660D electrochemical workstation with a potential window of 2.0–4.5 V (vs Li/Li+) and a scan rate of 0.001 V s−1. The cell assembly consisted of a lithium metal foil (2.2 mm thickness) functioning as the counter electrode and reference electrode, paired with an oxygen electrode as the working electrode. Galvanostatic cycling tests were carried out on a battery tester (LAND CT2001A) at current densities ranging from 100 to 1,000 mA g−1. The charge and discharge cutoff voltages were set to 4.5 and 2.0 V, respectively. Electrochemical impedance spectroscopy (EIS) measurements were conducted using a CHI660D electrochemical workstation. EIS was detected under 0.1 Hz-100 kHz at open-circuit voltage.
-
Figure 1b–g and Supplementary Fig. S2 show the morphologies and self-assembled structures of the ZDC, ZWC, and ZMC materials. ZDC was composed of 500-nm-long sheets, and the surface was covered with pores with diameters ranging from 40 to 120 nm (Fig. 1b, c). ZWC (Fig. 1d, e) and ZMC (Fig. 1f, g) possessed nanorod-assembled architectures composed of interconnected nanorods ranging from 100 to 250 nm in diameter, with nanoscale crevices formed among neighboring nanorods. Compared to ZMC, ZWC exhibited a more loosely packed structure, which provided enhanced mass transport pathways through larger and more channels. The corresponding EDS elemental mapping images of ZDC (Supplementary Fig. S3) showed the uniform distribution of C, N, and O on the material surface and the absence of zinc species. The preliminary characterization results confirmed the successful synthesis of N-doped hierarchical porous carbon materials characterized by self-assembled architectures. Additionally, the distinct morphologies and assembly structures could be attributed to the differential interactions between ZIF precursors and the various solvents, with vacuum conditions amplifying these differences.

Figure 1.
(a) Schematic diagram of the preparation process for ZDC, ZWC, and ZMC, in which the corresponding precursors were synthesized using N,N-Dimethylformamide (DMF), deionized water, and methanol as solvents, respectively. SEM images of (b), (c) ZDC, (d), (e) ZWC, and (f), (g) ZMC at different magnifications.
To further investigate the influence of these interactions on the self-assembly outcomes, pore structure characterization was conducted. N2 adsorption–desorption isotherms of ZDC, ZWC, and ZMC (Fig. 2a) showed type IV isotherms with H4-type hysteresis loops, indicating that ZDC, ZWC, and ZMC all possessed meso-microporous structures[28]. The BET analysis demonstrated that ZWC possessed the highest specific surface area (933 m2 g−1) and pore volume (100 m2 g−1), significantly exceeding those of ZDC (554.37, 57 m2 g−1) and ZMC (253.26, 26 m2 g−1) (Supplementary Table S1). The BJH pore size distribution of ZDC, ZWC, and ZMC is shown in Fig. 2b. All samples exhibited a characteristic most probable pore size of ~3.7 nm, which is conducive to enhanced mass transport and improved electrolyte wettability[29,30]. Notably, ZDC and ZWC displayed an additional pore distribution in the 5–10 nm range, which was not observed in ZMC. Non-local density functional theory (NLDFT) further confirmed that ZDC and ZWC possess additional mesoporous channels beyond the 5 nm range (Supplementary Fig. S4). The pore structure differences could be attributed to the distinct solvent environments employed during ZIF synthesis, which modulated the self-assembly and pore formation processes. The enhanced porosity and surface area of ZDC and ZWC provided more active sites, while larger mesopores improved site accessibility, mass transfer, and discharge product accommodation, thereby improving cathode capacity and discharge performance[31−33]. Raman spectra revealed comparable ID/IG ratios of ZDC, ZWC, and ZMC, suggesting similar graphitization degrees and structural defect levels (Fig. 2c). Thus, electronic conductivity was not the primary factor affecting the rate performance of the three electrodes[34−37].
