









-
Elephant grass (Pennisetum purpureum) and its hybrid with hybrid Pennisetum (Pennisetum purpureum × Pennisetum americanum) are high-yielding, high-quality perennial forage grasses in tropical and subtropical regions, playing a significant role in ensuring feed supply for livestock[1−3].
However, its germplasm resources have complex origins, its breeding cycles are long, and target traits such as stress resistance and yield have a highly complex genetic basis, which poses a significant challenge to efficient breeding[4]. Comprehensive genetic analysis is therefore essential to dissect the genetic basis of key agronomic traits, accelerate breeding cycles, and improve the efficiency of parental selection in hybridization programs[5,6]. Although studies analyzing genetic diversity based on traditional simple sequence repeat (SSR) markers have been carried out in elephant grass to some extent, their throughput is low and genome coverage is limited, making it difficult to comprehensively dissect the genetic mechanisms of complex traits[7]. On the other hand, although whole-genome resequencing can provide high-density information on genomic variation, the cost of resequencing large-scale germplasm resources remains high because of the large and complex genome of elephant grass, limiting its widespread application in breeding practices[8].
Single nucleotide polymorphism (SNP) markers have become an important tool in population genetics and molecular breeding because of their abundance, wide distribution, and high detection throughput[9−11]. Medium-density SNP arrays (e.g., 10,000) offer a good balance between cost and information density, making them suitable for large-scale genetic evaluation of germplasm resources[12]. Liquid-phase array technology, which relies on biotin-labeled probes for targeted capture and high-throughput sequencing, has been widely adopted in genetic studies of various crop species, including maize (Zea mays)[13], rice (Oryza sativa)[14], rapeseed (Brassica napus)[15], and tea plant (Camellia sinensis)[16]. However, SNP-based studies at the population level for elephant grass and hybrid Pennisetum are still scarce, and functional variations at the genome level remain inadequately explored.
In this study, we developed a custom 10K liquid-phase array based on the purple elephant grass reference genome and performed genotyping on 83 core accessions[8], aiming to (1) elucidate the population's genetic structure, genetic relationships, and genetic diversity of elephant grass and hybrid Pennisetum; (2) characterize the genome-wide SNP mutation spectrum; (3) mine candidate genes and pathways related to stress resistance, metabolism, and growth and development through functional enrichment analysis; and (4) establish a cultivar-specific molecular identification system based on core SNPs, providing technical support for germplasm identification, protection, and molecular breeding.
-
In total, 83 accessions of elephant grass and hybrid Pennisetum were used in this study, including major commercial cultivars (e.g., Guimu 1, Bangde 1), local landraces (e.g., mangong), and breeding lines (e.g., ZR series). Details and geographical origins of the materials are provided in Supplementary Table S1.
The 10K liquid-phase SNP array design and genotyping
-
The array's design was based on the purple elephant grass reference genome (GCA_028749085.1)[8]. SNP sources included (1) 504 functional loci previously screened for yield and quality traits and (2) 10,982 background loci selected from resequencing the data of 450 elephant grass individuals (PRJEB73794)[17]. Background loci were filtered with the following criteria: Sequencing depth > 5,000, minor allele frequency (MAF) > 0.05, missing rate ≤ 0.1, and approximately uniformly distributed every 4 kb across the genome. The final array contained 11,486 SNP loci.
Genotyping used a 'tiled' probe design and liquid-phase hybridization capture. The workflow included DNA fragmentation, end repair, adapter ligation, pre-polymerase chain reaction (PCR) amplification, probe hybridization capture, PCR enrichment, and PE150 sequencing on the MGISEQ-2000 platform. Raw data were quality-controlled with Fastp and aligned to the reference genome using BWA (v0.7.17). SNP calling was performed with GATK.
For downstream population genetics and fingerprinting analyses, only high-quality SNPs with a call rate ≥ 90% were retained. No imputation was performed for missing genotypes; instead, SNPs with excessive missing data were excluded to minimize potential bias.
SNP mutation types and functional enrichment analysis
-
The spectrum of nucleotide substitutions was comprehensively analyzed across all SNP sites. Gene Ontology (GO) functional enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the gene list corresponding to the SNPs on the array. Significance thresholds were set at false discovery rate (FDR)-adjusted p-values < 0.05.
