








-
Twentieth-century environmental degradation spurred renewable energy adoption (e.g., solar/wind)[1−4], yet their intermittency and instability have driven critical advances in high-energy-density storage systems to overcome energy transition challenges. Rechargeable lithium-ion batteries, which rely on embedded reaction, have been successfully commercialized and have dominated the market for energy storage batteries. However, conventional lithium-ion batteries composed of oxide-based cathodes and graphite anodes exhibit limited energy density, currently approaching their theoretical maximum (~420 Wh kg−1), and reaching a developmental plateau. In light of this, extensive efforts have been devoted to the development of new energy storage systems, among which, Li–S batteries have attracted widespread attention due to their high theoretical specific capacity (1,672 mAh g−1) and energy density (2,600 Wh kg−1). They are regarded as a prospective rechargeable technology for energy-density-sensitive applications, such as unmanned aerial vehicles and spacecraft.
Sulfur is a naturally abundant element, giving Li–S batteries significant advantages in terms of cost and environmental sustainability. Unlike the 'intercalation–deintercalation' mechanism of lithium-ion battery cathodes, sulfur-based cathodes store energy through multistep conversion reactions involving the cleavage and reformation of S–S bonds (Fig. 1a). The conversion from S8 ring-opening to long-chain LPSs, followed by their progressive transformation into short-chain, insoluble LPSs, involves a multiphase process characterized by a solid–liquid–solid transition. The multistep and multiphase conversion processes in Li–S batteries give rise to numerous challenges (Fig. 1b), including: (1) slow redox kinetics. The energy barrier associated with the liquid–solid conversion process is significantly higher than that for the dissolution of solid S8[5]. Moreover, the electronically insulating nature of Li2S2 and Li2S leads to the formation of a passivation layer on the cathode surface in the later stages of discharge, causing premature termination of the discharge, and limiting the accessible capacity[6−8]; (2) severe LPSs shuttle effect. Soluble LPSs shuttle from the cathode to the anode, leading to reduced Coulombic efficiency and pronounced capacity fade. More critically, the chemical reactions between LPSs and lithium metal result in the progressive thickening of a sulfide-rich passivation layer on the anode, ultimately causing battery failure[9]. In recent years, to address the poor electrical conductivity of the cathode, various porous carbon materials have been introduced by researchers. These materials not only provide conductive networks, but also physically confine the diffusion of LPSs. However, the non-polar sp2-hybridized carbon structure, lacking polar active sites, exhibits weak interactions with polar LPSs, resulting in ineffective LPS anchoring. Particularly under lean-electrolyte conditions, insufficient anchoring interactions exacerbate the heterogeneity of chemical reactions and result in markedly sluggish redox kinetics. Hence, heteroatom-doped (N, O, S) and metal-based adsorbents have been incorporated into carbon materials to enhance their polarity[10−12]. The strong binding affinity between polar materials and LPSs can suppress the shuttle effect to some extent. Nevertheless, it has gradually been recognized that increasing material adsorption alone is insufficient to address the root of the problem. As a result, LPS species trapped at the cathode continue to diffuse and are shuttle driven by concentration gradients. Therefore, the research emphasis has moved from inhibiting LPSs dissolution and diffusion toward facilitating the redox conversion between LPSs and Li2S2/Li2S, which is a key strategy for thoroughly overcoming the challenges in Li–S via catalytically active materials.
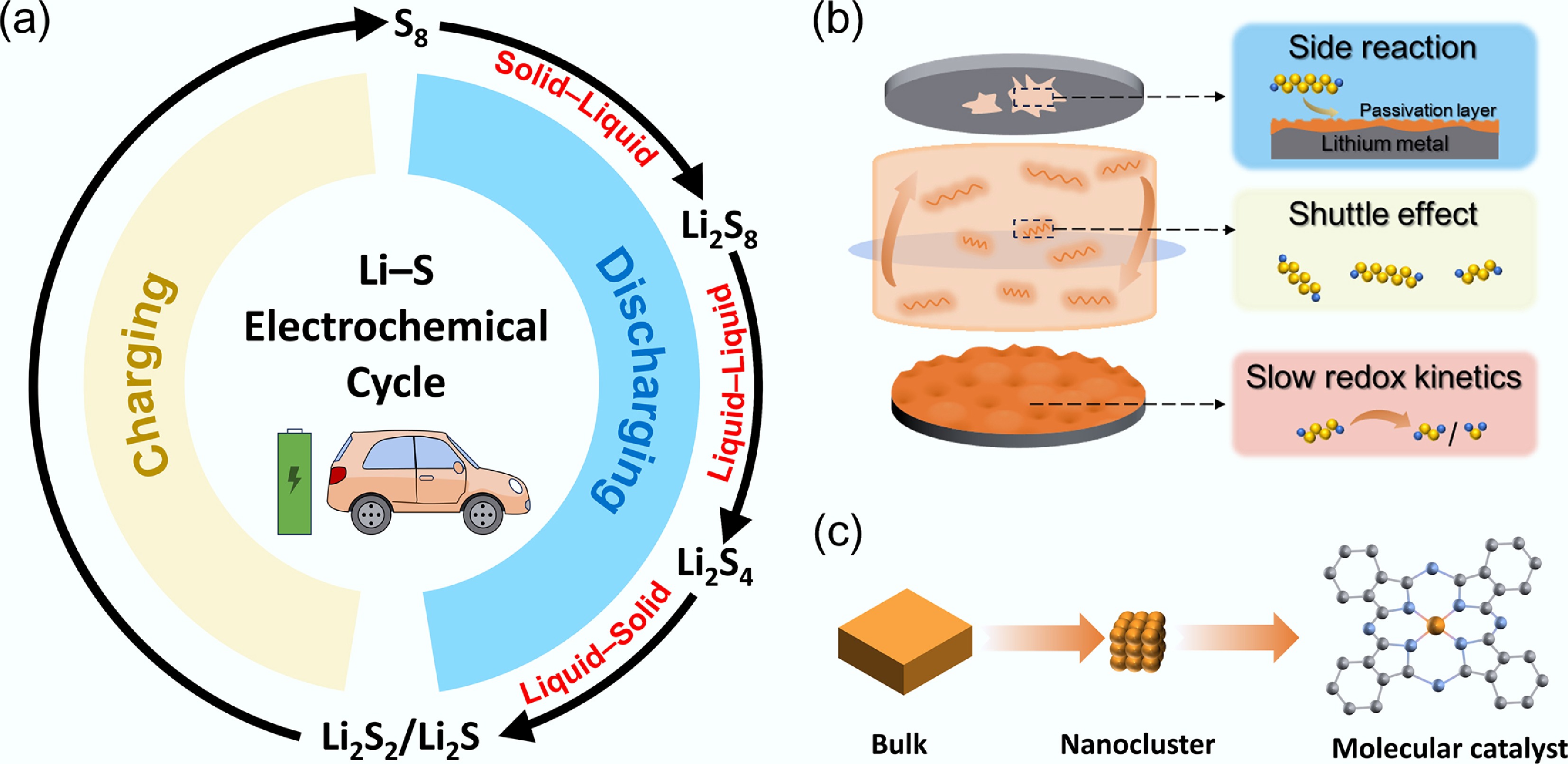
Figure 1.
