






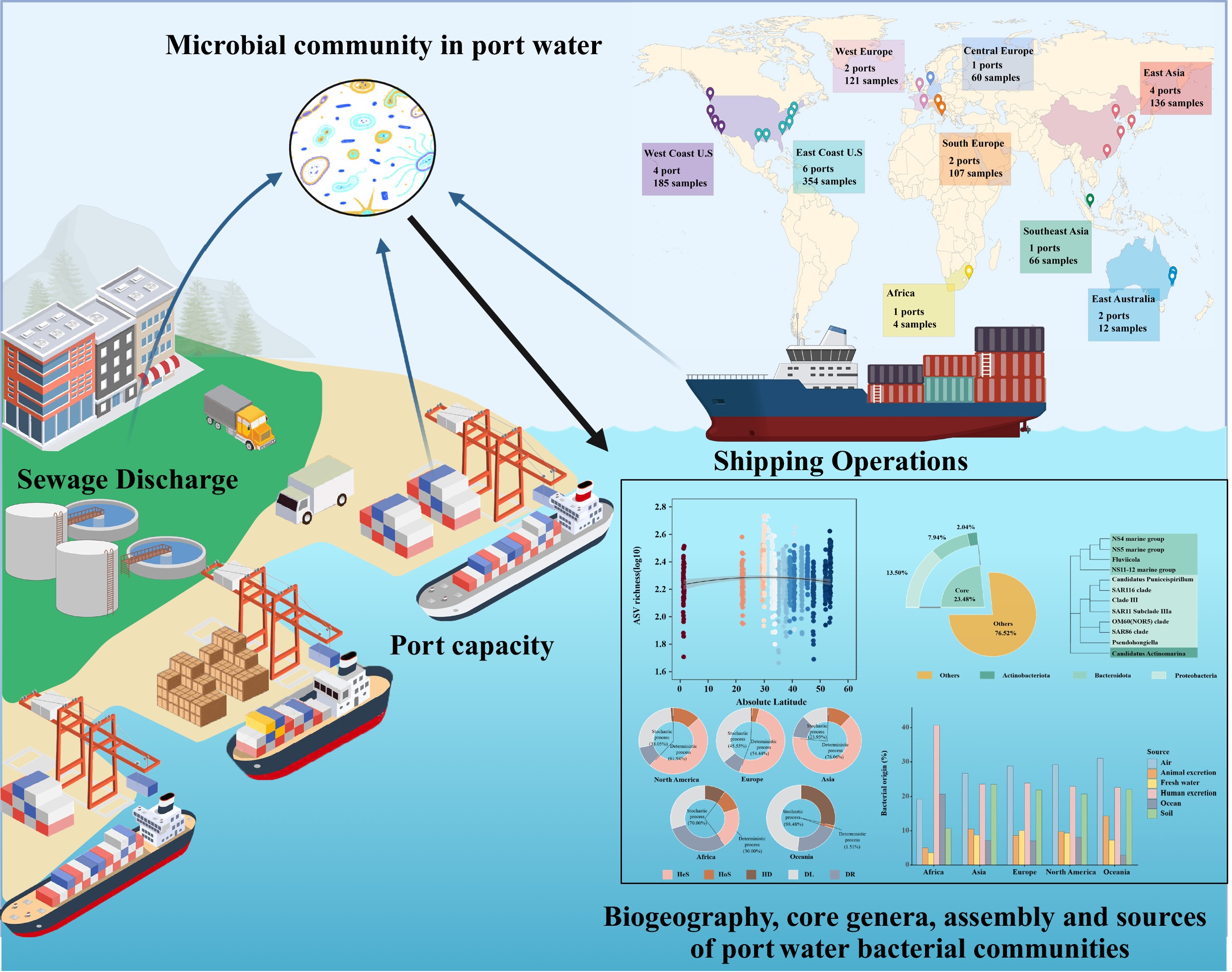



-
The globalization process has led to remarkable growth in trade between countries over the centuries. Over 80% of the international merchandise trade is transported via maritime routes[1]. Ports are recognized as crucial nodes in maritime logistics, playing a vital role in global trade[2]. However, increasing anthropogenic pressures from coastal urbanization, shipping, cargo handling, and terrestrial contamination pose unprecedented challenges to port water ecosystems. Bacteria are ubiquitous in the marine environment, including port ecosystems, where they play crucial roles in biogeochemical processes such as carbon and nutrient cycling[3]. These bacterial communities play a dual and interconnected role in port ecosystems. On the one hand, they actively drive changes in local environmental factors, including salinity, nutrient levels, and dissolved oxygen levels[1]. Concurrently, these altered conditions reciprocally shape bacterial communities, enhancing their responsiveness to both natural environmental gradients and anthropogenic pressures, such as wastewater discharge. This reciprocal interaction underscores their value as sensitive indicators of ecosystem change, and their composition is essential for monitoring the health of coastal ecosystems. Thus, knowledge of bacterial diversity and biogeographic distribution patterns in ports can provide insights into biogeochemical processes and port ecosystem functions.
Previous studies have invested significant effort into examining the composition, dynamics, and functional roles of bacterial communities in coastal and port waters. For example, Proteobacteria, Cyanobacteria, Actinobacteria, Pseudomonadota, and Bacteroidetes were identified as the dominant bacterial taxa in the coastal waters of China[4]. Various environmental factors could influence the diversity of bacterial communities. Analysis of samples from the Northwest Atlantic revealed that salinity and dissolved oxygen predominantly affected bacterial diversity, whereas temperature and oxygen concentrations contributed significantly to bacterial community similarity[5]. In addition to natural factors, anthropogenic activities are another critical factor influencing bacterial community structure in coastal ecosystems[6]. Dai et al. revealed that effluent discharged from wastewater treatment plants significantly reduces the diversity and network interactions of the bacterial community in coastal waters[7]. Meanwhile, the detection of human fecal bacteria in port and coastal waters further confirms pollution caused by human activities[8]. This contamination likely originates from untreated or inadequately treated sewage discharge. In such contaminated environments, pathogens such as Vibrio cholerae and Vibrio parahaemolyticus have been detected in tropical port waters[9]. These bacteria are recognized as causative agents of serious gastrointestinal illnesses in humans. Notably, pathogens present in ports can be transported globally via the ballast water of ships[10]. Nevertheless, existing knowledge remains largely constrained to small-scale analyses of port or coastal waters in specific regions[11,12]. Recent global-scale studies have identified the dominant taxa and revealed significant regional variations in port waters[13]. Moreover, shipping activity may represent a major yet consistently overlooked factor influencing the port water microbiome[14]. However, a comprehensive understanding of the overall characteristics of bacterial communities in port water and their interactions with anthropogenic activities remains insufficient. Addressing this gap is essential for understanding the biogeographic distribution of port water microbiomes and establishing a scientific foundation for port ecosystem management.