Genetic diversity and population structure analysis
-
Polymorphism information content (PIC), Nei's gene diversity index (GD), observed heterozygosity (Ho), and expected heterozygosity (He) were calculated using PowerMarker v3.25. Based on high-quality SNPs, a neighbor-joining phylogenetic tree was constructed with MEGA v5.2 (bootstrap = 1,000), and principal component analysis (PCA) was performed using Python.
Determination of genetic similarity thresholds and cultivar grouping
-
Nei's genetic similarity coefficient (GS) was calculated for all pairwise comparisons among accessions. To determine the optimal threshold for precise cultivar identification, we systematically compared grouping outcomes across a range of thresholds, including absolute values (0.95, 0.98) and percentile-based thresholds (the 95th, 98th, and 99th percentiles of the GS distribution). The selection criteria prioritized thresholds that maximized within-group genetic consistency while ensuring high discriminative power for cultivar separation. The optimal threshold was identified by evaluating the trade-off between group cohesion and the number of groups formed, with a focus on minimizing ambiguous assignments.
Core SNP selection and fingerprint construction
-
On the basis of stringent quality control criteria, we selected 50 high-quality SNP loci from the whole-genome SNP data to construct a fingerprint profile. The selection criteria included (1) call rate ≥ 90%, (2) MAF ≥ 0.30, and (3) heterozygosity ≤ 70%. Representative loci with the highest MAFs were selected from each chromosome, and additional high-MAF loci were included when necessary to meet the required number. A digital fingerprint profile was constructed, based on the genotype data for unique cultivar identification.
Statistical analysis
-
Statistical analyses were performed using R (v4.2.1). Descriptive statistics, correlation analyses, and threshold optimizations were conducted as described in the respective method sections. Enrichment analyses were performed with clusterProfiler (v4.4.4) using the default parameters and FDR correction for multiple testing.
-
Using the high-quality reference genome of purple elephant grass[12] (total length = 1.97 Gb, N50 = 150.59 Mb), we successfully designed and fabricated a custom 10K liquid-phase SNP array comprising 11,486 SNP loci (Fig. 1a). The array adopted a 'tiled' probe design, which effectively enhanced capture uniformity and genome-wide coverage. Sequencing of 83 samples yielded approximately 94.73 Gb of raw data. After stringent quality control, the effective data rate exceeded 99.80%, the average Q30 base ratio was above 92%, and the average GC content was ~52.8% (Supplementary Table S2). The clean data showed a high average mapping rate to the reference genome (97.85%), confirming the suitability of the reference genome for genotyping in this species.