(a) Electrochemical cycling process of Li–S batteries; (b) challenges of Li–S batteries; (c) the evolution from bulk materials to molecular catalysts.
To identify suitable catalytic materials, early studies primarily focused on bulk transition metal oxides, sulfides, and nitrides. Although these materials possess polar surfaces that can promote polysulfide conversion to a certain extent, their active sites are largely located within the bulk, resulting in low utilization efficiency and limited catalytic performance. Subsequently, with the advancement of nanotechnology, a series of metal nanoclusters were introduced, leading to significant improvements in rate capability under lean-electrolyte conditions. Generally, the activity and selectivity of an electrocatalyst correlate strongly with both the number of surface active sites and their intrinsic activity[13]. In this context, molecular catalysts—featuring nearly 100% utilization of catalytic centers—have attracted widespread attention (Fig. 1c). Molecular catalysts have been extensively explored in recent years in fields such as CO2 reduction and photocatalysis, and their application in Li–S batteries has also been steadily expanding[14−16]. The structural tunability of these systems endows them with broad applicability in Li–S electrocatalysis. Conventionally, catalysts are categorized as homogeneous or heterogeneous depending on their solubility in the electrolyte. From this perspective, this review highlights the intrinsic advantages of molecular catalysts in Li–S batteries and delineates their heterogeneous and homogeneous catalytic behaviors based on their respective operational mechanisms. Finally, we summarize and provide an outlook on the applications of molecular catalysts.
-
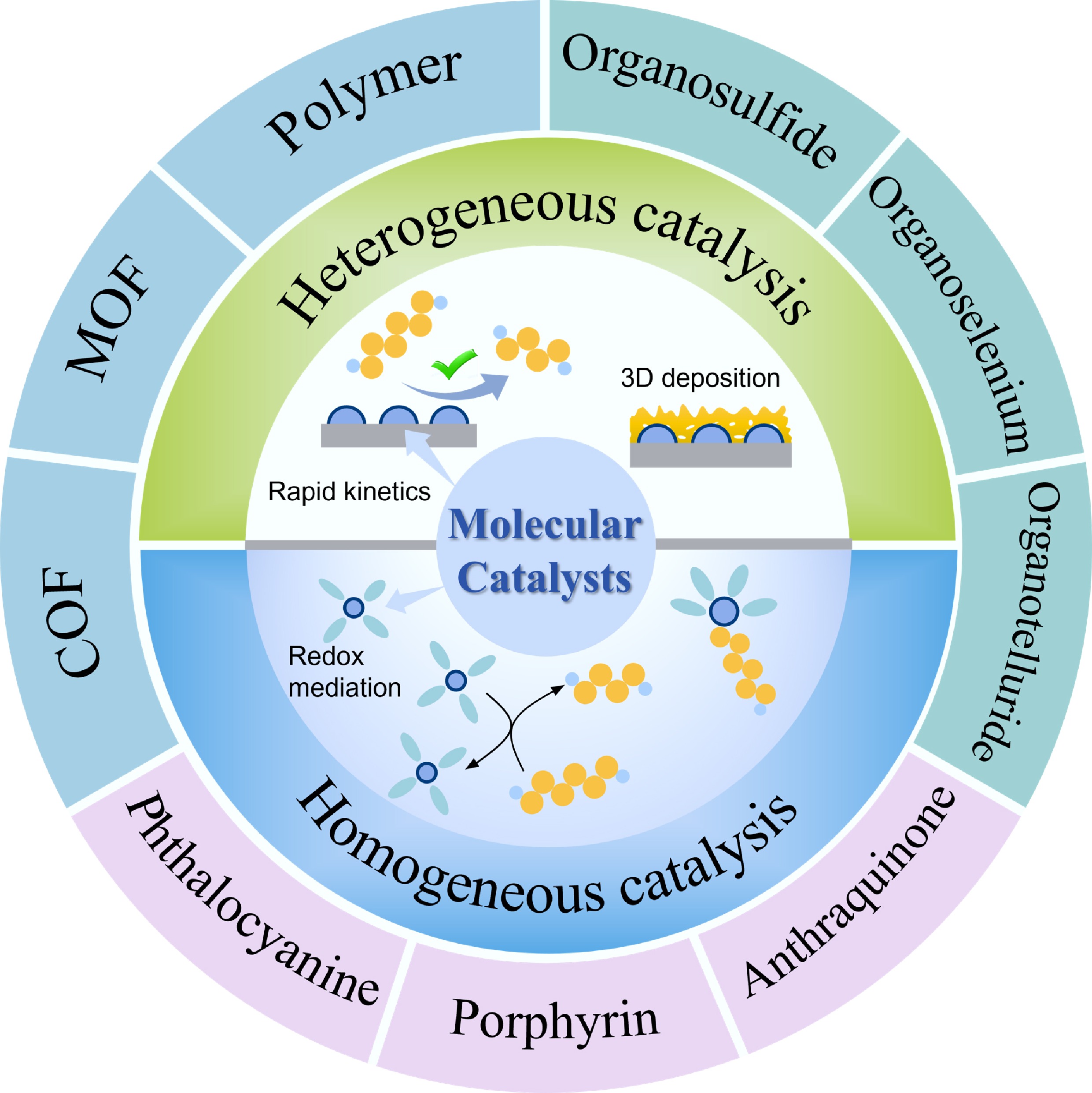
Molecular catalysts primarily refer to metal complexes composed of a central metal active site coordinated by surrounding organic ligands[14]. In addition, in Li–S batteries, certain metal-free organic molecules with intrinsic redox activity have also been reported to exhibit catalytic behavior. Existing studies have shown that a variety of molecular catalysts can participate in LPSs conversion and reaction regulation, including macrocyclic complexes such as phthalocyanines and porphyrins, as well as organic catalysts such as quinones and organochalcogen compounds. On this basis, the key advantages of molecular catalysts in electrochemical energy storage systems are outlined below (Fig. 2).