With recent advances in high-throughput sequencing technologies, microbial features have been intensively studied across a diverse range of environments, including the human gut[15], wastewater treatment plants[16], air[17], soil[18], lake[19], drinking water[20], and oceans[21]. These studies revealed unique microbiomes in each ecological habitat, where microbial biodiversity and community structures often exhibit geographic distribution patterns[22]. Port ecosystems experience dual stress from terrestrial and shipping-derived anthropogenic activities at the human-marine interface. Nevertheless, a global perspective on how anthropogenic activities alter these communities and the underlying mechanisms remains incomplete. Although previous studies have verified the presence of pathogenic bacteria in diverse ports[9], it remains largely unclear whether geographical patterns and core bacteria or pathogens exist in port water worldwide. Addressing these knowledge gaps is crucial both for evaluating the role of ports as potential hotspots in the global dissemination of microbes, and for developing effective monitoring and management strategies.
To address these knowledge gaps, a comprehensive dataset was compiled by collecting port water samples from 23 ports across eight countries on five continents. First, the biodiversity and distribution of the bacterial community was investigated, and core bacteria and potential pathogens were identified in port water worldwide. Second, the assembly mechanisms shaping bacterial community composition were investigated. Finally, the relationships among the port water bacterial community, pathogens, and various anthropogenic sources were examined. The findings from this study systematically clarify the composition and distribution characteristics of port bacterial communities, providing essential data to support the protection and sustainable development of the port ecosystem.
-
To provide a global perspective on the port water bacterial community, 16S rRNA gene sequence data were collected from publicly available datasets at the National Center for Biotechnology Information (NCBI,
https://www.ncbi.nlm.nih.gov ) and the European Nucleotide Archive (ENA,https://www.ebi.ac.uk/ena ) based on the references[13,23,24]. To ensure data quality and consistency, the following filtering criteria were applied: (i) Only raw paired-end sequencing reads in FASTQ format were included for analysis, while single-end sequencing data were excluded. (ii) The 16S rRNA V3~V4 region generated exclusively by the Illumina sequencing platform was retained due to its maturity and widespread application. (iii) High-quality sequences (above 400 bp) were retained for subsequent analysis. Samples lacking metadata (e.g., sample details and geographic coordinates) were removed to ensure dataset integrity and reliability.Evidence increasingly indicates that anthropogenic activities profoundly shape aquatic microbial communities across a broad spectrum of ecosystems[25,26]. For example, socioeconomic indicators, such as wastewater discharge, GDP, and population density, exhibit potential correlations with microbial community structure[26,27]. Therefore, basic sample metadata were also collected in this study, including geographical information: continent, country, city, average annual temperature, longitude, and latitude; socioeconomic factors: population density (PD), gross domestic product (GDP), population growth rate (PGR), sustainable development goals index (SDGI), human development index (HDI), wastewater discharge (WWD), environmental performance index (EPI); marine and maritime related indices: port capacity, port liner shipping connectivity index (PLSCI), marine protected area (MPA), marine trophic index (MTI), fish stock status (FSS). All data were retrieved from the World Bank (worldbank.org), NASA Earthdata (earthdata.nasa.gov), UNCTAD (unctad.org), and the maritime authority websites of the respective countries or regions. Port capacity was classified based on the global container throughput ranking. Ports within the top 20 were classified as 'high-capacity', while those ranked from 21st to 100th were designated as 'low-capacity'. Samples were categorized by geographic latitude: high (> 50° N or > 40° S), middle (20° S−40° S, 30° N−50° N), and low (20° S−30° N)[28,29]. Details of the samples are described in Supplementary Tables S1 and S2.
Processing of sequencing reads
-
All 16S rRNA gene sequencing data were analyzed using Quantitative Insights into Microbial Ecology (QIIME2 v. 2022.8). Primer sequences were first removed using Cutadapt[30]. Sequence quality control and feature table construction were performed using the DADA2 denoise-paired pipeline[31]. The reads with a quality score < Q20 were removed. The remaining reads were denoised to generate amplicon sequencing variants (ASVs). Taxonomy classification was performed using a Naive Bayes classifier trained on the SILVA database (v. 138.1) via QIIME's feature-classifier classify-sklearn[32]. This method leverages the comprehensive and curated reference sequences in the SILVA database to accurately assign taxonomic labels to ASVs. ASVs assigned to Archaea and singletons were excluded from subsequent analyses. The ecologically relevant functions of the bacterial community in port water were predicted using the Functional Annotation of Prokaryotic Taxa (FAPROTAX)[33]. Pathogens were screened against a comprehensive database of known pathogenic sequences using the multiple bacterial pathogen detection pipeline (
https://github.com/LorMeBioAI/MBPD ). This pipeline is designed to detect multiple bacterial pathogens simultaneously and categorize them into distinct classes (animal, plant, and zoonotic pathogens) based on their potential impact[34].Core bacteria identification
-
In this study, core bacterial taxa were identified based on their abundance and occurrence frequency within the sampled regions. Firstly, ASVs exhibiting an occurrence frequency > 60% across all samples were retained to form the widely distributed bacterial community. Then, taxa with a relative abundance > 0.1% within this group were classified as the core bacterial community[16].
Assembly processes and source tracker analysis
-
The null model analysis was conducted using the 'picante' package in R to measure the ecological processes shaping the bacterial community. The assembly processes were quantified using phylogeny-based metrics (β-nearest taxon index, βNTI) and taxonomic β-diversity measures (based on Bray-Curtis' Raup-Crick, RCbray values). Briefly, |βNTI| > 2 indicated that a deterministic process dominated the construction of microbial communities, and could be further divided into homogeneous selection (βNTI < −2) and heterogeneous selection (βNTI > 2). |βNTI| < 2 indicated that random processes dominated the construction of microbial community, which was further divided into homogenizing dispersal (RCbray < −0.95) and dispersal limitation (RCbray > 0.95) and undominated processes (|RCbray| < 0.95)[35]. To explore the sources of bacteria in port water, Bayesian methods were employed to compare 16S rRNA sequences from port water samples (designated 'sink') with reference sequences from the Earth Microbiome Project (EMP,
ftp.microbio.me/emp/ ). The reference samples included data from a variety of environments (soil, freshwater, air, ocean, human- and animal-associated habitats)[36]. The 'SourceTracker' package in R was then applied to estimate the proportion of the bacterial community in each port water sample attributable to potential sources using the Bayesian algorithm[37].Statistical analysis
-
Alpha diversity indices were calculated using the alpha diversity.py in QIIME2. The Kruskal‒Wallis test was applied to determine significant differences in alpha diversity indices between groups of samples. To evaluate β-diversity and compositional differences in bacterial communities across port water samples, Bray–Curtis dissimilarity was calculated as the distance metric. Principal coordinate analysis (PCoA) was subsequently conducted based on the distance matrix using the vegan package in R (v. 4.1.3). Permutational multivariate analysis of variance (PERMANOVA) was employed to assess the statistical significance of observed differences. To investigate the distance-decay relationship (DDR) in bacterial community composition, linear regression of community similarity was performed as a function of geographic distance. This analysis was conducted using the geosphere package in R (v. 4.1.3)[38]. Canonical correspondence analysis (CCA) was performed using the vegan package in R (v. 4.1.3) to assess the effects of anthropogenic and environmental factors on microbial communities[39]. The contribution degrees of various factors to the microbial community structure were quantified using the Hierarchical partitioning (HP) by the rdacca.hp package in R[40]. Mantel tests were used to link the bacterial community composition with social indicators using the linkET package (v. 0.0.7.4) in R. Statistical significance was defined as a p-value of less than 0.05.