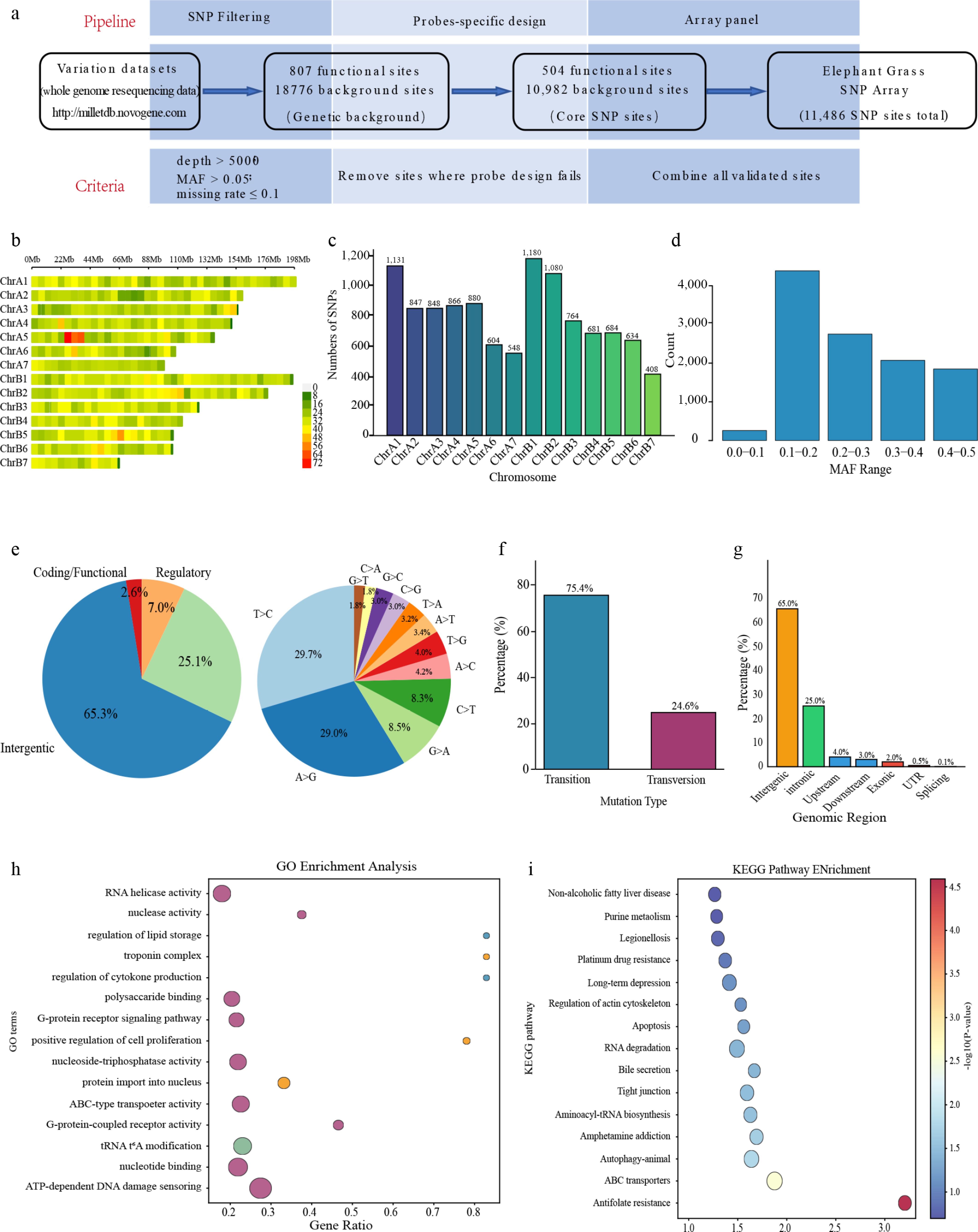
Figure 1.
Genome-wide SNP analysis based on the 10K liquid-phase array. (a) Schematic overview of the array's design and workflow. (b) Distribution of SNP loci across the 14 chromosomes. (c) Correlation between chromosome length and number of SNPs. (d) MAF distribution of the SNP set. (e) Spectrum of SNP mutation types. (f) Proportion of transition (Ts) and transversion (Tv) events. (g) Genomic annotation of SNP locations. (h) GO enrichment analysis of genes associated with SNPs. (i) KEGG pathway enrichment analysis.
The performance of capturing the target regionwas robust, with an average depth exceeding 200×, a SNP call rate > 97%, and with more than 96% of the target region covered by at least 10 reads (≥ 10× coverage; Table 1). These metrics demonstrate the high quality and reliability of the genotyping data obtained from the liquid-phase array system. The 11,486 SNPs were distributed across all 14 chromosomes, providing genome-wide coverage (Fig. 1b), with the longest chromosome (ChrB1) harboring the most SNPs and the shortest chromosome (ChrB7) the fewest (Fig. 1c). This even distribution strategy minimized genomic coverage bias and provided balanced marker density for subsequent genetic analyses. The MAF distribution showed that the majority of SNPs exhibited moderate to high polymorphism, with 68.4% of loci having MAF ≥ 0.2 and 31.6% with MAF < 0.2, indicating a broadly informative marker set for analyses of the genetic diversity (Fig. 1d, where the y-axis represents the count of SNPs). Analysis of the SNP mutation spectrum revealed that transition events predominated, with A > G and C > T accounting for 29.0% and 29.7%, respectively, totaling approximately 58.7% of all mutations (Fig. 1e, f). The majority of SNPs were located in intergenic regions (~65%), followed by introns (~25%), upstream/downstream regions (~7%), and exonic regions (~2%) (Fig. 1g). Functional enrichment analysis of genes associated with the SNP set revealed significant GO terms related to adenosine triphosphate (ATP) binding, RNA helicase activity, ATP-binding cassette (ABC) transporter activity, DNA damage sensing, and proton transmembrane transport (Fig. 1h). KEGG pathway analysis also highlighted enrichment in ABC transporters, purine metabolism, and steroid biosynthesis (Fig. 1i). The enrichment in ABC transporters is particularly noteworthy, as this gene family is known to play critical roles in tolerance to abiotic stress by facilitating the transport of diverse substrates across cellular membranes. These enriched pathways therefore represent promising candidate systems for further functional validation and targeted improvement in Pennisetum breeding programs.