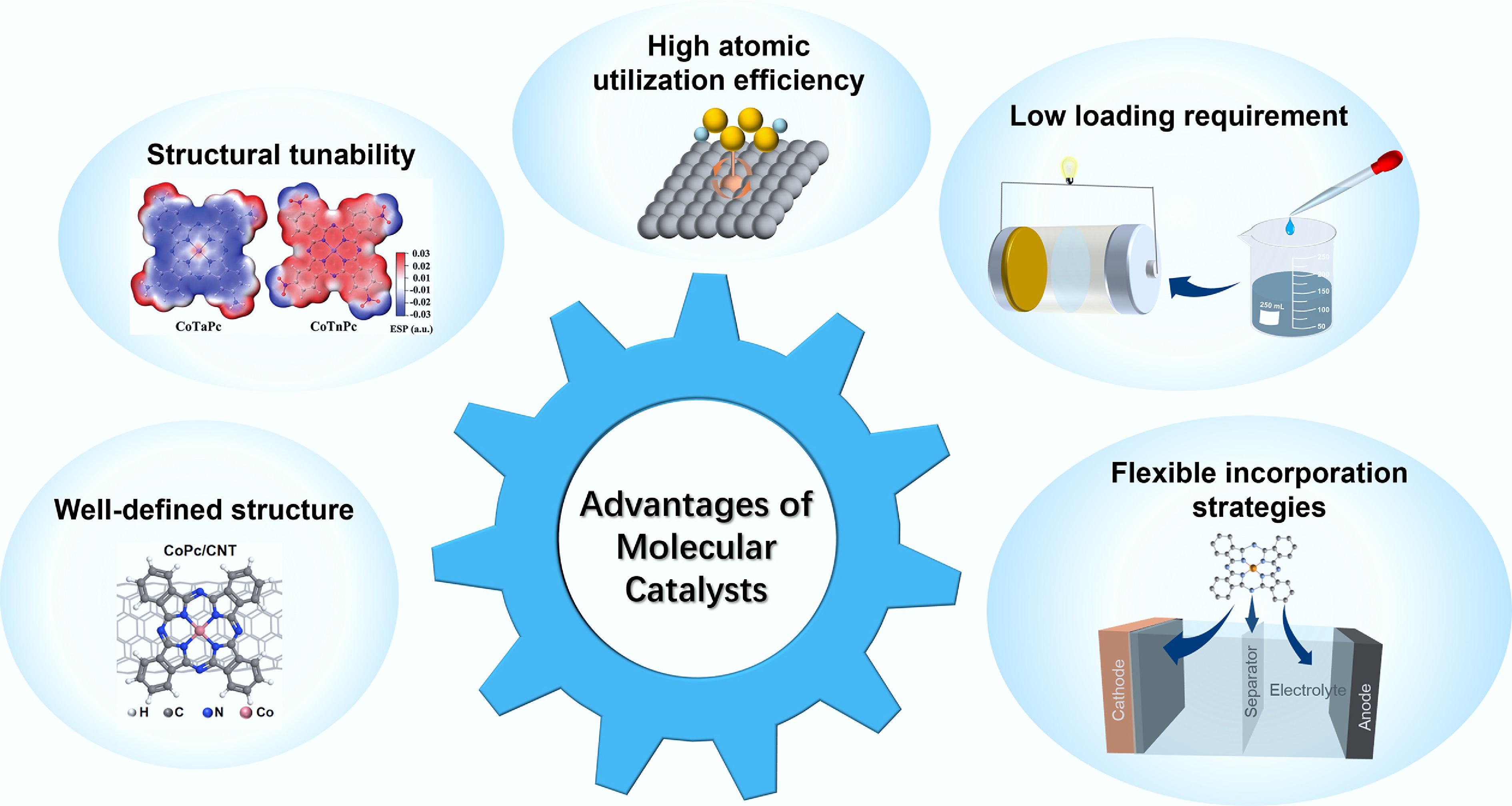
(1) Well-defined structures and clearly identifiable catalytic sites. Molecular catalysts typically possess well-defined molecular frameworks and coordination environments, whose structural information can be reliably elucidated using conventional characterization techniques such as nuclear magnetic resonance (NMR) spectroscopy, X-ray absorption spectroscopy (XAS), infrared (IR) spectroscopy, Raman spectroscopy, and X-ray diffraction (XRD). More importantly, the potential active centers of molecular catalysts can often be explicitly identified. Such structural advantages mainly apply to the molecular design and initial-state characterization of molecular catalysts, whereas their actual coordination environments under working conditions may still evolve and require further verification by operando techniques. Combined with the relatively well-defined initial structures of molecular catalysts, such operando characterization enables more reliable identification and systematic comparison of catalytic processes under realistic battery conditions.
(2) Structural tunability. The complex chemical and electrochemical processes involved in Li–S batteries require multifaceted considerations in reaction regulation. For example, although suppressing the dissolution of LPSs can mitigate the adverse effects associated with the shuttle phenomenon, a reduced LPS concentration inevitably leads to sluggish reaction kinetics[19,20]. Similarly, excessively strong interactions between catalysts and LPSs may result in poisoning of the active sites, whereas overly weak interactions lead to inefficient anchoring of LPSs[21−23]. A moderate adsorption strength is required to facilitate both adsorption and desorption. In this context, structurally tunable catalytic systems that can adapt to diverse reaction conditions are highly desirable. Molecular catalysts offer a unique advantage in this regard, as their electronic structures and spatial configurations can be precisely tailored by modulating the metal centers and coordination environments. This tunability provides a favorable platform for simultaneously balancing suppression of the LPSs shuttle and enhancement of reaction kinetics.
(3) Lightweight characteristics and high atomic utilization efficiency. Molecular catalysts typically contain only a single metal atom as the active center, thereby imposing a minimal mass burden on the battery system. In addition, compared with metal compounds, in which only a small fraction of metal species can participate in catalytic reactions[24], the metal active sites in molecular catalysts are highly dispersed within well-defined coordination environments. This single-molecule catalytic architecture enables maximal utilization of the catalytic centers.
(4) Low loading requirement. Owing to the intrinsically poor electrical conductivity of molecular catalysts, excessive loading may induce molecular aggregation, which in turn hinders electron transfer at the catalytic interface[25]. However, benefiting from their high intrinsic catalytic activity, molecular catalysts typically require only a low loading amount to achieve excellent catalytic performance. This feature is particularly important for the practical application of noble-metal-based catalysts (e.g., Pd, Pt, and Ru). In addition, the reduced catalyst loading alleviates the overall battery mass, which is favorable for the development of high-energy-density Li–S batteries.
(5) Flexible incorporation strategies. Depending on their incorporation location, molecular catalysts can exert distinct functions. Catalysts introduced into the cathode mainly regulate polysulfide conversion and Li2S deposition behavior, while those in the separator control polysulfide diffusion and adsorption, and catalysts dispersed in the electrolyte can further modulate solvation structures and reaction pathways of polysulfide species.
-
In view of the multiphase reaction characteristics of Li–S systems, the catalysis-enhanced reaction kinetics can be classified into liquid–liquid conversion and liquid–solid conversion. For liquid–liquid conversion, electrochemical impedance spectroscopy (EIS) results indicate that accumulation of LPSs leads to increased electrolyte resistance, thereby impeding their conversion pathway[26]. Some transition metal compounds, such as CoS2[27], have been demonstrated to effectively catalyze and accelerate the liquid–liquid conversion of LPSs.
During discharge, the liquid–solid conversion (typically associated with the conversion from Li2S4 to Li2S2/Li2S) contributes approximately 70% of the theoretical capacity, making it critical for fully unleashing the potential of Li–S batteries. However, a growing body of studies suggests that the conversion from soluble LPSs to solid sulfur species presents the largest kinetic barrier, serving as the crucial rate-determining step of the sulfur reduction reaction (SRR)[28,29]. This phenomenon becomes even more pronounced in lean electrolyte systems[20], where the reduced ionic conductivity and increasingly non-uniform electrochemical reactions further slow down the deposition kinetics of solid sulfur species, leading to a continuous decline in the lower discharge plateau. Chen et al.[29] employed electrochemical impedance spectroscopy combined with galvanostatic intermittent titration technique (EIS–GITT) to decompose the overall cathodic polarization (ηtotal) during discharge into activation polarization (ηac), concentration polarization (ηcon), and ohmic polarization (ηohm). Specifically, the instantaneous voltage jump at the end of a discharge pulse reflects the sum of ηohm and ηac, while the relaxed equilibrium potential during the subsequent rest period represents ηcon; EIS tests performed before each discharge further permit the determination of ηohm. Based on this analytical framework, the authors demonstrated that Li2S nucleation contributes the largest ηtotal, with ηac being identified as the key kinetic limiting factor during the Li2S nucleation stage, which dominates the sluggish kinetics and performance degradation observed in lean-electrolyte Li–S batteries. Therefore, targeting the key kinetically limiting steps and leveraging the advantages of molecular catalysts, a series of homogeneous and heterogeneous molecular catalysts have been proposed.