-
A total of 16,575,450 high-quality 16S rRNA gene sequences were obtained from 1,045 port water samples spanning 23 ports in eight countries (Supplementary Fig. S1), yielding 23,302 ASVs across five continents. The bacterial datasets from five continents exhibited considerable variation in the number of observed ASVs. Specifically, 3,923, 6,140, 7,581, 1,086, and 1,183 ASVs were identified in the samples from Europe, Asia, North America, Oceania, and Africa, respectively. Chao1 and Shannon indices were calculated to assess the impact of geographic location on the alpha diversity of the port water bacterial community (Fig. 1). The results revealed significant variation in α-diversity across continents and geographic latitudes. Both the Chao1 and Shannon indices were highest in samples from Africa, followed by Oceania, while the port water bacterial community from Asia demonstrated the lowest α-diversity. Additionally, samples from mid-latitudes exhibited a broader range of α-diversity values than those from low- and high-latitudes (Fig. 1).
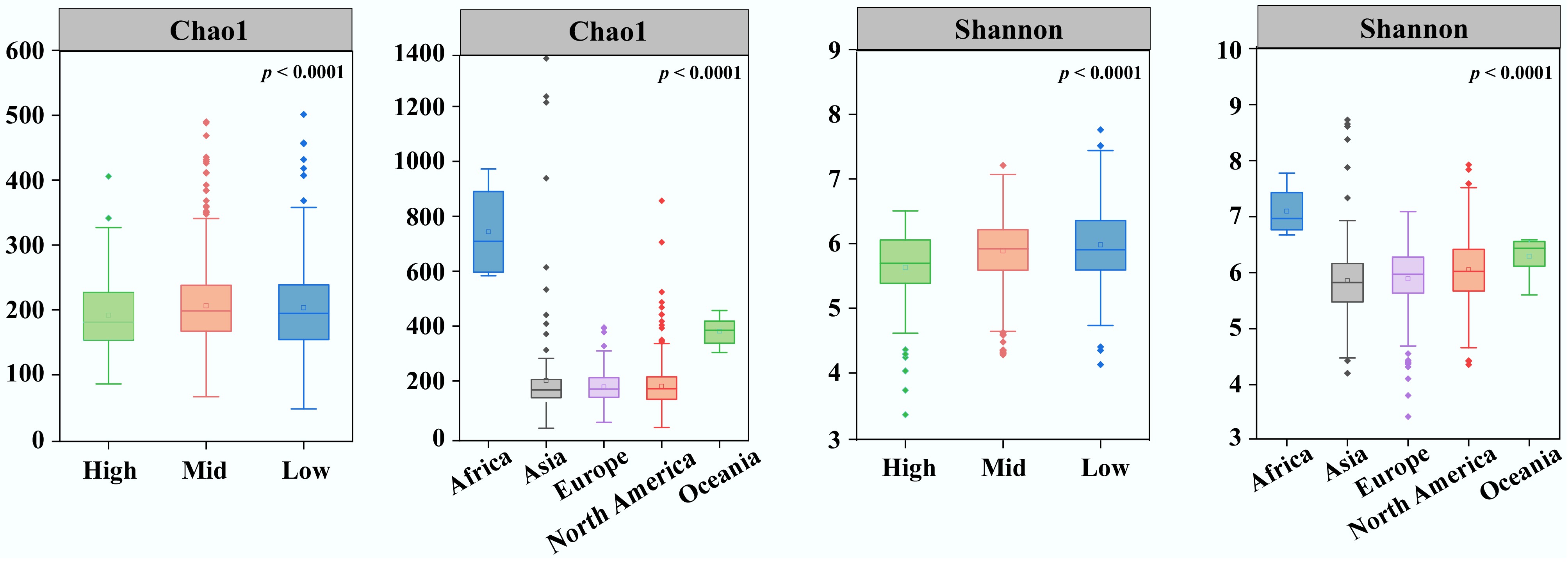
Figure 1.
Alpha diversity indices Chao1 and Shannon of port water from different latitudes and continents.
The latitudinal diversity gradient (LDG) and DDR were utilized to explore the biogeographic patterns of the port water bacterial community. Although statistically significant (p < 0.01 and p < 0.001), the observed relationships between latitude and two key metrics of bacterial diversity (ASV richness and Shannon diversity) were relatively weak (Fig. 2a, b). These results suggested that latitude accounts for a limited proportion of the total variation in diversity across the studied ports, and the typical LDG pattern was not perfectly suitable in port water bacterial communities at a global scale. Moreover, the bacterial community in port waters exhibited a significantly negative correlation between geographical distance and Bray-Curtis similarity (p < 0.0001; Fig. 2c), reflecting a clear distance-decay pattern. Notably, this distance–decay relationship varied with port capacity. Low-capacity ports exhibited a steeper decline in community similarity with geographic distance (k = –0.0439) compared to the high-capacity ports (k = –0.0295) (Supplementary Fig. S2). Analysis of community structure (β-diversity) is essential for understanding the biogeographic distribution of global port water bacteria. Our result revealed that bacterial communities in port water differed significantly across continents based on Bray-Curtis similarity (p < 0.01; Fig. 2d). Despite being located on the same continent, bacterial communities in the US East Coast (Atlantic) and West Coast (Pacific) ports exhibited significant differences (PERMANOVA, p < 0.05), reflecting the distinct oceanographic regimes (Fig. 2d). Furthermore, bacterial communities in port waters also varied significantly across latitudinal gradients (Supplementary Fig. S3).