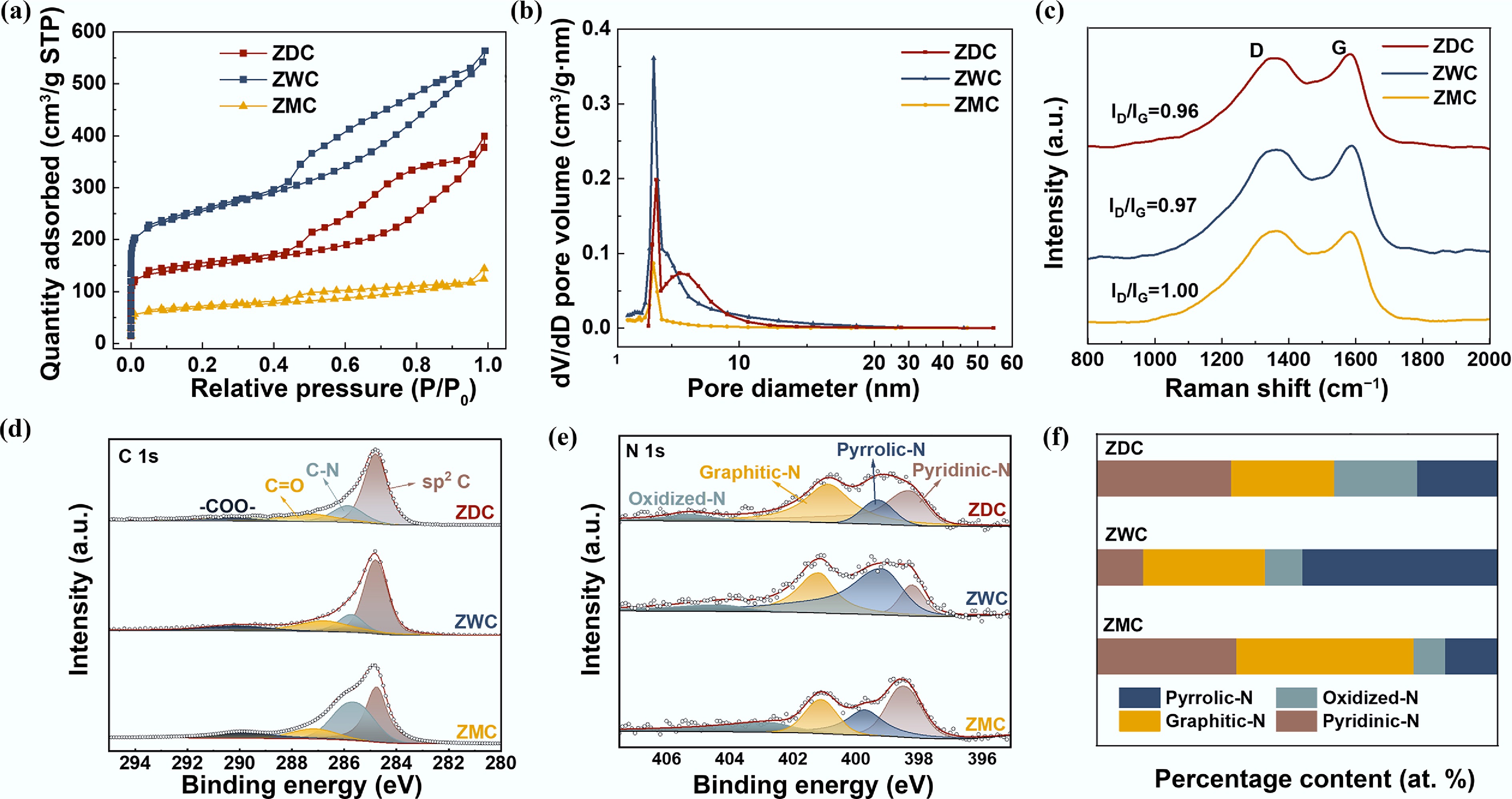
Figure 2.
Characterization of ZDC, ZWC, and ZMC. (a) N2 adsorption–desorption isotherms. (b) Pore size distributions. (c) Raman spectra, high-resolution XPS spectra of (d) C 1s, (e) N 1s, and (f) corresponding N species content.
The distinct chemical environments of ZDC, ZWC, and ZMC were investigated by XPS analysis. Supplementary Fig. S5 reveals N doping in ZDC, ZWC, and ZMC carbon frameworks and the absence of zinc species, consistent with the EDS results. The high-resolution C 1s spectra (Fig. 2d) were deconvoluted into four peaks at 284.8, 285.9, 287.3, and 289.9 eV, corresponding to the sp2 C, −C−N, −C=O, and −COO− bonds, respectively[38,39]. Supplementary Table S2 reveals comparable nitrogen contents of ZDC (5.53 at.%), ZWC (6.52 at.%), and ZMC (5.74 at.%), suggesting similar nitrogen functional group concentrations. As shown in Fig. 2e, high-resolution N 1s spectra were fitted into four components: pyridinic-N (398.8 eV), pyrrolic-N (399.2 eV), graphitic-N (400.8 eV), and oxidized-N (405.0 eV)[40,41]. Figure 2f and Supplementary Table S3 shows the corresponding nitrogen species distribution in three samples. ZDC contained 34.97% pyridinic-N, 12.98% pyrrolic-N, 44.15% graphitic-N, and 7.90% oxidized-N. ZWC exhibited 11.73% pyridinic-N, 48.54% pyrrolic-N, 30.34% graphitic-N, and 9.39% oxidized-N, while ZMC showed 33.66% pyridinic-N, 20.66% pyrrolic-N, 25.63% graphitic-N, and 20.66% oxidized-N. Notably, ZDC and ZMC, which can be derived from precursors synthesized in solvents (DMF or methanol) with stronger coordination ability and a lower dielectric constant than water, exhibited higher surface pyridinic-N content, in contrast to ZWC, which was rich in pyrrolic-N. Previous studies[38,42,43] have demonstrated that pyridinic-N enhances oxygen reduction reaction (ORR) activity by inducing high spin and charge density on surrounding carbon atoms, resulting in high electron-donating capability, reduced energy barriers, and lower charge transfer resistance at the cathode triple-phase interface. Therefore, the microstructural construction and optimization of surface catalytically active sites in N-doped porous carbon materials for enhanced ORR/OER catalysis were achieved by tailoring the synthesis solution environment.
Electrochemical performance
-
To evaluate the electrochemical performance, Li−O2 batteries were assembled using ZDC, ZWC, and ZMC as cathodes, respectively (Li | 1 M LiTFSI/TEGDME | ZDC-O2 cell, Li | 1 M LiTFSI/TEGDME | ZWC-O2 cell, Li | 1 M LiTFSI/TEGDME | ZMC-O2 cell). The cyclic voltammogram (CV) curves (Fig. 3a) revealed that ZDC exhibited more negative oxidized peaks (Epa1 ~3.29 V, Epa2 ~4.17 V), and a more positive reduction peak (Epc ~2.50 V), indicating enhanced ORR and OER catalytic performances. Notably, compared to ZDC and ZMC, ZWC exhibited only one significant oxidation peak (~3.29 V), which could be attributed to the low content of pyridinic-N moieties and relatively lower OER catalytic kinetics[44,45]. However, galvanostatic charge-discharge tests at 100 mA g−1 (Fig. 3b) demonstrated a significantly higher discharge capacity of ZDC (21,395 mAh g−1) and ZWC (13,719 mAh g−1) compared to ZMC (3,225 mAh g−1). The unexpectedly low specific capacity stems from the restricted mesopore dimensions and lower specific surface area of ZMC, which are critical factors determining Li–O2 battery discharge capacity[46,47].