Homogeneous and heterogeneous catalysis of molecular catalysts
-
Conventionally, catalysts are classified as either homogeneous or heterogeneous based on whether the catalytic mediator and the substrate are in the same phase. In a typical Li–S battery, the reaction substrate, namely LPSs, is always present in the liquid phase. Therefore, heterogeneous molecular catalysts are typically embedded within conductive substrates, whereas homogeneous molecular catalysts are dispersed in the electrolyte. This classification provides a useful conceptual framework for understanding their distinct catalytic pathways and structure–performance relationships in Li–S chemistry.
Figure 3 illustrates the distinct catalytic mechanisms of homogeneous and heterogeneous molecular catalysts. The incorporation of catalytic molecules into the cathode or separator enables heterogeneous catalysis of LPSs, which can directly regulate the nucleation and decomposition kinetics of Li2S. However, the deposition of an insulating sulfide layer during the late stage of discharge covers the catalytic sites and prevents direct contact between LPSs and the catalysts, thereby hindering the catalytic function of molecular catalysts. Furthermore, the effective operational region of heterogeneous catalysis is confined to a narrow region perpendicular to the catalyst surface, rendering the reaction capacity dependent on the specific surface area of the cathode or separator. In contrast, uniformly dissolving catalyst molecules in the electrolyte increases the collision frequency between reactants and catalyst molecules, thereby overcoming the limitation imposed by a finite active surface area. The mechanistic roles of molecular catalysts in the electrolyte can be categorized into redox mediation, and modulation of the chemical states of LPS intermediates. The mechanism of redox mediation can be summarized as follows: during discharge, the catalyst molecules diffuse to the cathode surface and undergo electrochemical reduction to form the reduced-state redox mediator (RM). Subsequently, the reduced RM chemically reacts with LPSs to generate short-chain LPSs, while itself being oxidized and diffusing back into the electrolyte to participate in the next cycle. Catalyst molecules dispersed in the electrolyte can also facilitate the formation of a more stable solid electrolyte interphase (SEI). These advantages have enabled the widespread application of molecular catalysts in Li–S batteries. Based on this classification, the following sections discuss the mechanistic roles of heterogeneous and homogeneous molecular catalysts in Li–S batteries in greater detail.
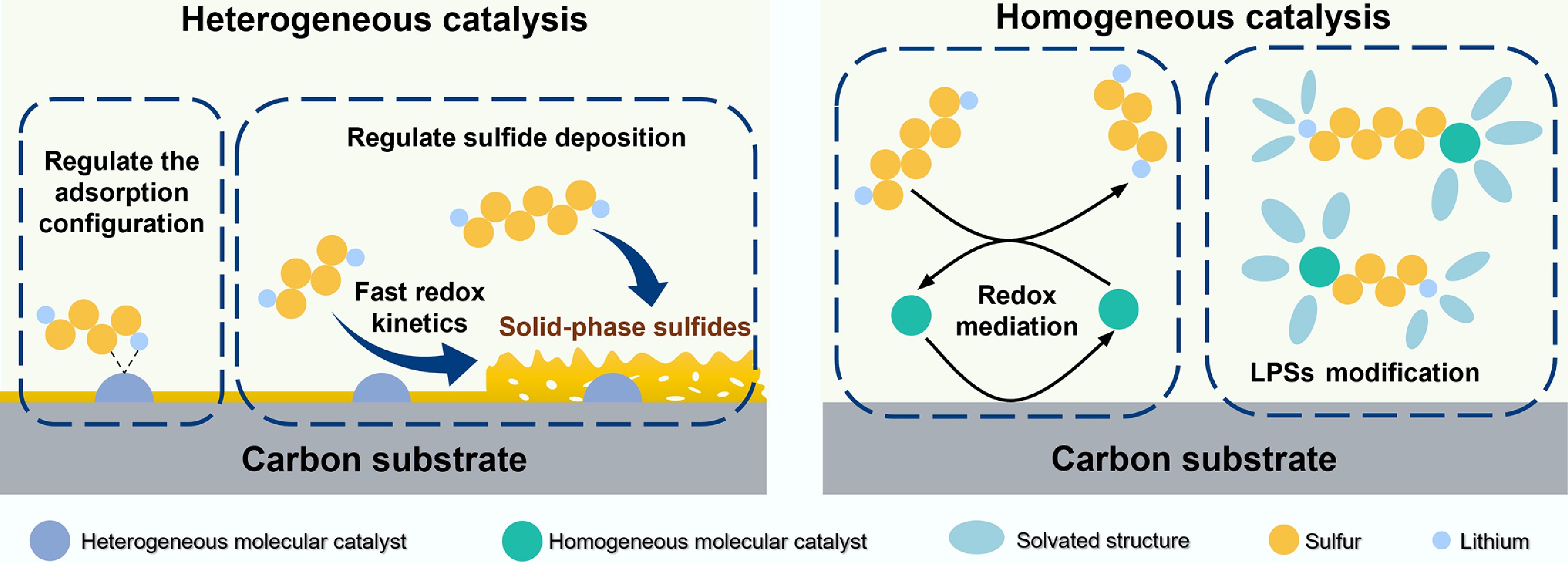
Figure 3.
Homogeneous and heterogeneous molecular catalytic roles in Li–S batteries.
-
In Li–S batteries, heterogeneous molecular catalysts regulate the adsorption and conversion of LPSs by constructing a stable solid–liquid interface. Distinct from homogeneous systems, the catalytic function of heterogeneous molecular catalysts primarily relies on the modulation of interfacial adsorption and reaction kinetics. Overall, molecular catalysts can synergistically optimize sulfur conversion processes and electrochemical performance through multiple mechanisms, including the regulation of adsorption configurations, control of deposition morphology, and modulation of reaction pathways. Accordingly, this section systematically discusses the roles of molecular catalysts from these three aspects.
Adjustment of the adsorption configuration
-
In heterogeneous molecular catalytic systems, the construction and regulation of adsorption configurations are among the key factors governing catalytic performance. The adsorption configuration of polysulfides at active sites not only determines their binding strength, but also directly influences electron transfer and reaction energy barriers, thereby regulating sulfur conversion kinetics. Among various molecular catalysts, macrocyclic conjugated molecules represented by phthalocyanines and porphyrins possess stable metal–ligand coordination structures and tunable electronic environments. They feature highly tunable structures, with a central cavity capable of coordinating nearly 70 different metal elements[30]. Even metal-free phthalocyanine (H2Pc) has been demonstrated to be effective in capturing LPSs (Fig. 4a)[31]. Moreover, the C–N, Fe–N, and C–N=C bonds in the phthalocyanine skeleton also contribute to the catalysis of LPSs[32]. To date, metal centers employed in phthalocyanines and porphyrins for Li–S batteries include Co, Fe, Ni, Cu, In, and Mn, among others. Owing to the generally low solubility of most metal phthalocyanines in conventional solvents[33], they can be introduced into the cathode and separator as heterogeneous catalysts.