Table 1. Performance metrics of the custom 10K liquid-phase SNP array for genotyping Pennisetum germplasm.
Category Metric Value Notes Sample and design Samples with full quality control (QC) 83 Total 83 accessions were genotyped. Total SNPs on the array 11,486 504 functional + 10,982 background SNPs. Sequencing quality Average effective rate > 99.85% – Average Q30 > 94.79% – Average mapping rate 97.85% – Target capture Average SNP call rate > 97% – Average depth on the target > 200× – ≥10× coverage rate > 96% – Reproducibility Replicates' concordance 96.17% From prearray validation. Data integration Total samples in the final analysis 83 All passed genotype quality filters. Overall, these results validate the robustness and efficiency of the custom 10K liquid-phase array for genotyping in elephant grass. The high-quality SNP dataset generated here provides a solid foundation for subsequent investigations into population genetics, germplasm identification, and the discovery of functional genes associated with key agronomic traits in elephant grass and hybrid Pennisetum.
Genetic diversity and population structure
-
To further explore the genetic basis of the germplasm collection and facilitate its efficient utilization in breeding, we assessed the genetic diversity and population structure of the 83 accessions using the high-quality SNP dataset. Genetic diversity analysis revealed an average polymorphism information content (PIC) of 0.288 (Fig. 2a). According to common classification criteria, this average PIC value indicates a moderate to high level of marker polymorphism within this collection. Nei's GD averaged 0.360, with a wide range (0.014−0.500), confirming substantial genetic variation among the accessions (Fig. 2b). The observed heterozygosity (Ho = 0.354) was consistently higher than the expected heterozygosity (He = 0.290) (Fig. 2c), and the average inbreeding coefficient (Fis) was –0.366 (Fig. 2d), suggesting a pronounced excess of heterozygotes in the population. This pattern is likely attributable to frequent hybridization events or natural gene flow, which is characteristic of outcrossing species such as elephant grass. Pairwise comparisons among these four genetic indices (PIC, Nei's GD index, Ho, and He) revealed strong positive correlations (r > 0.69 for all comparisons), indicating high consistency among different measures of genetic diversity (Fig. 2e). Collectively, these results demonstrate that the population possesses moderate to high genetic diversity with a clear heterozygote advantage, providing a valuable genetic basis for future germplasm utilization and breeding programs.