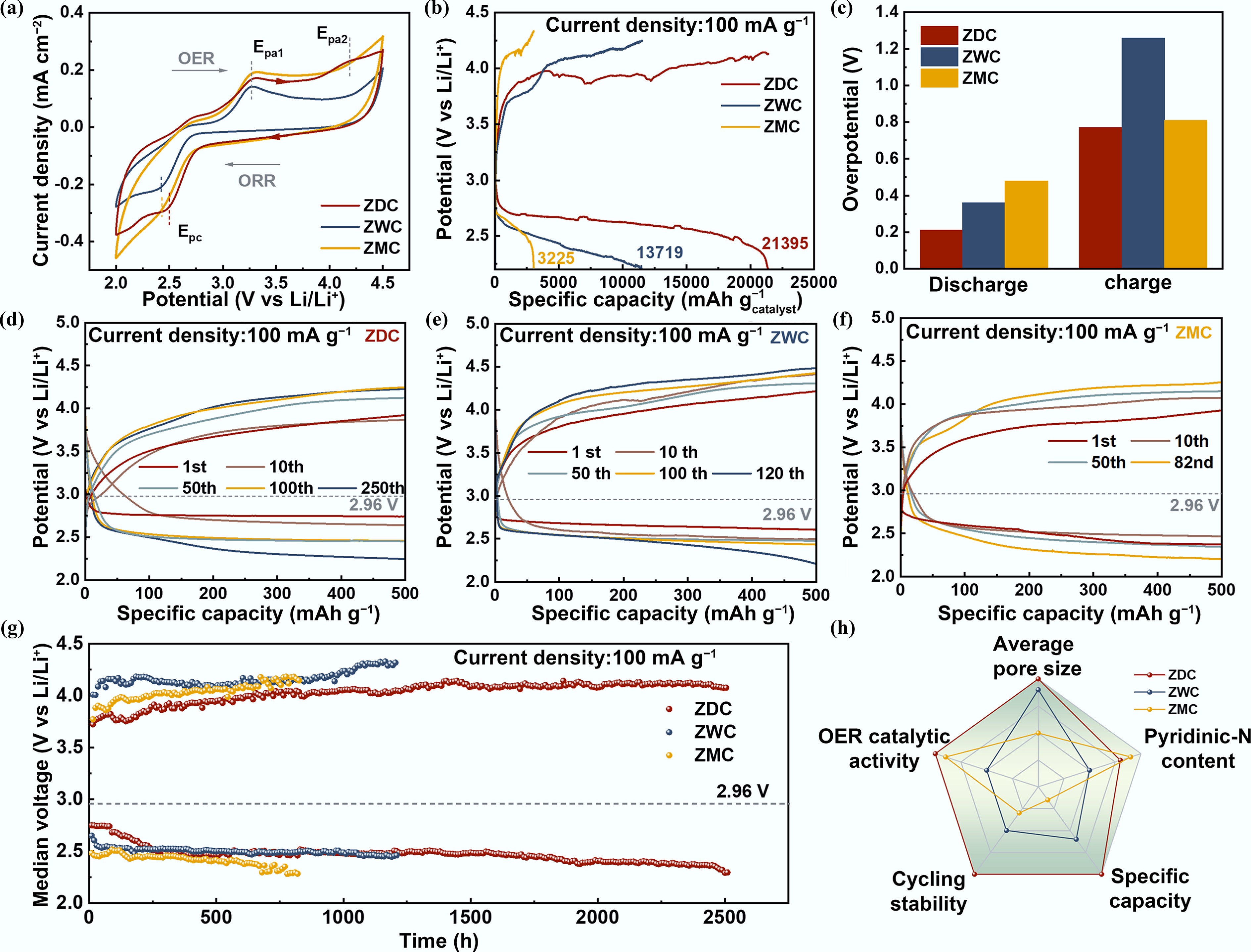
Figure 3.
Electrochemical performance of Li–O2 batteries with ZDC, ZWC, and ZMC cathodes. (a) Cyclic voltammetry curves. (b) Galvanostatic discharge-charge curves (at 100 mA g−1). (c) Comparison of discharge and charge overpotentials at the fifth cycle of LOBs with ZDC, ZWC, and ZMC cathodes. The selected cycling curves of LOBs with cathode (d) ZDC, (e) ZWC, and (f) ZMC. (g) The Medium voltage-time curves of LOBs with ZDC, ZWC, and ZMC cathodes. (h) A radar map comparing the materials properties and electrochemical performances of LOBs with ZDC, ZWC, and ZMC cathodes.
Figure 3c presents the discharge and charge overpotentials of ZDC, ZWC, and ZMC after five cycles, revealing distinct polarization characteristics among the three samples. Notably, ZDC exhibited the lowest discharge and charge overpotentials (0.21 and 0.77 V). This outstanding performance is primarily attributed to its superior ORR and OER electrocatalytic activities, which stem from enhanced mass transport enabled by additional large-size mesopores (6–30 nm) and the intrinsically high ORR catalytic activity conferred by its pyridinic-N-rich surface. ZWC and ZDC demonstrated discharge overpotentials of 0.36 and 0.48 V, representing intermediate and maximum polarization degrees among the three samples, respectively. The reduction of discharge overpotential for ZDC and ZWC could be attributed to additional mesopores and channels, leading to enhanced mass transfer[23,48,49]. In contrast to the discharge overpotentials, the charge overpotentials followed a reversed order. ZWC exhibited the highest charge overpotential (1.26 V), while ZMC showed an intermediate value (0.81 V), nearly identical to that of ZDC. This inverse relationship with surface pyridinic-N content indicates that higher pyridinic-N concentration promotes OER kinetics, thereby reducing charge polarization. The observed trend aligns well with previous reports, further confirming the pivotal role of pyridinic-N in enhancing OER activity[43,50,51]. During the charging process, the decomposition of Li2O2 was significantly enhanced by the pyridinic-N moiety, which functioned as electron-rich Lewis-base sites to boost the activation of Li2O2, resulting in accelerated OER reaction kinetics[52]. Figure 3d–f shows the selected charge-discharge curves of ZDC, ZWC, and ZMC. The discharge and charge overpotentials of the three samples at selected cycle numbers revealed a consistent performance trend with Fig. 3c, further validating the crucial influence of large mesopores in facilitating discharge reaction kinetics, as well as the function of pyridinic-N as active sites for enhancing charge reaction kinetics in LOBs. The charge overpotential of ZDC was slightly lower than that of ZMC, which may be associated with improved active-site accessibility and more efficient mass transport during the charging process.