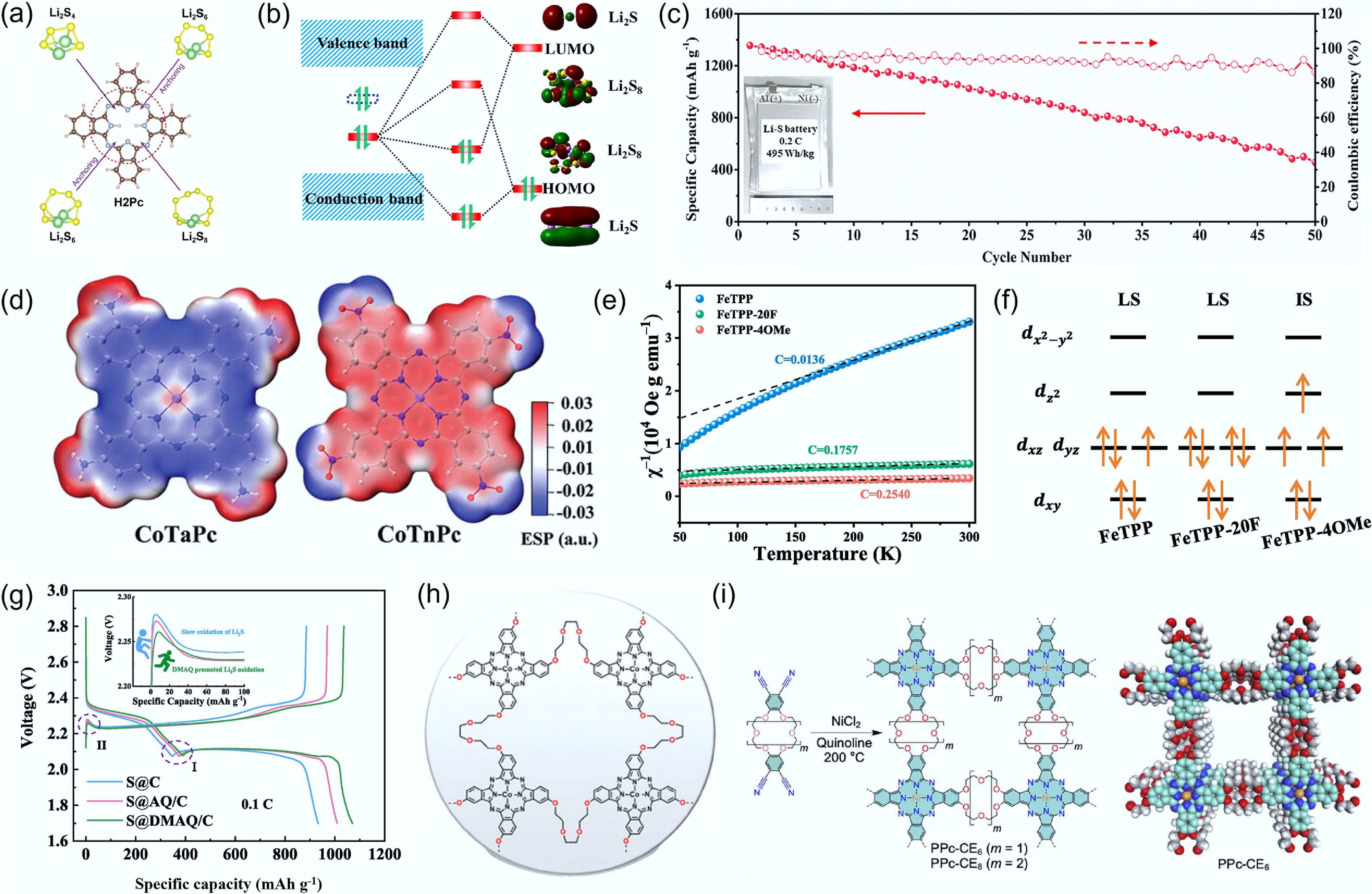
Figure 4.
(a) Anchoring effect of H2Pc on LPSs. Reproduced with permission[31]. Copyright 2019, Elsevier. (b) Fermi level shift induced by the introduction of In. Reproduced with permission[34]. Copyright 2023, John Wiley & Sons. (c) Cycling performance of pouch cells employing separators integrated with SAIn@CNT. Reproduced with permission[35]. Copyright 2023, RSC Publishing. (d) Electrostatic potential plots of CoTaPc and CoTnPc. Reproduced with permission[18]. Copyright 2023, John Wiley & Sons. (e) The temperature dependence inverse susceptibilities, and (f) electronic configurations of FeTPP, FeTPP-4OMe, FeTPP-20F. Reproduced with permission[36]. Copyright 2024, Elsevier. (g) Charge–discharge profiles of S@C, S@AQ/C, and S@DMAQ/C at 1 C. Reproduced with permission[37]. Copyright 2024, Elsevier. (h) Molecular structure of the PCT[38]. Copyright 2023, John Wiley & Sons. (i) Schematic illustration of the synthesis procedure of PPc-CE and the space-filling model of its repeating unit. Reproduced with permission[39]. Copyright 2025, John Wiley & Sons.
To achieve stable and controllable adsorption configurations, it is first necessary to effectively immobilize the catalyst molecules onto conductive substrates to prevent their aggregation and migration during the reaction process. However, phthalocyanines and porphyrins tend to aggregate due to intermolecular π–π interactions, which reduces the utilization efficiency of the active sites[40]. Ordinary mechanical stirring and ball milling are ineffective in breaking this aggregation, while ultrasonication induces the rapid collapse of microbubbles in solution, generating high-energy shockwaves that disrupt nanoparticle clusters. Additionally, the cavitation effect produces high temperatures, which can break the van der Waals forces between carbon nanotubes and the interactions between the catalyst molecules[41,42]. Several studies have successfully used ultrasonic dispersion of carbon nanotubes and phthalocyanines in N, N-dimethylformamide (DMF) solution, which provides high polarity and favorable solvation for both carbon nanotubes and molecular catalysts, to break these interactions, achieving molecularly dispersed phthalocyanines on the carbon matrix[43−45].