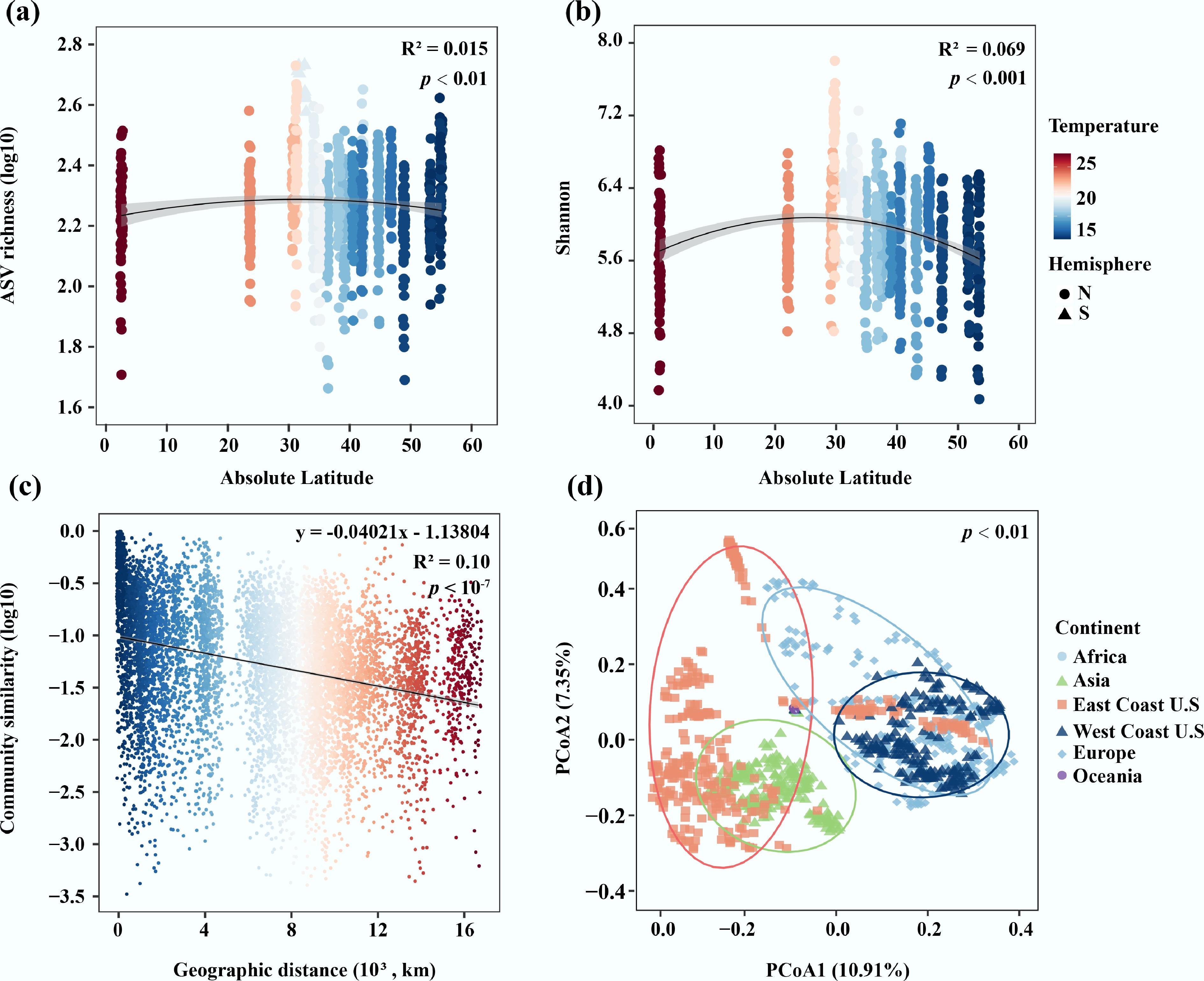
Figure 2.
(a) Relationship between ASV richness and absolute latitude in port water. (b) Relationship between Shannon index and absolute latitude in port water. (c) The DDRs based on Bray-Curtis similarity of port water bacterial community. (d) The β-diversity based on the Bray-Curtis distance of the bacterial community in port water.
A total of 45 phyla were identified in the port water bacterial community. Despite geographical variation, the dominant phyla were consistent across continents. Proteobacteria dominated all sampled regions (relative abundance, 37.2%−57.5%), with the highest value in samples from Asia (Fig. 3a). Bacteroidota and Actinobacteriota were also prominent components in port water. Notably, Nitrospirota was detected exclusively in North American and Asian samples, while Armatimonadota was restricted to European samples (Supplementary Table S3). Likewise, bacterial compositions also varied with latitude. Cyanobacteria showed higher abundance in low- (12.5%) and mid-latitude (11.8%) than in high-latitude (0.3%) ports (Fig. 3c). SAR324 showed a strict mid- and low-latitude distribution. At the same time, Armatimonadota was confined to high latitudes (Supplementary Table S4). At the genus level, 551, 716, 258, 740, and 338 bacterial genera were detected in Europe, North America, Oceania, Asia, and Africa, respectively. The distribution of bacterial genera also varied both across continents and with latitude. Among them, Cyanobium PCC-6307 (10.6%), SAR11 subclade IIIa (9.6%), NS5 marine group (4.9%), Planktomarina (3.7%), and HgcI clade (3.2%) were the dominant taxa in the port water samples from North America, Europe, Asia, Oceania, and Africa, respectively (Supplementary Fig. S4a). Several bacterial genera exhibited continent- and latitude-specific distributions. Pseudomaricurvus, Tateyamaria, and Microbacteriaceae DS001 were detected exclusively in European samples. Litoribrevibacter and Bermanella were solely present in Asian samples (Fig. 3b). Cyanobium PCC-6307 was the dominant bacterial genus in port water samples from mid- (11.0%) and low-latitude (14.7%). In comparison, Planktomarina dominated in high- (7.2%) and mid-latitude (4.5%) samples (Supplementary Fig. S4b).
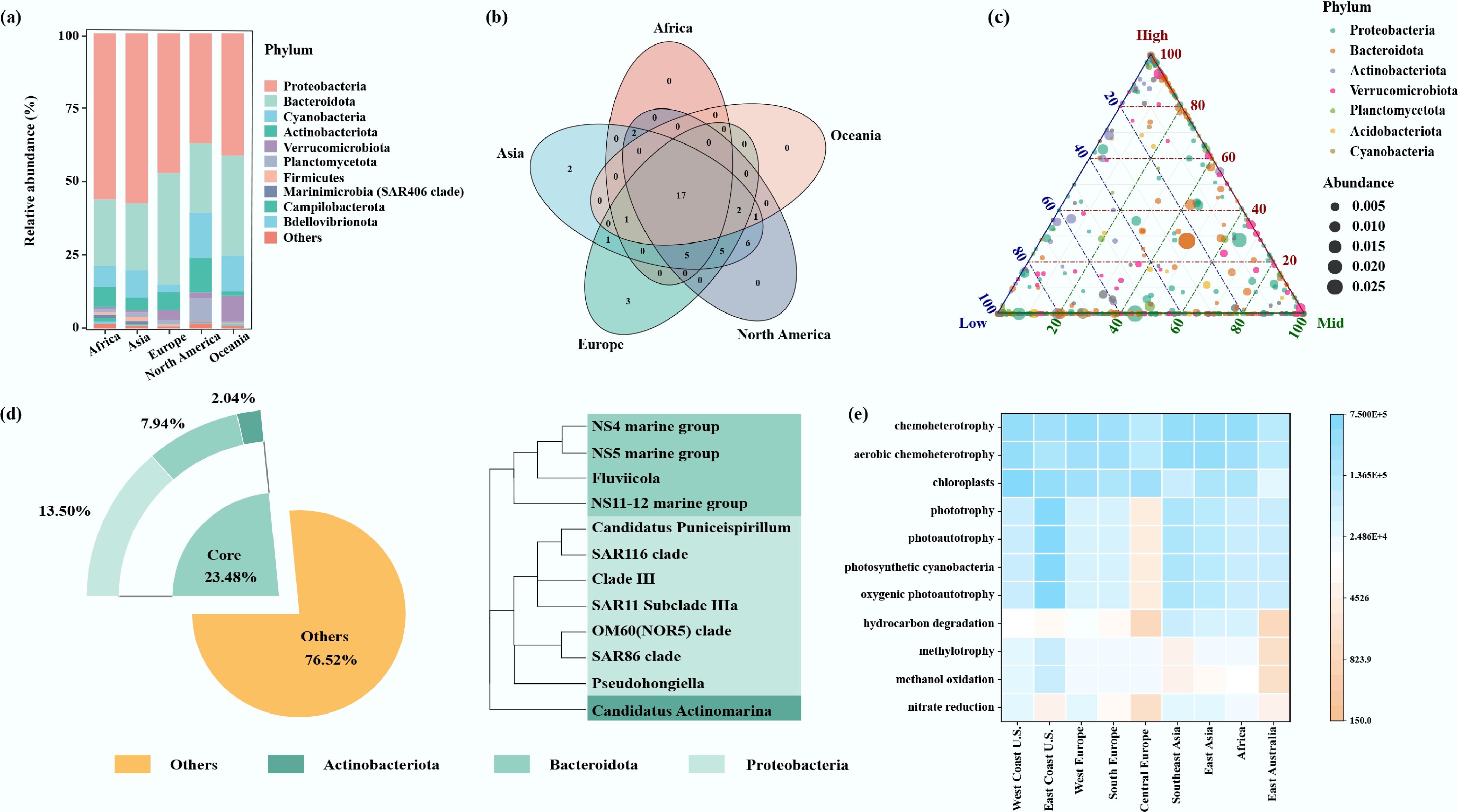
Figure 3.