To further investigate the effects of porosity and pyridinic-N content on discharge/charge polarization and the resulting cycling life, we compared the cycling performance and overpotential evolution during cycling. Figure 3g shows the cycling stability of ZDC (over 2,500 h), ZWC (1,250 h), and ZMC (800 h) at a current density of 100 mA g−1 and a fixed capacity of 500 mA h g−1. Based on the evolution of the median voltage during cycling, the stability of ZWC and ZMC was limited by high charge overpotential and low capacity, which led to the irreversible decomposition of the discharge product and the passivation of the cathode. Figure 3h evaluates the electrochemical performance and material properties of ZDC, ZWC, and ZMC. In summary, the superior stability and minimal overpotentials of ZDC can be attributed to the synergetic effects of its hierarchical mesoporous architecture and abundant pyridinic-N sites. Mesopores exceeding 6 nm in diameter significantly enhanced the cycling stability by providing abundant channels that facilitated efficient O2 and Li+ transport, while simultaneously boosting specific capacity by increasing the accessibility of active sites[30]. Simultaneously, the pyridinic-N moiety optimized the charge transfer kinetics by lowering the energy barrier for Li2O2 decomposition. ZMC possessed a pyridinic-N-dominated surface and exhibited a low charge overpotential. However, its cycling life was limited to 800 h due to its restricted mesopore dimensions, characterized by the absence of large mesopores and pore sizes approaching the micropore regime (~3.7 nm). In such confined pores, the accumulation of insulating discharge products (e.g., Li2O2) readily blocks O2 and Li+ transport pathways, severely degrading battery reversibility, despite an initially low charge overpotential. Notably, although its pyrrolic-N dominated surface limited the charge reaction kinetics, ZWC exhibited more stable cycling performance than ZMC, which can be attributed to its pore architecture that mitigates cathode surface passivation by discharge products. These findings underscored the dominant role of mass transport kinetics in ORR, whereas charge transfer kinetics dominated the OER process.
The effects of porosity and pyridinic-N content on the rate performance of LOBs were further examined. Figure 4a displays the cycling performance of ZDC, ZWC, and ZMC at 200 mA g−1 with a fixed capacity of 500 mA h g−1. The charge voltages of ZDC and ZMC were remarkably lower than those of ZWC, confirming the excellent catalytic activity of pyridinic-N[51]. Despite ZMC having more pyridinic-N sites than ZDC, ZDC presented a longer cycling life. The charging overpotentials of ZMC were lower than those of ZDC for the first eight cycles, which originated from its massive catalytic sites, pyridinic-N. However, after a few cycles, the overpotentials of ZMC increased and exceeded those of ZDC, which could be ascribed to the progressively deteriorating mass transport kinetics and accessibility of the active sites of ZMC as cycling proceeded. Thus, large pore size and abundant pyridinic-N sites imparted ZDC with both optimized catalytic activity and enhanced catalytic active-site accessibility, which improved both charging kinetics and cycling reversibility. As current density increased, discharging and charging overpotentials of ZDC increased slightly (both by less than 100 mV), indicating its excellent high-rate cycling performance (Fig. 4b). LOB with ZDC cathode delivered a stable high-rate cycling performance at 1,000 mA g−1 (Fig. 4c). Furthermore, ZDC exhibited optimized charging kinetics and cycling performance among LOBs with carbon-based cathodes, and even surpassed metal-coated carbon cathodes (Fig. 4d and Supplementary Table S4). The synergistically improved performance of ZDC originated from its optimized mass transport and charge transfer kinetics, which mitigated discharging-charging overpotentials and enhanced high-rate cycling durability.