It has also been reported that molecules can be grown on carbon nanotube surfaces via liquid-phase self-assembly. For instance, Guo et al.[34] employed this strategy to deposit InPc-containing catalysts onto the separator. Benefiting from the unique coordination environment, the incorporation of In induces modifications in the electronic structure (Fig. 4b). As a result, both the Gibbs free energy of the exothermic conversion from S8 to Li2S, and the energy barrier for Li2S decomposition are significantly reduced, leading to a decrease in polarization voltage from 0.21 to 0.16 V. In another study[35], compared with In–N4, the In–N3 configuration exhibits a Fermi level closer to the conduction band, enabling faster electron transfer kinetics and stronger interactions with LPSs. As a result, an exceptional pouch cell energy density of 495 Wh kg−1 at 0.2 C was achieved, along with stable cycling over 50 cycles (Fig. 4c). In addition to defect-coordinated metal–N3 structures, metal–N5 coordination has also been demonstrated to exhibit excellent catalytic activity. Zhang et al.[46] independently synthesized a nitrogen–phosphorus co-doped porous carbon material. Their results revealed that, beyond the coordination between Fe atoms and the nitrogen atoms in the phthalocyanine ring, Fe atoms also coordinate with nitrogen species in the carbon matrix. Moreover, electron-rich phosphorus species can regulate the electronic properties of the carbon framework, enabling enhanced coupling between nitrogen and iron sites and improving reaction kinetics.
Molecular catalysts can also regulate adsorption configurations by tuning different metal centers. For example, Li et al.[47] synthesized a series of porphyrin-based metal organic–framework (MOF) nanosheets, denoted as PCN-222(M)-NSs (M = Fe3+, Co2+, Ni2+, and Cu2+). The interactions between these MOF nanosheets and LPSs were strongly dependent on the nature of the metal centers. Specifically, the Co–N4 coordination configuration exhibited the strongest chemisorption toward LPSs, whereas the Cu–N4 sites more readily facilitated S–S bond cleavage, resulting in higher catalytic activity. Consequently, the cell incorporating a PCN-222(Cu)-NS interlayer achieved a high areal capacity of 5.3 mAh cm−2 at a sulfur loading of 5.7 mg cm−2. In addition to varying the central metal, the phthalocyanine macrocycle can also be functionalized with diverse substituents, enabling the rational design of catalysts with tailored functionalities. For example, sulfonic acid groups introduced into phthalocyanine molecules can directly anchor LPSs, thereby influencing the overall adsorption–catalysis process[48]. The influence of substituents on catalytic performance urgently requires the introduction of unified descriptors to enable more efficient screening of molecular catalysts. Among various descriptors, electron-withdrawing and electron-donating substituents have attracted widespread attention. For instance, Zhao et al.[18] compared the effects of electron-withdrawing nitro groups and electron-donating amino groups (Fig. 4d) on the SRR. It was found that cobalt tetraaminophthalocyanine (CoTaPc) preferentially forms covalent bonds with the terminal sulfur atoms of LPSs, whereas cobalt tetranitrophthalocyanine (CoTnPc) tends to form covalent bonds with the bridging sulfur atoms of LPSs. The resulting adsorption geometries give rise to distinct catalytic behaviors, with electron-withdrawing nitro groups exhibiting a superior ability to weaken bridging S–S bonds and thereby promote the liquid–solid conversion of LPSs.
In contrast to electron-withdrawing groups, which enhance catalytic performance by inducing interactions between the metal center and bridging sulfur species, electron-donating groups can also promote the formation of more favorable adsorption configurations by tuning the electronic environment of the central atom. For example, Wang et al.[36] synthesized graphene-supported fluorinated tetraphenylporphyrin iron (FeTPP-20F) and methoxy-substituted tetraphenylporphyrin iron (FeTPP-4OMe). 57Fe Mössbauer transmission spectra indicate that the introduction of the –OMe group induces a transition of the Fe center from a low-spin state to an intermediate-spin state, accompanied by the excitation of more electrons to higher energy levels. Consequently, FeTPP-4OMe exhibits a higher density of unpaired electrons, as further confirmed by temperature-dependent inverse magnetic susceptibility measurements (Fig. 4e). This ultimately leads to a reconfiguration of the Fe 3d electron spin states, providing an increased number of free valence electrons (Fig. 4f). Therefore, batteries based on FeTPP-4OMe exhibit the lowest activation energy, higher Li2S nucleation capacity, and faster nucleation kinetics.
The behavior of electron-donating substituents in anthraquinone derivatives differ from those in macrocyclic systems such as phthalocyanines and porphyrins. In general, electron-donating groups such as –OH increase the electron density on the carbonyl oxygen atoms of anthraquinone, thereby strengthening their interaction with LPSs[49]. By comparing the UV–vis spectra of 2,6-dimethoxyanthraquinone (DMAQ) before and after interaction with carbon materials, Gao et al.[37] found that the p orbitals of oxygen atoms in methoxy groups form p–π conjugations with the aromatic rings of the carbon substrate, thereby strengthening intermolecular interactions and effectively suppressing the dissolution in organic electrolytes. In addition, the carbonyl oxygen in DMAQ exhibits a more negative electrostatic potential, leading to stronger adsorption toward LPSs and reduced energy barriers for Li2S nucleation and decomposition (Fig. 4g).
However, single-molecule adsorption sites are often unstable, as they can be readily covered by deposited sulfur species and are prone to migration, redistribution, or loss of the originally designed adsorption configuration during electrochemical operation, thereby weakening their effectiveness in catalytic regulation. To achieve more stable adsorption configurations, a series of polymeric and framework structures has been proposed. Kim et al.[38] synthesized a novel triethylene glycol (TEG)-functionalized polymeric cobalt phthalocyanine and subsequently loaded it onto multiwalled carbon nanotubes (MC) (Fig. 4h). Benefiting from its unique polymeric architecture, the polymeric cobalt phthalocyanine (TCP) layer uniformly coats the surface of MC without noticeable aggregation of phthalocyanine units. This composite material not only mediates the reaction kinetics of LPSs but also provides additional adsorption sites for lithium ions through the TEG linker groups. As a result, sulfur cathodes based on this material deliver a capacity retention of 81.5% after 200 cycles, and even at a high sulfur loading of 6.6 mg cm−2, an initial discharge capacity of 1,032 mA h g−1 is achieved at 0.2 C. Moreover, covalent organic frameworks (COFs) formed through the covalent linkage of organic building units have also been widely applied. For instance, when employed as a separator modification material, porphyrin-based conductive COF with 3,4-ethylenedioxythiophene unit (EDOT-Por-COF) exhibits superior mechanical robustness and tensile strength[50]. Benefiting from these advantages, separators incorporating EDOT-Por-COF enable stable cycling over 2,000 cycles at 1 C. In another work, a two-dimensional phthalocyanine-based COF, termed PPc–crown ether (PPc–CE), has been constructed by linking nickel phthalocyanine (NiPc) units with crown ether (CE) moieties and employed as a separator coating (Fig. 4i)[39]. This COF architecture creates a supramolecular confined space, in which the CE linkers effectively adsorb LPSs. Meanwhile, the NiPc units act as catalytic sites to enhance the redox kinetics of LPSs. As a result, efficient sulfur utilization under lean-electrolyte conditions is achieved, delivering a high areal capacity of 5.32 mAh cm−2 at high sulfur loading. In summary, these framework structures provide more stable adsorption sites for LPSs and offer greater flexibility for tuning the adsorption configuration.