(a) Relative abundances of the dominant phyla in port water. (b) Venn diagram of bacterial communities' interactions among different continents. (c) Ternary plots showing the distribution of dominant bacterial phyla across different latitudes. Each circle corresponds to one phylum, its position is determined by its relative abundance. (d) Relative abundance and phylogenetic tree of core ASVs in port water. (e) Heatmap of bacterial community functions in port water.
Notably, 12 core genera were detected in global port water samples based on their abundance and occurrence frequency, collectively representing 23.5% of the bacterial community (Fig. 3d). More than 50% of core members belonged to the phylum Proteobacteria, with the remainder attributable to Bacteroidota and Actinobacteriota. The most abundant genus was SAR11 subclade IIIa, which was present in 82.6% of the samples and accounted for 7.2% of the port water bacterial communities. The second-most-abundant genus was the NS5 marine group (3.7%). Other prominent core bacterial genera included Candidatus Actinomarina, SAR86 clade, NS4 marine group, NS11-12 marine group, Fluviicola, Clade III, Pseudohongiella, SAR116 clade, and OM60 (NOR5) clade (Supplementary Table S5).
Prediction of the metabolic function of bacterial communities
-
Functional annotation using FAPROTAX predicted 77 distinct metabolic and ecological functional groups across port water bacterial communities (Supplementary Table S6), highlighting their diverse roles in biogeochemical cycling. Chemoheterotrophy and aerobic chemoheterotrophy were the predominant functional groups. Additionally, phototrophic processes emerged as key metabolic features, encompassing oxygenic photoautotrophy (primarily mediated by photosynthetic cyanobacteria), chloroplast-associated phototrophy, and related light-dependent energy acquisition pathways. It should be noted that functional structure also varied across samples from different regions (Fig. 3e) and latitudes (Supplementary Fig. S5). For example, functional groups such as phototrophy, photoautotrophy, photosynthetic cyanobacteria, and oxygenic photoautotrophy were significantly higher in samples from the US East Coast than those from other regions, especially Central Europe. Hydrocarbon degradation pathways were more abundant in Asian and African ports than in other regions. Nitrate respiration and nitrogen respiration showed higher abundance in mid-latitude ports, while hydrocarbon degradation pathways were more abundant in low-latitude ports.
Potential pathogens in global port water
-
A better understanding of pathogen distribution is crucial for evaluating the potential risks associated with port water. In this study, a total of 295 potential pathogenic bacteria (6% of the total sequences) were identified in port water samples (Fig. 4a). Among them, 226 were classified as animal pathogens, and 60 were zoonotic pathogens, capable of being transferred between animals and humans (Fig. 4b). These pathogen-related taxa were distributed among nine phyla and 11 classes, with over 97% belonging to the phylum Proteobacteria (Supplementary Fig. S6).
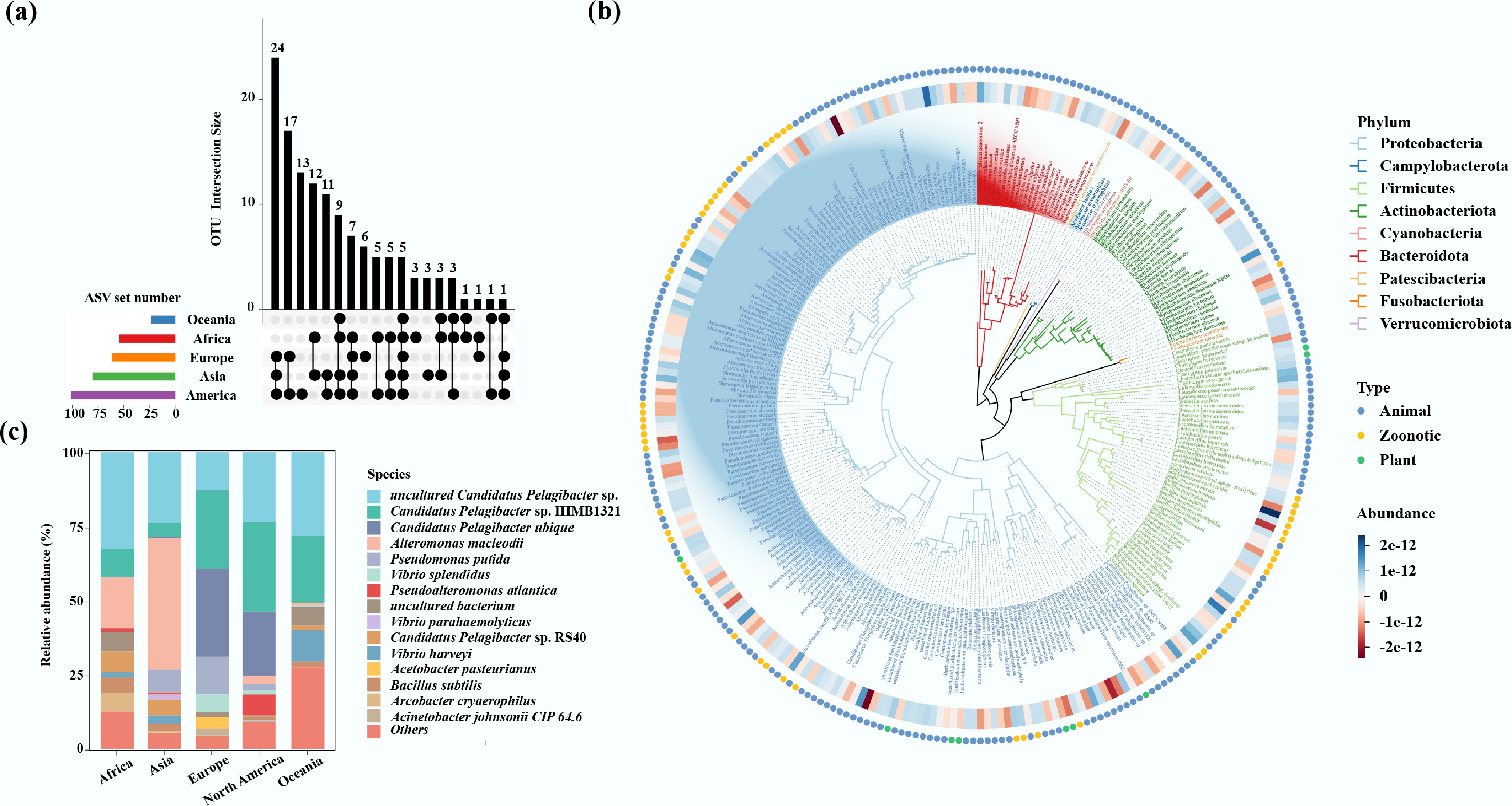
Figure 4.