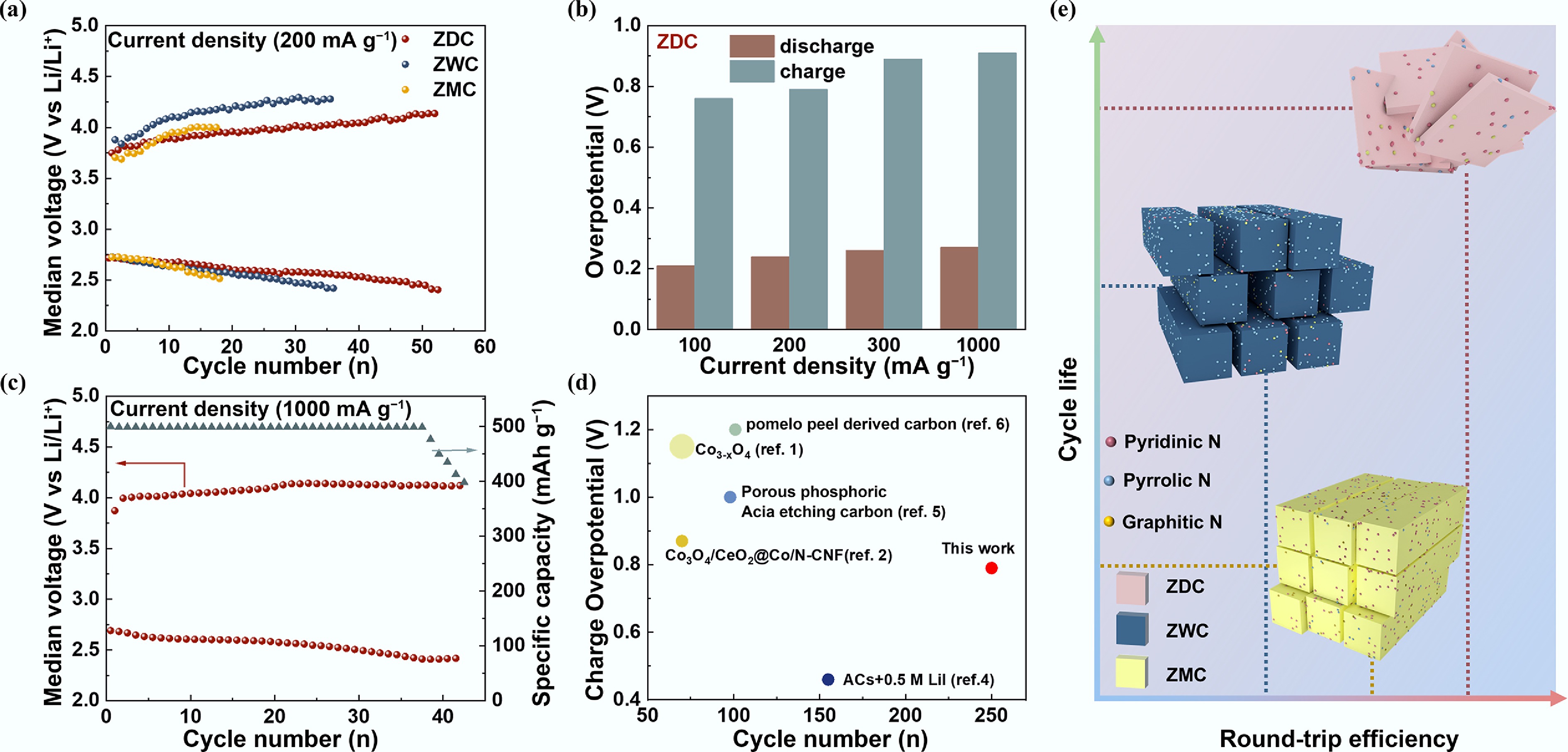
Figure 4.
Overpotentials and cycling performance of ZDC, ZWC, and ZMC cathodes under different current densities. (a) Median voltage–cycling number curves of LOBs with ZDC, ZWC, and ZMC cathodes at 200 mA g−1 and a limited capacity of 500 mA h g−1. (b) Median voltage–cycling number curve at 1,000 mA g−1 and a discharge depth of 500 mA h g−1 of LOB with ZDC cathode. (c) Discharge/charge overpotentials of LOB with ZDC cathode at 100, 200, 300, and 1,000 mA g−1 and a limited capacity of 500 mA h g−1. (d) Comparison of charge overpotentials and cycle number of this work with recent literature reported. (e) Schematic diagram illustrating the relative relationship between the round-trip efficiency and cycling life of LOBs with ZDC, ZWC, and ZMC cathodes.
To further elucidate the relationship between material properties and LOB performance, we illustrated the correlations among cycling life, round-trip efficiency, and carbon materials with distinct porous structures and N moieties. Figure 4e illustrates the correlation between the structural and compositional features of ZDC, ZWC, and ZMC and key electrochemical performance metrics of LOBs. ZWC possessed a low pyridinic-N content and large pore size, exhibiting the lowest round-trip efficiency and a moderate cycling life. Endowed with numerous pyridinic-N active sites, ZMC exhibited enhanced catalytic activity, resulting in higher round-trip efficiency. However, the cycling life of ZMC was limited by insufficient mass transport due to restricted pore dimensions. LOB with the ZDC cathode achieved a concurrent enhancement in round-trip efficiency and cycling life. ZDC possessed an optimized catalytic activity and pore structure. These features led to a synergetic effect of abundant active sites and enhanced mass transport kinetics, thereby improving the passivation resistance of the highly efficient active sites. Consequently, the ZDC cathode enabled an increased round-trip efficiency and prolonged cycling life.
Electrochemical mechanism
-
The synergistic effects of N moieties and porous structures were examined by analyzing the composition and morphology of the discharge products, which appeared to significantly influence the discharge and charge behaviors of LOBs. Figure 5a–c shows the morphologies of the discharge products of ZDC, ZWC, and ZMC, respectively. XPS of the high-resolution Li 1s spectra confirmed that the domain discharge products were Li2O2 (Fig. 5d). No detectable amounts of side products (e.g., Li2CO3, LiOH) were detected, demonstrating that the discharge capacity was primarily attributed to the electrons produced during the Li2O2 formation process. Figure 5e, f presents the EIS measurements of ZDC before discharging, after discharging, and after charging. The equivalent circuit of ZDC after discharging is depicted in the inset of Fig. 5f, corresponding to the two parallel charge-transfer paths at the electrode/Li2O2/electrolyte and Li2O2/electrolyte interfaces[19]. After charging, the charge-transfer impedance of LOB with the ZDC cathode recovered, illustrating the highly reversible decomposition of Li2O2 on the ZDC cathode. DRT analysis (Supplementary Fig. S6) provided further insights into the changes of electrochemical interfaces in the system. The τ1 region was associated with the contact resistance and the resistance of the SEI layer to Li+ transport. τ2 and τ3 in the range of 10−4–10−1 s were attributed to the Rct of the electrode. After discharge (Stage II), a prominent peak appeared at longer relaxation times (τ3 region), indicating sluggish interfacial charge transfer kinetics of Li2O2. This peak intensity decreased after charging, indicating enhanced kinetic pathways as the Li2O2 interface decomposed. τ4 (~10−1–100 s), which was typically associated with oxygen diffusion and electrolyte-mediated species transport, became prominent after discharge (Stage II), indicating strong transport-related impedance growth induced by Li2O2 accumulation within the porous electrode. The increased τ4 intensity after discharge suggested pore blockage, whereas its decrease after charging reflected the partial recovery of mass transport pathways as Li2O2 decomposed. The concurrent evolution of τ4 with τ2 and τ3 indicates a coupled involvement of charge transfer and mass transport processes[53]. The reversible decomposition of the discharge product accounted for the highly stable cycling performance of ZDC.