Regulation of the product morphology
-
The deposition of solid sulfur species, as a critical step in the sulfur reduction process, follows a 'nucleation–proliferation–growth' mechanism during the discharge process. First, solid-phase sulfide nucleates on the cathode substrate by overcoming the high interfacial impedance. This high interfacial impedance mainly originates from the nucleation and interfacial energy barriers associated with the liquid–solid conversion, as well as the poor electronic and ionic conductivity of Li2S/Li2S2, which hinders interfacial charge transfer. Subsequently, due to the attraction between similar substances, LPSs are adsorbed onto the formed solid-phase sulfide nuclei and are simultaneously deposited in the form of solid-phase sulfide. As discharge proceeds, the number of solid-phase sulfide nuclei increases rapidly, preparing for the later growth of these nuclei. It is demonstrated that solid-phase sulfide prefers to grow adjacent to the solid-phase sulfide nuclei along the interface, forming solid-phase sulfide islands. Finally, these islands will proliferate into a thin layer as the battery discharges. Such deposition morphology evolution plays a critical role in battery performance, in which more dispersed and uniformly anchored structures are beneficial for sulfur utilization and cycling stability. An important approach for characterizing deposition morphology is based on the analysis of four classical nucleation models derived from the Bewick–Fleischmann–Thirsk (BFT) and Scharifker–Hills (SH) models. Xu et al.[51] employed this analytical framework to investigate the morphology regulation capability of N-skeleton-substituted cobalt tetrapyridinoporphyrazine-loaded reduced graphene oxide (CoTAP/rGO). Benefiting from the rapid nucleation and growth kinetics of solid-phase sulfides, the deposition behavior on CoTAP/rGO follows a hybrid mode consisting of three-dimensional (3D) spherical growth and two-dimensional (2D) sheet-like Li2S deposition (Fig. 5a). Compared with rGO, CoTAP/rGO exhibits a higher nucleation rate, leading to the formation of a more porous deposition layer (Fig. 5b). Molecular catalysts also play a critical role in stabilizing morphology during cycling. For instance, Yang et al.[52] modified the cathode with cobalt(II) tetraaminophthalocyanine (TaPcCo)-functionalized multiwalled carbon nanotubes (MWCNTs). After 200 cycles, in contrast to the pure CNF electrode, where solid sulfides detach from the conductive substrate, the solid sulfur species in the TaPcCo-MWCNT cathode remain uniformly embedded within the conductive network. Moreover, the solid-phase sulfide nucleation capability of the TaPcCo-MWCNT-based cell is nearly doubled, which can be attributed to the unoccupied d orbitals of the Co centers that significantly promote nucleation kinetics.
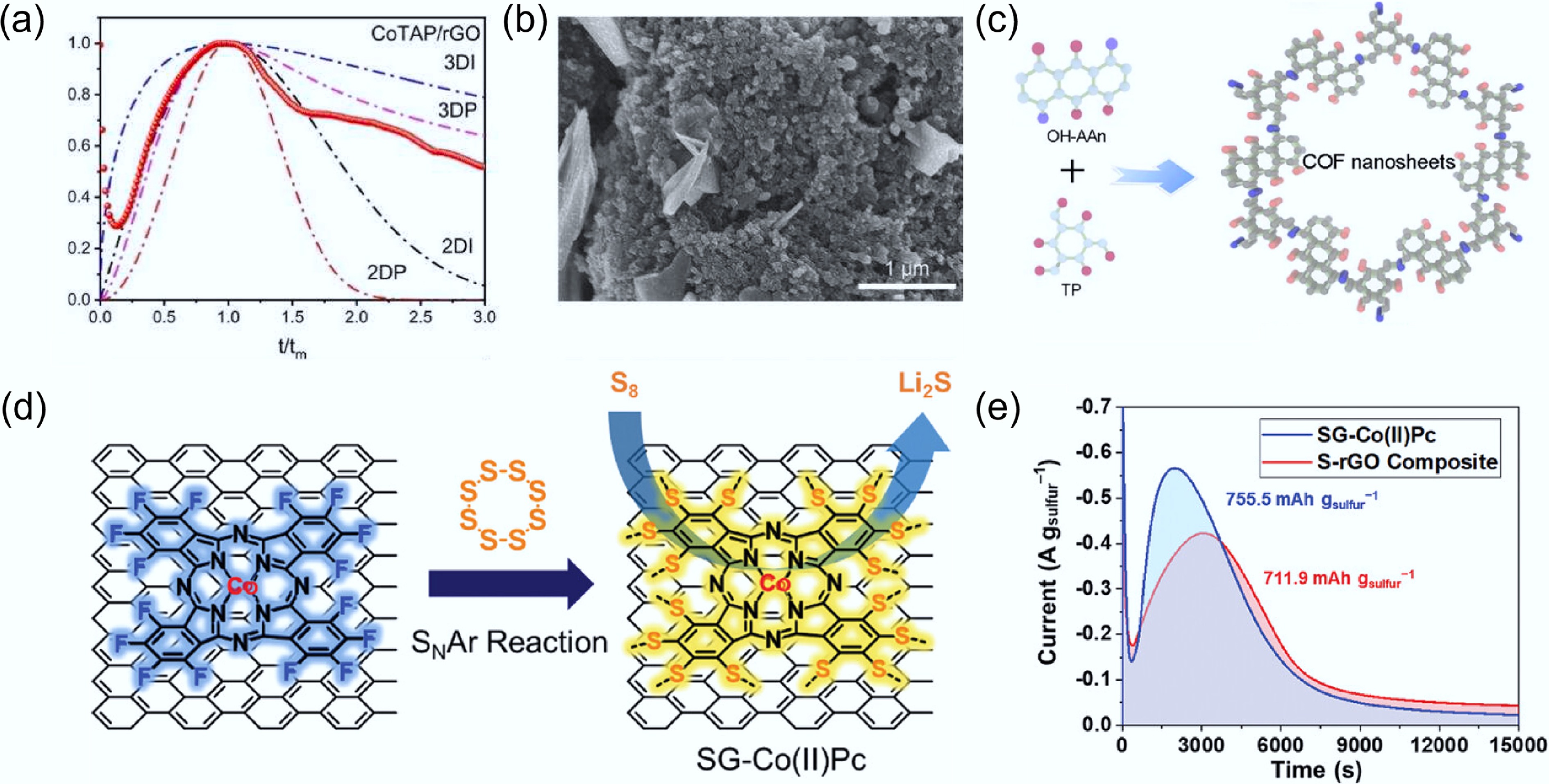
Figure 5.
(a) Comparison between the dimensionless current-time transient curves of the CoTAP/rGO separator and the theoretical model. (b) Final deposited Li2S morphology with the CoTAP/rGO-modified separator. Reproduced with permission[51]. Copyright 2024, Elsevier. (c) Schematic illustration of the OH-AAn-COF structure. Reproduced with permission[53]. Copyright 2025, John Wiley & Sons. (d) Nucleophilic substitution reaction between rGO/F-Co(II)Pc and S8. (e) Potentiostatic discharge curves using SG-Co(II)Pc and S-rGO. Reproduced with permission[54]. Copyright 2021, John Wiley & Sons.