(a) Upset plot of potential pathogenic bacteria among different continents. Overlapping regions between columns indicate the shared pathogens among continents. (b) The phylogenetic tree of potential pathogenic bacteria. (c) The pathogenic bacterial community composition in port water.
Potential pathogenic bacteria varied significantly across different continents and latitudes. Specifically, port water samples from Europe, North America, Oceania, Asia, and Africa contained 103, 173, 27, 139, and 113 potential pathogenic bacteria, respectively (Fig. 4a). Moreover, 28, 26, 18, 10, and 1 unique pathogenic bacteria were identified in samples from Africa, North America, Asia, Europe, and Oceania, respectively (Supplementary Fig. S7a). For example, sequences related to Arcobacter cryaerophilus were detected exclusively in samples from Africa and Asia. In contrast, sequences associated with V. parahaemolyticus were found solely in Asian samples (Fig. 4c). In addition, European (13.7%) and Asian (10.4%) samples showed a significantly higher abundance of potential zoonotic pathogens compared to North American samples (4.4%; K-W test, p < 0.05). Meanwhile, North American samples were dominated by potential animal pathogens (95.2% prevalence) (Supplementary Fig. S7b). Furthermore, 4, 36, and 18 unique potential pathogenic bacteria were identified in the samples from high-, mid-, and low-latitude regions, respectively (Supplementary Fig. S7c). Samples from mid- (11.95%) and low-latitude (14.79%) regions exhibited a higher abundance of potential zoonotic pathogens than those from high-latitude (1.04%) regions, whereas high-latitude samples exhibited a greater prevalence of potential animal pathogens (98.7%) (Supplementary Fig. S7d).
Four pathogens, including uncultured Candidatus Pelagibacter sp., Candidatus Pelagibacter sp. HIMB1321, Candidatus Pelagibacter ubique, and Alteromonas macleodii were particularly prevalent in global port water bacterial communities. Furthermore, 24 potential pathogenic bacteria, such as uncultured Candidatus Pelagibacter sp., Candidatus Pelagibacter sp. HIMB1321, Pseudomonas putida, A. macleodii, and Vibrio splendidus were present simultaneously in samples from Asia, Europe, and North America. Notably, their abundance still varied across these continents. For example, sequences related to A. macleodii were substantially more prevalent in Asian samples (83.8%) than in those from other continents. Sequences related to V. splendidus (82.4%) and P. putida (64.0%) exhibited considerably higher abundance in European samples compared to other continents (Fig. 4c).
Assembly and sources of port water bacterial communities
-
The ecological processes governing the assembly of bacterial communities in port water were assessed using a null model. Deterministic processes (54.1%) drove bacterial community assembly in port water worldwide. This trend was particularly pronounced in Asia (74.1%), North America (67.9%), and Europe (51.3%), especially the process of homogeneous selection (Fig. 5a). Conversely, stochastic processes played a more crucial role in port bacterial community assembly in Africa (66.0%) and Oceania (98.1%) (Fig. 5a). The SourceTracker results indicated that the potential sources of bacterial makeup in port water were relatively consistent across continents and latitudes (Fig. 5b and Supplementary Fig. S8). Air was identified as the primary potential microbial source (26.9%) for port water bacterial communities, and the contribution from human excretion (26.6%) was also notable. Freshwater sources contributed the least to the bacterial communities observed in port water, accounting for only 7.7% of the overall composition.
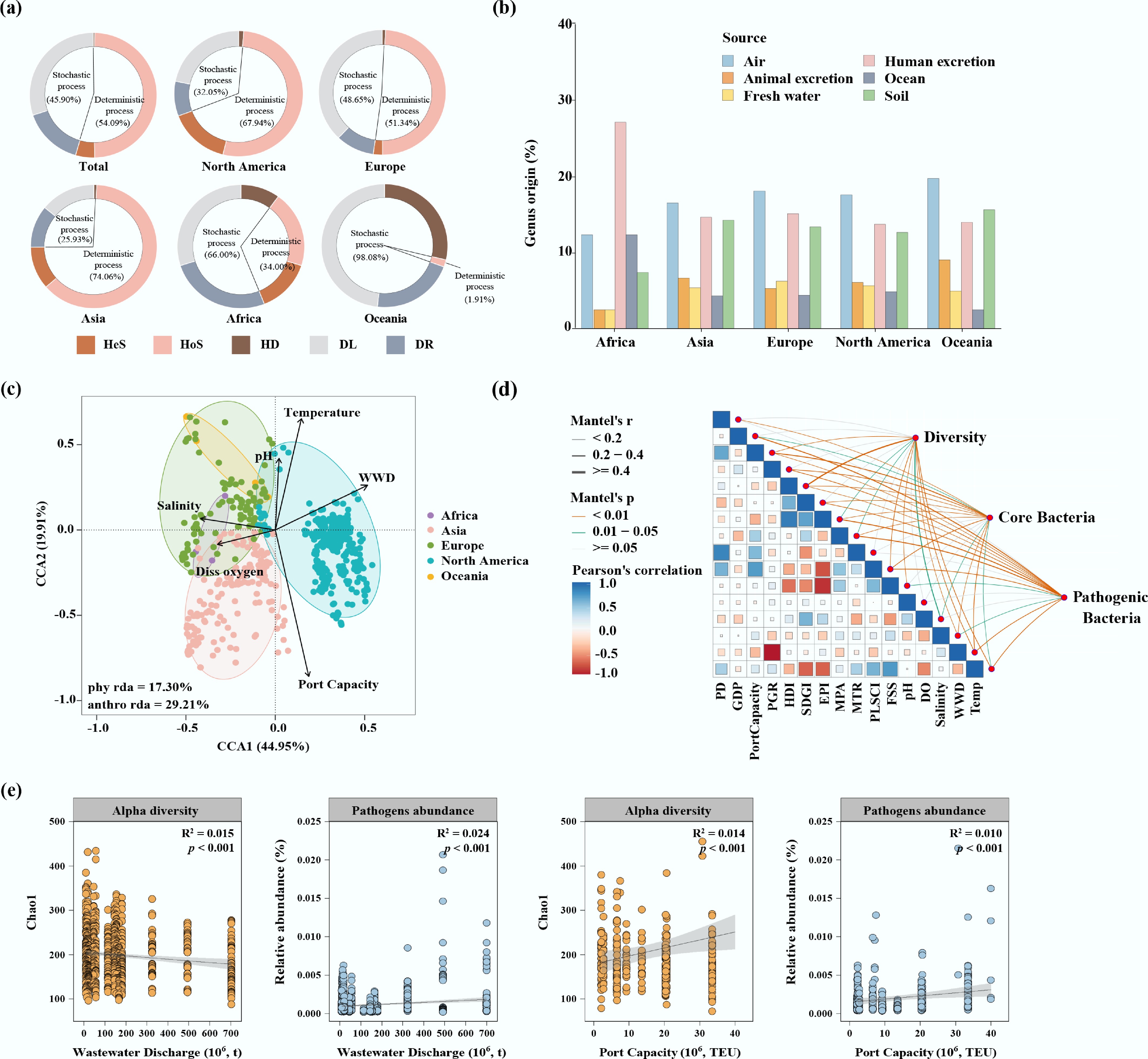
Figure 5.