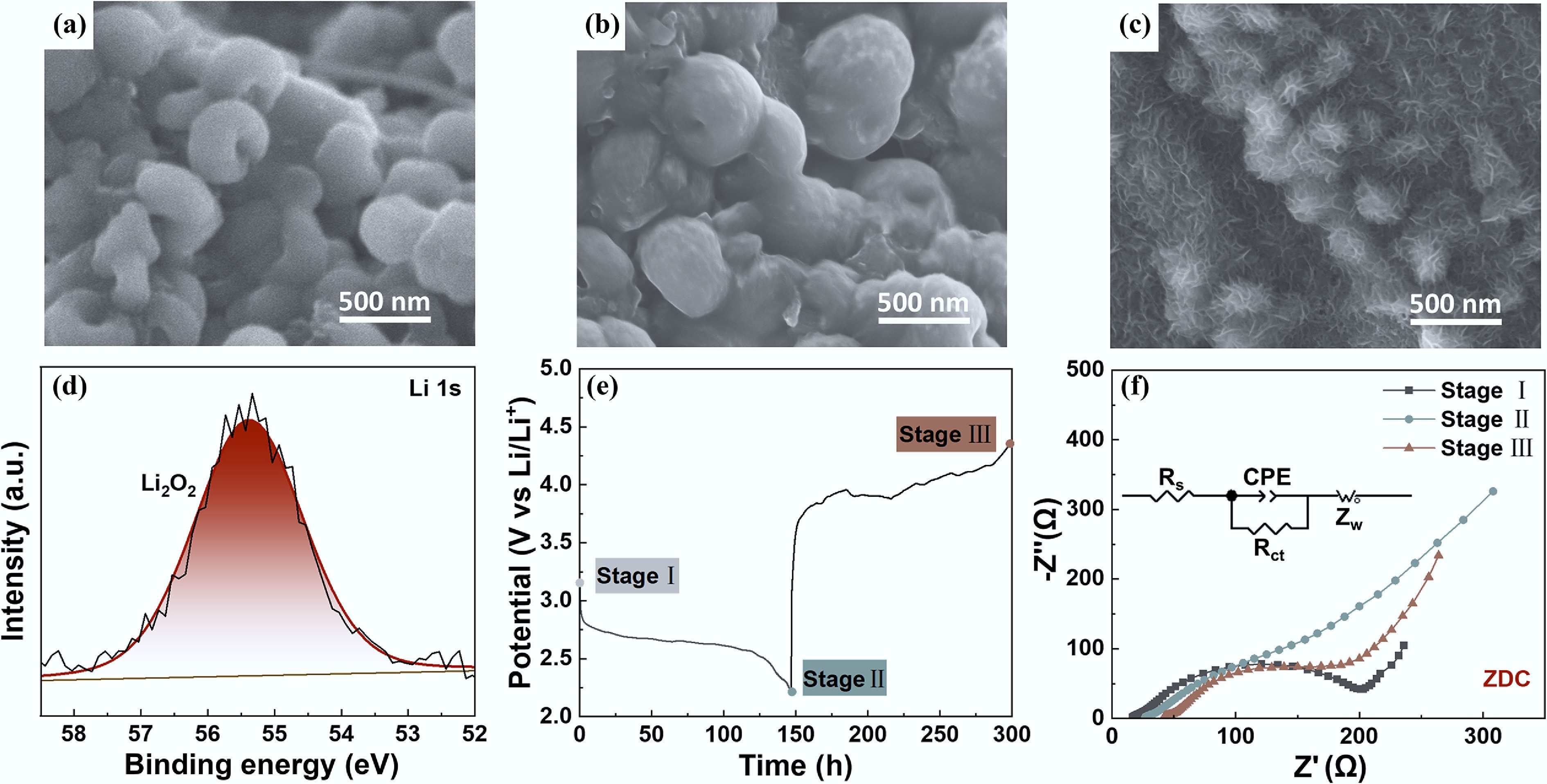
Figure 5.
SEM images of (a) ZDC, (b) ZWC, and (c) ZMC after first discharge. (d) High-resolution XPS spectrum of Li 1s of the ZDC cathode after first discharge. (e) The galvanostatic discharge-charge curve and corresponding Nyquist plots before discharge (stage I), after discharge (stage II), and after charge (stage III) (inset: the equivalent circuit model of stage II).
The morphologies of the discharge products on ZDC, ZWC, and ZMC cathodes exhibited marked differences. ZDC and ZWC (Fig. 5a, b) exhibited toroidal Li2O2 particles, while ZMC exhibited flower-like Li2O2 particles. Both morphologies originated from the solution-based routes of discharge product nucleation[54]. The flower-like Li2O2 morphology on ZMC cathode may be attributed to the dynamic competition between solution- and surface-mediated pathways during Li2O2 growth[16,19,54,55]. As Li2O2 nuclei grew larger through the solution route, the dominant growth pathway gradually switched to the surface pathway, followed by a 2D growth model[19,23,27]. Ultimately, the ZMC discharge product exhibited a morphology of particles covered with flake-like Li2O2. The toroidal Li2O2 formed on ZDC and ZWC cathodes indicated that the solution route dominated the growth pathway of Li2O2, which was consistent with their nucleation pathway. Additionally, the slightly smaller toroidal Li2O2 on ZDC cathode may be attributed to the enhanced ORR catalytic activity of ZDC, resulting from its high content of pyridinic-N species[42]. The enhanced catalytic activity facilitated the charge transfer kinetics during discharge, promoting a faster nucleation rate of Li2O2. Therefore, the discharge product particle size on ZDC was comparatively smaller than that on ZWC. Theoretically, the decomposition of flower-like Li2O2 is favored to occur at a lower overpotential due to its loose structure and greater contact with the cathode. The decomposition of toroidal Li2O2 typically requires a higher charge overpotential, resulting from the insulating nature of bulk Li2O2. However, ZDC with toroidal Li2O2 exhibited an optimized performance with reduced charge polarization and prolonged cycling life, surpassing the performance of ZMC with flower-like Li2O2, which seemed to be inconsistent with previous studies[19,56−58].