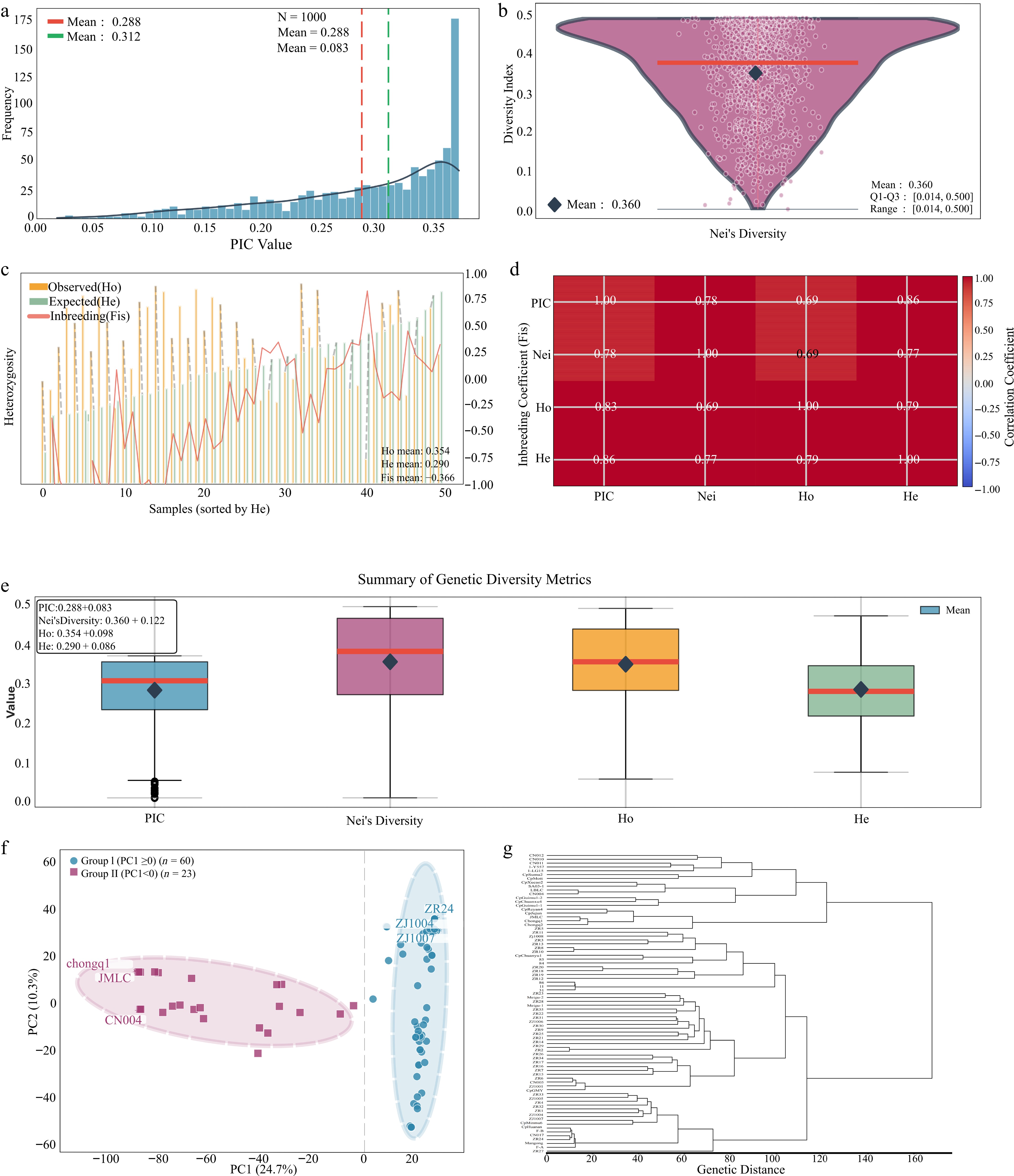
Figure 2.
Genetic diversity and population structure analysis. (a) PIC distribution. (b) Nei's GD. (c) Comparisons of Ho and He. (d) Inbreeding coefficient (Fis). (e) Correlation matrix among genetic indices (PIC, Nei's GD, Ho, and He). The color scale represents the Pearson correlation coefficients (r), with red indicating strong positive correlations and blue indicating weaker correlations. All correlations were statistically significant (p < 0.001). (f) PCA plot. (g) Phylogenetic tree.
Analysis of the population structure based on PCA and phylogenetic reconstruction revealed clear genetic differentiation within the population. PCA along the first principal component (PC1, explaining 24.7% of the variance) clearly separated the materials into two major groups: Group I (PC1 ≥ 0, n = 60) and Group II (PC1 < 0, n = 23) (Fig. 2f). Phylogenetic analysis yielded highly consistent results with the PCA grouping (Fig. 2g): The 60 accessions from PCA Group I formed a major, relatively homogeneous clade, whereas the 23 accessions from PCA Group II clustered into a distinct separate clade. Notably, all interspecific hybrid materials (P. purpureum × P. americanum) and several phenotypically distinct breeding lines (e.g., purple-stemmed accessions) were exclusively clustered within Group II, whereas Group I primarily consisted of elephantgrass accessions with relatively homogeneous genetic backgrounds. This clear genetic separation underscores the distinct ancestry and hybridization history between pure elephant grass and hybrid Pennisetum germplasm.