(a) Assembly processes of the port water bacterial communities. (b) Contributions of various environments to port water bacterial communities determined by SourceTracker. (c) Canonical correspondence analysis (CCA) of environmental factors and anthropogenic factors on bacterial communities. (d) Relationships between port water bacterial communities and socioeconomic factors: population density (PD), gross domestic product (GDP), population growth rate (PGR), sustainable development goals index (SDGI), human development index (HDI), wastewater discharge (WWD), environmental performance index (EPI), dissolved oxygen (DO), port capacity, port liner shipping connectivity index (PLSCI), marine protected area (MPA), marine trophic index (MTI), fish stock status (FSS). (e) Linear relationships of wastewater discharge and port capacity with bacterial diversity and pathogen abundance in port water.
Linking port water bacterial communities and anthropogenic factors
-
In light of the above findings regarding microbial source and assembly, the potential influence of human imprints, including demographic, economic, and port development factors on port water bacterial communities was further investigated. The CCA results indicated that both anthropogenic and physicochemical factors significantly impacted the bacterial communities (Fig. 5c). Among all individual factors, port capacity exhibited the strongest correlation with bacterial community (Mantel test, R2 = 0.32), followed by temperature (Mantel test, R2 = 0.15; Supplementary Table S7). Further HP analysis showed that port capacity and FSS independently explained 18.98% and 20.42% of the variation in bacterial communities, respectively. Compared with terrestrial human activities, shipping-related factors (such as port capacity) had a greater influence on bacterial community structure (Supplementary Fig. S9). Moreover, there was a significant association between bacterial diversity (including core and pathogenic bacteria) and port development factors such as port capacity and PLSCI (Mantel test, p < 0.05; Fig. 5d). In addition, wastewater discharge intensity was significantly negatively correlated with bacterial diversity (Chao1 index, R2 = 0.015, p < 0.001), yet positively associated with total pathogen abundance (R2 = 0.024, p < 0.001). Furthermore, port capacity exhibited significant positive correlations with both bacterial diversity (Chao1 index, R2 = 0.014, p < 0.001) and total pathogen abundance (R2 = 0.010, p < 0.001; Fig. 5e).
-
Ports serve as critical hubs in global maritime networks, playing an essential role in international trade and economic development[2]. However, increasing anthropogenic pressures from coastal urbanization and maritime activities present unprecedented challenges to port water ecosystems. Characterizing fundamental ecological patterns and quantifying anthropogenic impacts on bacterial communities are essential for the effective management of port ecosystems. This study provides a global overview of the general principles shaping bacterial communities in port waters. The LDG typically describes a decline in species richness with increasing latitude, a well-established pattern in plant and animal biogeography[41]. Nevertheless, the bacterial communities in global port waters did not closely follow this pattern. Instead, bacterial community richness peaked in mid-latitude regions rather than tropical zones. This unimodal distribution aligns with observations from other microbial habitats (marine, terrestrial, and atmospheric ecosystems)[17,18,42], potentially driven by the balanced thermal conditions and moderate nutrient levels of intermediate latitudes[43]. It should be noted that the overall latitudinal trend in ports is relatively weak, likely due to human-driven homogenization from shipping and other anthropogenic activities.
The DDR analysis revealed the bacterial communities in port water exhibit a clear spatial turnover, with greater compositional similarity observed among geographically proximate locations. This finding is consistent with observations from other environments such as lakes, air, and even wastewater treatment plants[16,17,44]. Further analysis indicated that port capacity might mediate this distance-decay relationship. Frequent shipping activities in high-capacity ports likely facilitate microbial dispersal through ballast water discharge, thereby weakening the distance-decay effect[45]. Moreover, the intracontinental β-diversity was significantly lower than intercontinental variation (Supplementary Fig. S10), indicating strong geographic clustering of bacterial communities. A striking divergence was observed within North American ports, where bacterial communities differed significantly between the US East Coast and West Coast sites. These findings confirmed the influence of geographic and oceanographic factors on shaping port microbiomes, and indicated that microorganisms encounter substantial dispersal barriers at macroecological scales. Accordingly, this has critical implications for maritime transport and marine biosecurity, as ballast water exchange enables the circumvention of natural dispersal barriers between geographically distant ports.
Global core bacterial communities and pathogens
-
Consistent with prior investigations, port water harbors a diverse range of bacterial communities[13]. A set of core bacterial communities were also identified across port waters worldwide. Proteobacteria and Bacteroidetes were the dominant phyla in port water, which aligns with earlier findings[46,47]. Notably, SAR11, a member of the Proteobacteria phylum, was the most abundant genus in global port water. SAR11 is ubiquitously distributed across the oceans and accounts for approximately 30% of bacterial communities in marine environments[48]. As an aerobic, free-living chemoheterotrophic bacterium, it plays a significant role in the global carbon cycle[49]. NS5 marine group was also highly prevalent in port water, corroborating prior findings of its dominance in estuarine environments[50]. The NS5 marine group could degrade high-molecular-weight organics, such as algal detritus, chitin, and even terrestrial inputs, a metabolic trait that may be particularly relevant given the escalating anthropogenic pollution in offshore port waters[50,51]. Additionally, Pseudohongiella, a genus capable of denitrification under low-oxygen conditions, was identified as a core bacterial genus, suggesting its significant role in nitrogen cycling within ports[52]. Similarly, as a keystone taxon in port ecosystems, Candidatus Actinomarina may mediate the cleavage of dimethylsulfoniopropionate (DMSP) to dimethyl sulfide (DMS), thereby influencing the sulfur cycling in port waters. These findings align consistently with the functional predictions derived from FAPROTAX, corroborating the inferred metabolic potential of port microbial communities. Furthermore, the functional structure exhibited marked regional differentiation. Photosynthetic functions were significantly more abundant in North America, which may be linked to the predominance of Cyanobacteria in this region[53]. The higher abundance of hydrocarbon-degradation pathways in Asian and other mid-latitude ports can be attributed to polycyclic aromatic hydrocarbon (PAH) pollution. Studies indicated that PAH concentrations in Asian port waters were generally high, primarily originating from the incomplete combustion of petroleum and coal, as well as urban wastewater discharge[54]. Such pollutants might promote the functional microbial groups, including Pseudomonas and Acinetobacter[55]. These findings highlight the complex metabolic functional potential of bacterial communities in port environments. Accordingly, characterizing these functional attributes provides a scientific foundation for evaluating the ecological health of port ecosystems.