The formation and decomposition processes of the discharge product Li2O2 are shown in Fig. 6a, b, respectively. During the formation process (Fig. 6a), a solution route was operative in ZDC, ZWC, and ZMC due to their relatively low adsorption energy of intermediates on the active sites on N-doped carbon[16]. As the Li2O2 grains increased in size and amount, inherently limited pore size distribution of ZMC caused progressively deteriorating mass transport kinetics. Additionally, ZMC experienced a higher local current density than ZDC and ZWC at the same apparent current density due to less accessible active sites. Consequently, the solution route of Li2O2 formation on ZMC shifted as discharge proceeded due to the reduced O2 transport and increased current density. The surface route became competitive and emerged as the dominant pathway due to the requirement for electron transfer, which resulted in the flower-like morphology of Li2O2. LSV measurements were further conducted to evaluate the influence of charge transfer and mass transport kinetics on the discharge process (Supplementary Fig. S7a). The ORR curves exhibited broad peaks without a well-defined limiting current plateau, suggesting mixed kinetic control involving charge transfer and mass transport, rather than a single rate-limiting process. Notably, according to galvanostatic charge-discharge and LSV tests, ZMC, with its limited pore structure, displayed markedly higher polarization. This behavior indicated that restricted pore accessibility amplifies transport limitations during discharge, leading to a process influenced by mass transport rather than being purely controlled by charge transfer. In the charging process, the effective decomposition factors were collectively determined by the charge transfer and mass transport kinetics at the two interfaces, electrode/Li2O2/electrolyte triple-phase interface and Li2O2/electrolyte interface[19], denoted as Site A and B, respectively, in Fig. 6b. ZWC, which featured toroidal Li2O2 and efficient transport channels yet possessed a low pyridinic-N content, exhibited the highest charging polarization. This indicated the dominant influence of charge transfer kinetics in the charge process. LSV measurements provided further evidence for the enhanced catalytic activity of ZDC and ZMC during the charge process (Supplementary Fig. S7b). Similar to the discharge process, no well-defined diffusion-limited plateau was observed. Instead, multiple distinct slope regions were observed below 3.5 V, which were attributed to the decomposition of Li2O2 with distinct interfaces and structures. These features indicated that the charging reaction proceeded through multi-stage interfacial oxidation rather than a single charge-transfer step. Notably, a distinct oxidation feature centered at ~3.2 V was clearly observed for ZDC and ZMC, despite their distinct Li2O2 morphologies (toroidal for ZDC and flower-like for ZMC), whereas ZWC displayed a much weaker response in the same potential region. This interfacial behavior suggested that a favorable local electronic environment, imparted by pyridinic-N sites, played a crucial role during the early-stage decomposition of Li2O2. Consequently, this enhanced early-stage oxidation is closely linked to the reduction of charge polarization. ZDC exhibited a lower charging potential than ZMC, owing to its superior active site accessibility and more efficient mass transport. The limited charging performance of ZMC compared with ZDC was attributed to its lower electrochemically active surface area, which was corroborated by its lowest current density in the OER LSV test.
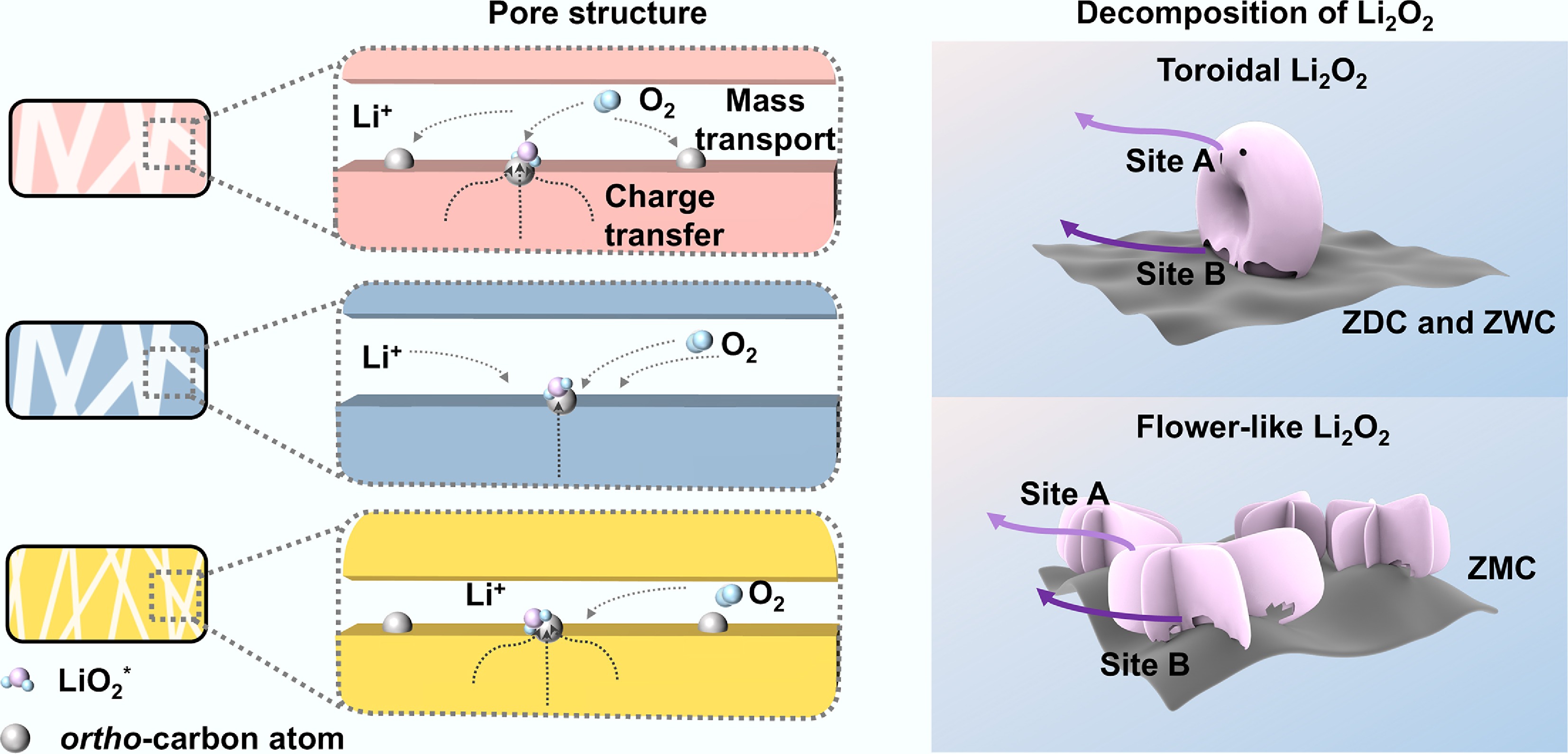
Figure 6.