Cultivar identification system and core SNP fingerprint
-
Given the substantial genetic diversity and clear population structure revealed above, we next sought to develop a practical tool for cultivar identification and germplasm management. To this end, we constructed a core SNP-based fingerprinting system for the 83 accessions. On the basis of the genome-wide SNP data, we first calculated the pairwise GS among the accessions, with an average and standard deviation of 0.727 ± 0.117 (Fig 3a). To determine a GS threshold that could strictly distinguish different cultivars, we evaluated grouping effects under a series of thresholds ranging from the 90th to the 99th percentile of the GS values. This analysis indicated that using the 98th percentile (GS ≥ 0.949) as the threshold provided the highest confidence for cultivar identification. This threshold represents the point where within-group genetic similarity is maximized while maintaining strict between-group distinctions. Under this stringent criterion, the 83 accessions were divided into 58 genetic groups (Fig 3b).
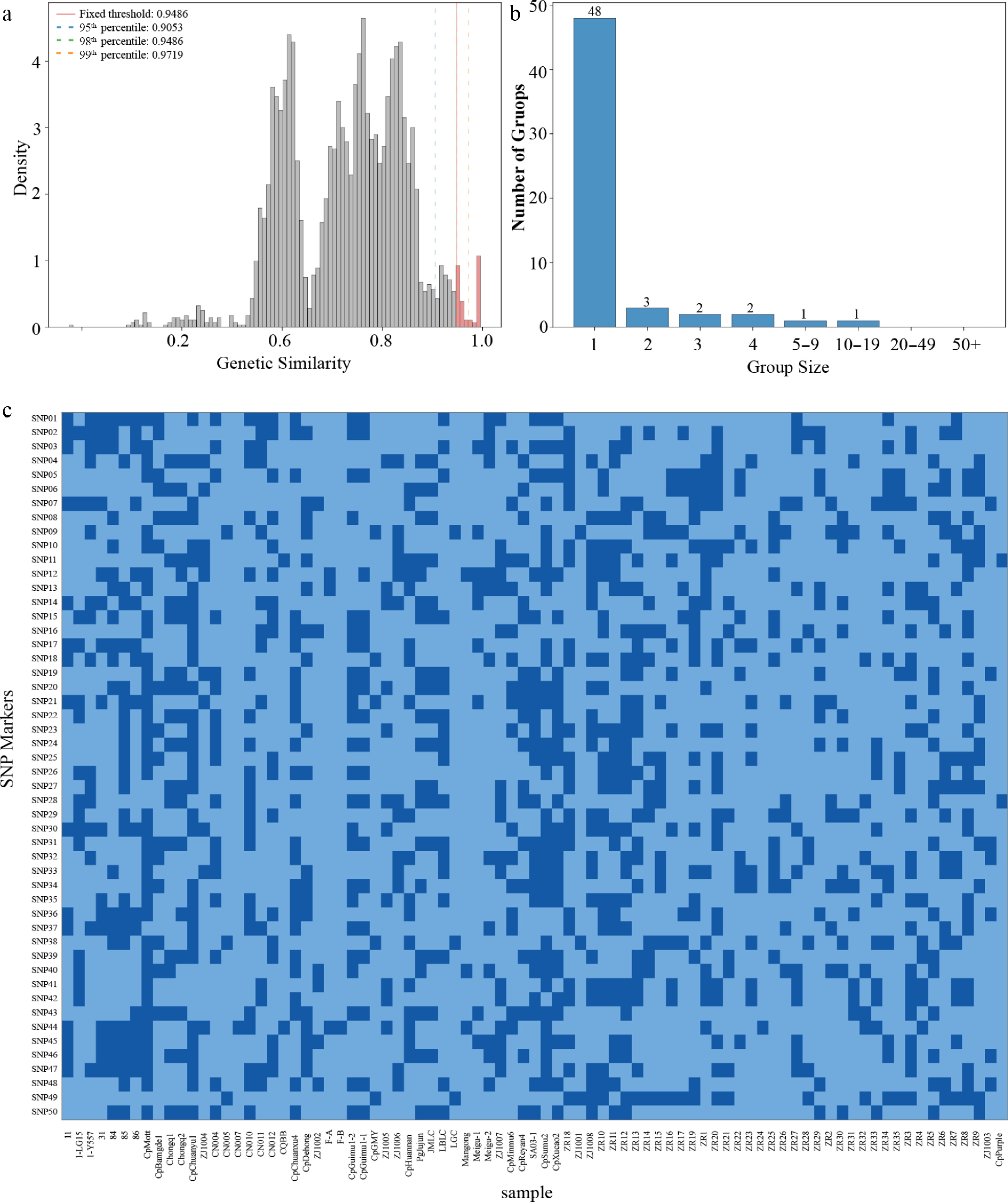
Figure 3.