Pathogenic bacteria were widely distributed in port waters, with 295 potential pathogenic species identified across the samples. The composition of primary pathogenic bacteria exhibited marked geographic variation among continents. Vibrio parahaemolyticus was detected only in samples from Asia, suggesting a potential geographic origin of its common ancestor[56]. Arcobacter cryaerophilus was detected exclusively in samples from Asia and Africa, its distribution appears to be associated with anthropogenic factors such as sewage discharge[57,58]. Pseudoalteromonas atlantica was detected at higher rates in North American samples compared to other continents. As this bacterium is recognized as a pathogen linked to mortality in marine crustaceans, its enrichment may represents a potential threat to aquaculture systems[59,60]. Moreover, the African ports showed the highest relative abundance of potential pathogens among all continents. For instance, Aeromonas caviae, detected at Durban port, is an important etiological agent of human intestinal infections[61]. This pathogen commonly occurs in a range of aquatic environments, including brackish water and sewage systems[62]. Its prevalence likely reflects inadequate wastewater treatment in the region[63]. Direct or indirect exposure to pathogenic bacteria in port waters can cause a spectrum of illnesses, including gastrointestinal disorders, respiratory complications, and dermatological infections[64]. Notably, V. parahaemolyticus is a halophilic bacterium frequently associated with shellfish and other seafood. Accordingly, the consumption of raw or undercooked contaminated seafood can lead to acute gastroenteritis[65]. Critically, these pathogens can achieve long-distance dispersal via ship ballast water, thereby introducing health risks to distant regions.
Assembly and anthropogenic impacts on global port water bacterial communities
-
Quantifying the relative contributions of deterministic vs stochastic processes in community assembly is essential for elucidating how bacterial diversity is maintained in port ecosystems[66]. In this study, deterministic processes dominated the bacterial community assembly in port water at the global scale. Nevertheless, previous investigations of airborne[17], desert[67], lake[18], activated sludge[68], and open marine[69] microbiomes have revealed that stochastic processes largely govern community turnover in those systems. The semi-enclosed nature of ports, coupled with steep environmental gradients (e.g., salinity and temperature) and anthropogenic pressures, appear to strengthen niche-based selection over stochastic processes in structuring microbial assemblages. The CCA further corroborated that physicochemical factors, including salinity, pH, temperature, and dissolved oxygen could significantly affect bacterial communities in port waters, supporting the predominance of deterministic processes in community assembly.
The bacterial community in port water originates from a variety of environmental sources, with human-associated sources emerging as a potentially significant contributor. Specifically, sewage discharge volume serves as a quantifiable metric of terrestrial human activity, demonstrating significant negative correlations with bacterial alpha diversity, while positively correlating with pathogen abundance. Given that sewage often contains elevated concentrations of pathogenic microorganisms, its discharge into marine environments accordingly increases the pathogen loads in coastal waters. As a key quantitative indicator of shipping intensity and maritime trade volume, port capacity exerted a greater influence on bacterial community structure than terrestrial human activity. In contrast to the Southern Ocean (k = –0.0398), the DDR in high-capacity ports (k = –0.0295) examined here was substantially attenuated[70]. This discrepancy suggests a possible link between shipping activity and port bacterial communities. Notably, port capacity was also significantly correlated with bacterial diversity. Greater port capacity is generally associated with more intensive shipping operations, which amplify anthropogenic pressures on coastal ecosystems. These maritime operations might promote microbial dissemination via ballast water and ship-fouling organisms[45], leading to an increase in pathogens[71]. This likely reflects the heightened risk of biological invasions from shipping activities rather than an improvement in ecosystem health.
-
Coastal ecosystems are vital to human society, with nearly 40% of the global population living within 100 km of the coast. Ports are critical hubs within coastal urban environments where human activities and marine environments intensely interact[72]. This investigation provides a systematic global characterization of port water bacterial communities, identifying universal ecological regularities and highlighting the influence of human activity. The observed decline in bacterial diversity and enrichment of pathogens associated with wastewater discharge underscore the substantial negative impacts of terrestrial anthropogenic activities on port ecosystems. The findings also highlight the influence of maritime activities on port water bacterial communities, where shipping may contribute to microbial dispersal. Nonetheless, further research is certainly needed to validate this assumption. The biogeographical patterns of microbes in port waters offer a scientific foundation for mitigating biosecurity challenges, especially those related to ballast water management. For ballast water originating from ports with high pathogen risk, maritime authorities should strengthen surveillance and implement strict disinfection protocols. This study also reveals that the composition and functional attributes of port bacterial communities are closely associated with regional socioeconomic activities and anthropogenic pressures. This relationship underscores the potential of microbial indicators as sensitive proxies for future assessment of the health status of port ecosystems. Although this study provides a global snapshot of port water bacterial communities, it is acknowledged that the scope is still constrained by specific countries. Future studies would benefit from large-scale, standardized sampling to reduce the potential for procedural biases. Moreover, the adoption of more comprehensive analytical approaches, such as metagenomics, full-length 16S rRNA sequencing, and single-cell sequencing, is recommended to precisely identify the bacteria and pathogens that colonize port water. Nonetheless, the insights from this study could guide the development of a more strategic approach to port environmental protection from a macroecological perspective.
-
It accompanies this paper at: https://doi.org/10.48130/biocontam-0026-0005.
-
Not applicable.
-
The authors confirm their contributions to the paper as follows: Baoyi Lv: methodology, validation, writing − review & editing. Qitong Zhang: investigation, visualization, writing − original draft & editing. Tingxuan An: investigation, validation, formal analysis. Shenglong Mei: resources, methodology. Guolin Kan: resources, investigation. Dong Wu: methodology, formal analysis. Jianhong Shi: writing − review & editing, resources. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets used or analyzed during the current study are available from the corresponding author on reasonable requests.
-
This work was financially supported by the National Science Foundation of China (Grant Nos 42477445, 42307503, 42377425), and the Shanghai Sailing Program (Grant No. 23YF1415900). We gratefully acknowledge the researchers who generated and submitted the sequencing data to the NCBI and ENA, which was instrumental for this study.
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
Full list of author information is available at the end of the article.
- The supplementary files can be downloaded from here.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Lv B, Zhang Q, An T, Mei S, Kan G, et al. 2026. A global synthesis of port water microbiome biogeography and anthropogenic associations. Biocontaminant 2: e008 doi: 10.48130/biocontam-0026-0005 

A global synthesis of port water microbiome biogeography and anthropogenic associations
- Received: 29 October 2025
- Revised: 26 January 2026
- Accepted: 03 April 2026
- Published online: 01 May 2026
Abstract: Ports are key nodes in global maritime transport networks. Microorganisms in port ecosystems are crucial for biogeochemical cycling and ecological stability, while their community structure and geographic distribution at the global scale remain inadequately understood. This study conducted a meta-analysis of bacterial communities in port water collected from 23 cities across five continents. Analysis of more than 16 million 16S rRNA gene sequences revealed that bacterial communities in port waters exhibited a clear distance-decay relationship, with species richness peaking at mid-latitudes. Moreover, global port water bacterial communities were characterized by 12 dominant genera, with the SAR11 subclade IIIa representing the most abundant lineage, followed by the NS5 marine group. A total of 295 distinct pathogenic ASVs (6% of the total sequences) were detected, and their prevalence varied significantly among regions, with Africa showing the highest abundance. Deterministic processes were identified as the dominant assembly forces in port waters worldwide. Source tracking linked the port water bacteria primarily to air and human-associated sources. Anthropogenic and shipping activities (as reflected in port capacity) showed a pronounced association with variations in bacterial community structure. Overall, the findings advance the understanding of diversity and biogeography within port water bacterial communities, and provide significant implications for managing human activities and ensuring sustainable port ecosystem operations.
-
Key words:
- Bacterial community /
- Global scope /
- Port water /
- Biogeography /
- Pathogens /
- Anthropogenic impacts