Coupled effects of mass transport and charge transfer on discharge product morphologies and subsequent decomposition processes. (a) Schematic illustration of the differences in the electrochemical process on the cathode surface of ZDC, ZWC, and ZMC. (b) Different decomposition mechanisms of discharge products Li2O2 with toroidal and flower-like morphologies.
-
In summary, this work developed a metal-free N-doped carbon catalyst for Li–O2 batteries via a dual-modulated synthesis strategy that integrated solvent-mediated nitrogen functionalization with vacuum-assisted heat treatment. This approach enables the co-regulation of two pivotal properties: (1) catalytically active N moieties (primarily pyridinic-N) that contribute to charge transfer kinetics, (2) a microscale-tunable pore architecture that regulates mass transport. The synergy between these optimized transport and kinetics promotes the formation of a toroidal Li2O2 discharge product with enhanced decomposition efficiency, thereby breaking the trade-off between specific capacity and round-trip efficiency. The optimal cathode ZDC featuring additional large pore size (exceeding 6 nm) and high content of pyridinic-N species (~37%) delivers a super high specific capacity of 21,396 mAh g−1, low charge overpotential of 0.76 V (100 mA g−1) and 0.90 V (1,000 mA g−1). This study establishes a versatile materials design platform and provides a promising strategy for probing the dynamic structural and chemical evolution of porous carbon-based cathodes under operating conditions.
The use of the Swagelok cell was supported by Cunzhong Zhang at the Beijing Institute of Technology.
-
It accompanies this paper at: https://doi.org/10.48130/scm-0026-0016.
-
The authors confirm their contributions to the paper as follows: Xinrong Hu: investigation, methodology, formal analysis, writing-original draft; Meiling Wang: conceptualization, resources; Feiyang Yang: conceptualization, resources; Zhaolin Gou: writing-review, editing; Ziyi Chen: data curation; Cunzhong Zhang: funding acquisition, supervision; Yuefeng Su: funding acquisition, supervision; Ying Yao: funding acquisition, supervision. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
This work was supported by the National Natural Science Foundation of China (NSFC No. 22579017).
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
Full list of author information is available at the end of the article.
- The supplementary files can be downloaded from here.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Hu X, Wang M, Yang F, Gou Z, Chen Z, et al. 2026. Breaking the capacity-polarization trade-off in Li–O2 batteries through dual-modulation of hierarchical porosity and active site configurations in ZIF-derived electrocatalysts. Sustainable Carbon Materials 2: e021 doi: 10.48130/scm-0026-0016 

Breaking the capacity-polarization trade-off in Li–O2 batteries through dual-modulation of hierarchical porosity and active site configurations in ZIF-derived electrocatalysts
- Received: 01 January 2026
- Revised: 18 February 2026
- Accepted: 26 March 2026
- Published online: 21 April 2026
Abstract: Lithium–oxygen (Li–O2) batteries offer high theoretical energy density among battery systems, yet their practical application is hindered by poor cycling stability, low round-trip efficiency, and poor high-rate performance. These limitations originate from the insulating and insoluble nature of the discharged Li2O2, which progressively accumulates in electrode pores, passivates the active surface, and severely deteriorates both mass transport and charge-transfer kinetics during cycling. Herein, we report a metal-free N-doped porous carbon catalyst synthesized via a novel dual-modulation strategy that integrates solvent-mediated surface functionalization with vacuum-assisted thermal treatment, enabling the precise regulation of surface nitrogen species and micropore architecture. The modulated micropores and nitrogen species predominantly influence mass transport and charge-transfer processes, respectively, allowing targeted investigation of their synergistic effects on Li2O2 formation and subsequent decomposition. The optimized cathode, featuring large pore sizes (3–30 nm) and high pyridinic-N content (~37%), delivers an ultrahigh specific capacity (21,395 mAh g−1) and a prolonged cycling life of 2,500 h at 100 mA g−1. Moreover, it promotes the toroidal Li2O2 formation and a remarkably low charge overpotential (0.77 V at 100 mA g−1), which is lower than that associated with the conventional flower-like Li2O2 (0.81 V), despite the latter being widely reported to exhibit a lower decomposition overpotential than toroidal Li2O2. Consequently, the optimized electrode effectively breaks the capacity-polarization trade-off typically associated with toroidal discharge products. This work provides a generalizable synthetic and design framework for systematically investigating the influence of dynamically evolving porosity and active sites on discharge products and Li–O2 battery performance.