Cultivar identification and fingerprint construction. (a) Distribution of genetic similarity coefficients. (b) Cultivar grouping under the GS ≥ 0.949 threshold. (c) Heatmap of 50 core SNP fingerprints.
To establish an efficient, standardized cultivar identification tool, we selected 50 core SNPs with high polymorphism (average PIC > 0.35) and high discriminative power from the genome-wide set to construct a digital fingerprint (Fig. 3c). The selection strategy prioritized representative loci with the highest MAF from each of the 14 chromosomes, ensuring even genomic distribution and minimizing chromosomal bias. This approach yielded a set of 50 core SNPs that is genomically representative and suitable for reliable cultivar discrimination. The combined probability of identity for this 50-SNP set was estimated to be less than 10–15, indicating an extremely low likelihood of two distinct accessions sharing an identical genotype by chance. The clustering pattern derived from the core SNP fingerprint closely matched the 58 genetic groups defined by the GS ≥ 0.949 threshold, successfully reproducing the major cultivar groups. This concordance validates the representativeness of the selected core SNP set. This core SNP set combines high discriminative power (capable of distinguishing most cultivars with quantitative confidence), high reproducibility, and low cost, and can be widely applied in cultivar authenticity verification, seed purity testing, moitoring breeding processes, and digital management of germplasm resources.
-
Our 10K liquid-phase SNP array represents one of the first medium-density, function-integrated genotyping platforms developed specifically for perennial forage grasses. SNP arrays have been widely used in major crops such as maize and wheat (Triticum aestivum) for genomic selection and diversity analyses[13,17]. For example, the MaizeGerm50K array provides high-density coverage for maize genetic studies[13], and the GenoBaits WheatSNP16K array has been successfully applied in wheat breeding programs[17]. In comparison, our 10K array offers a balanced trade-off between marker density and cost, making it particularly suitable for large-scale germplasm evaluation in species with limited genomic resources. However, it is important to acknowledge that the marker density of our array is lower than that of the high-density arrays available for well-established model crops, which may constrain the resolution of fine-scale mapping studies. Previous studies on Pennisetum species have relied primarily on SSR markers or low-density SNP panels[7,17], which offer lower genomic coverage and limited ability to capture functional variation. Though previous studies on the Pennisetum genus have focused on specific gene families such as bHLH transcription factors in pearl millet (Pennisetum glaucum)[18,19], our study provides the first medium-density, function-integrated SNP array platform specifically designed for elephantgrass and hybrid Pennisetum. Our array, with its combination of 504 trait-associated loci and genome-wide background markers, provides a more comprehensive tool for both diversity assessment and breeding applications, bridging a methodological gap in forage grass genomics.
Functional implications of enriched pathways
-
The significant enrichment of SNPs in pathways related to ABC transporters, PI3K-Akt signaling, and purine metabolism provides mechanistic insights into traits that are relevant to improving forage. ABC transporters are known to contribute to abiotic stress tolerance in grasses[20]. PI3K-Akt signaling is a key regulator of nitrogen use efficiency (NUE), which is a critical determinant of forage development, as it governs the balance between vegetative growth (biomass yield) and protein content (nutritional quality)[21]. Although these pathways have been characterized in model plants and staple cereals, their specific functions and regulatory networks in Pennisetum species remain largely unexplored. Consequently, our findings highlight these pathways as promising candidate systems for targeted functional validation in the context of a forage grass.The GO and KEGG enrichment analyses are based solely on genes captured by the array, which may introduce bias caused by the SNP selection criteria. Future whole-genome resequencing studies could provide a more comprehensive assessment of functional variations.
Advancements in cultivar identification methodology
-
The cultivar-specific fingerprint system developed here improves upon previous identification methods for Pennisetum, which often relied on morphological descriptors or limited molecular markers[7,22]. By establishing a quantitative genetic similarity threshold and selecting high-discrimination core SNPs, we provide an objective, reproducible system for distinguishing cultivars. This approach addresses the longstanding issues of synonymy and genetic redundancy in forage germplasm collections, offering a model that could be extended to other perennial grass species.
-
This study systematically evaluated the genetic characteristics of core germplasm of elephant grass and hybrid Pennisetum using a 10K liquid-phase SNP array, clarifying their population structure, mutation spectrum, and functional variation features. The newly constructed cultivar-specific molecular fingerprints and the established identification threshold standards provide important technical support and gene resources for germplasm management, protecting cultivar rights, and molecular breeding. Furthermore, the high-quality SNP dataset and array platform developed here lay a solid foundation for future applications such as genome-wide association studies to dissect complex agronomic traits and genomic selection to accelerate breeding cycles in Pennisetum species.
-
The authors confirm contribution to the paper as follows: study conception and design: Jia J, Zhong Y; data collection: Zhong Y; analysis and interpretation of results: Jia J, Zhong Y, Jin Y, Yang D, Zou X, Ji S, Wang X, Zhang X, Yu S; draft manuscript preparation: Jia J, Zhong Y; supervised the project: Huang L, Yan H. All authors reviewed and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors thank the National Ministry of Agriculture for supporting the development of the elephant grass liquid-phase chip.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/grares-0026-0012.
-
# Authors contributed equally: Jiyuan Jia, Yun Zhong
- Supplementary Table S1 Sample information of 83 elephant grass (Cenchrus purpureus) and hybrid pennisetum accessions.
- Supplementary Table S2 Sequencing data quality control statistics for 69 samples, and capture statistics of the 10K liquid-phase SNP array for representative samples.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Jia J, Zhong Y, Yang D, Yu S, Jin Y, et al. 2026. A 10K liquid-phase single nucleotide polymorphism array for genetic diversity analysis and cultivar identification in elephant grass and its hybrid. Grass Research 6: e011 doi: 10.48130/grares-0026-0012 

A 10K liquid-phase single nucleotide polymorphism array for genetic diversity analysis and cultivar identification in elephant grass and its hybrid
- Received: 01 March 2026
- Revised: 03 April 2026
- Accepted: 08 April 2026
- Published online: 06 May 2026
Abstract: Elephant grass (Pennisetum purpureum) and its hybrid with hybrid pennisetum (Pennisetum purpureum × Pennisetum americanum) are important high-yield forage grasses in tropical and subtropical regions. To analyze their genetic diversity and mine breeding-related functional genes, we developed a custom 10K liquid-phase single nucleotide polymorphism (SNP) array containing 11,486 SNPs based on the purple elephant grass (Pennisetum purpureum 'Red') reference genome and performed high-throughput genotyping on 83 core accessions. Population structure analysis revealed that the tested materials could be clearly divided into two major groups which align with their distinct ancestry and hybridization history. Analysis of the SNP mutation spectrum showed that transition events (A > G, C > T) predominated (approximately 58.7% in total). Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses indicated that the captured variations were significantly enriched in functional categories related to stress resistance, signal transduction, and basic metabolism. On the basis of a genetic similarity analysis, we classified the germplasm into 58 genetic groups and selected 50 highly discriminative core SNPs to construct cultivar-specific fingerprints. This study established, for the first time in forage grasses, a medium-density liquid-phase array genotyping system integrated with functional loci, providing an efficient tool and gene resources for germplasm identification, resource management, and molecular breeding in elephant grass and hybrid Pennisetum.
-
Key words:
- Elephant grass /
- 10K liquid-phase SNP array /
- Hybrid Pennisetum /
- Genetic diversity /
- Molecular breeding