Framework structures constructed from molecular assemblies possess abundant active nucleation sites, which are of great significance for improving Li2S decomposition/deposition kinetics and promoting 3D sulfide growth. For example, Zhao et al.[55] reported a bimetallic two-dimensional MOF, in which transition metal centers (M = Mn, Fe, Co, Ni, and Cu) serve as coordination nodes linking 5,10,15,20-tetrakis(4-pyridyl)porphyrin cobalt (CoTPyP). The rich catalytic sites significantly reduce the charge-transfer resistance of LPSs, delivering the highest capacity and the shortest response time during solid-phase sulfide nucleation and dissociation. This bidirectional catalytic behavior enables stable cycling for over 1,000 cycles at a rate of 2 C. Covalent organic frameworks (COFs) also exhibit unique capabilities in regulating deposition morphology. Li et al.[53] constructed an anthraquinone-based COF (OH-AAn-COF) (Fig. 5c) and employed it as a cathode binder in Li2S-based Li–S batteries. The abundant micro/mesoporous structure and high density of active sites in OH-AAn-COF effectively reduce the activation overpotential of Li2S and suppress the shuttle effect.
Certain molecular catalysts can regulate the nucleation and growth of solid sulfur species by modulating the initial state of sulfur, thereby enabling higher sulfur utilization. For example, Kim et al.[54] employed a nucleophilic aromatic substitution (SNAr) reaction between fluorinated cobalt(II) phthalocyanine (F-Co[II]Pc) and S8 to form covalent bonds between sulfur and the pristine rGO/F-Co(II)Pc composite (Fig. 5d). Owing to the synergistic effect between the catalytic Co centers and the adsorption capability of nitrogen atoms, enhanced Li2S nucleation and growth currents were observed in potentiostatic discharge measurements (Fig. 5e). In another study, Shao et al.[56] proposed a regulation strategy from the perspective of initial sulfur microcrystals by integrating Fe–N4-doped porous carbon single-atom catalysts with anthraquinone. The unique structural design regulates the sulfur grain size on the (222) crystal facet, thereby generating a higher density of catalytically active sites. This effect becomes more pronounced with increasing current density, effectively mitigating sulfur aggregation and ultimately enhancing sulfur utilization. Overall, molecular catalysts can efficiently regulate the deposition kinetics of solid-phase sulfides, thereby promoting favorable deposition morphologies and improving sulfur utilization.
Modulation of the reaction pathway
-
Conventional charge/discharge reaction pathways are inherently constrained by their intrinsic activity. In recent years, it has been found that molecular catalysts can alter the LPS intermediates involved in the reaction through regulation of the metal centers. For example, Li et al.[57] encapsulated iron tetrakis(4-carboxyphenyl)porphyrin (Fe-TCPP) into the Cu-BTC metal–organic framework (MOF) to construct a biomimetic catalyst with enzyme-like catalytic performance. In this system, the dual Cu and Fe metal centers promote the homolytic cleavage of Li2Sn into LiSn/2, thereby changing the reaction pathway (Fig. 6a). The new discharge pathway follows classical Michaelis–Menten kinetics (Fig. 6b). Within this framework, the Michaelis constant (Km) reflects the substrate affinity of the biomimetic catalyst, whereas Vmax characterizes its intrinsic catalytic activity. Accordingly, Fe-TCPP@Cu-BTC exhibits the lowest Km value, indicating the strongest affinity toward polysulfides, and improves the sulfur conversion efficiency by nearly two orders of magnitude. These results demonstrate the great potential of molecular catalysts to provide alternative reaction pathways for faster and more efficient Li–S battery chemistry.
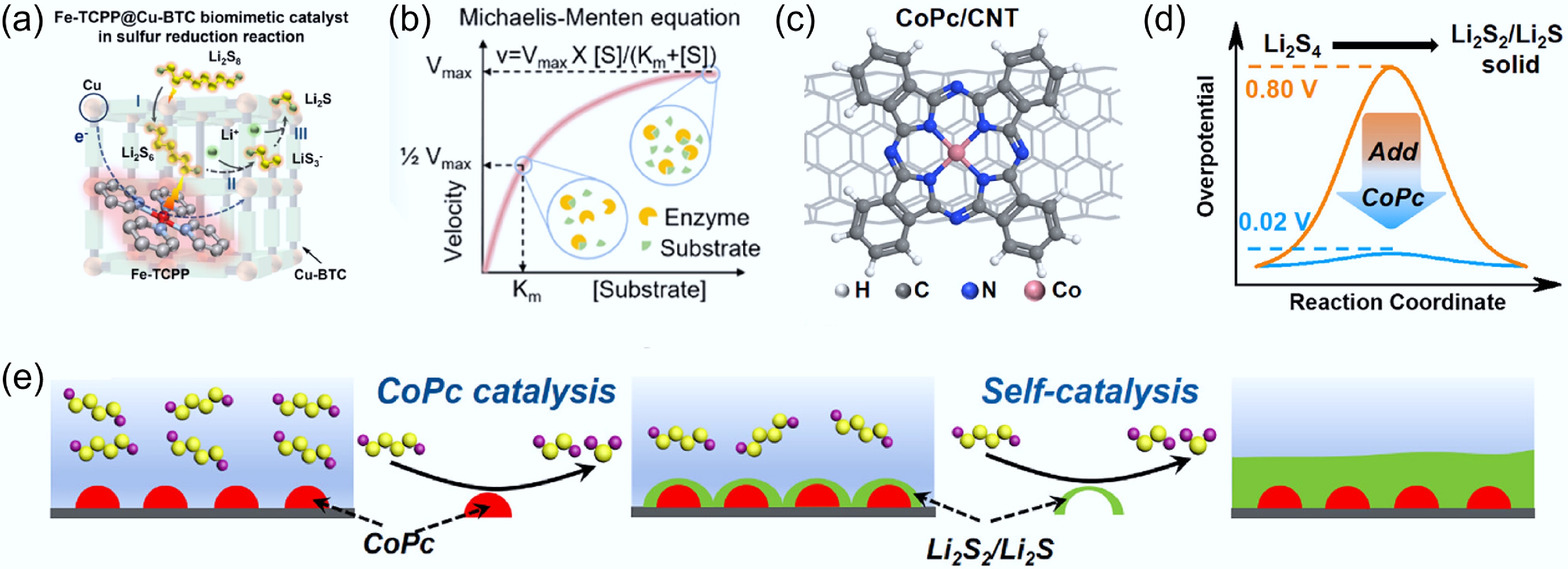
Figure 6.
(a) Pathway diagram for the catalytic conversion of LPSs by Fe-TCPP@Cu-BTC. (b) Schematic illustration of the Michaelis–Menten kinetics equation. Reproduced with permission[57]. Copyright 2025, American Chemical Society. (c) Molecular structure of the CoPc/CNT catalyst. (d) Reduction in reaction energy barrier upon CoPc incorporation. (e) Schematic illustration of solid sulfide deposition on CoPc/CNT. Reproduced with permission[17]. Copyright 2023, American Chemical Society.
The conventional view holds a rough picture that catalysts accelerate sulfur reduction/oxidation reactions by lowering the activation energy and facilitating rapid LPSs conversion. However, over the past decades, limited attention has been paid to how catalysis influences the sulfur redox pathways and how catalysis proceeds after heterogeneous active sites are covered by solid sulfides. In this context, Zhong et al.[17] conducted a model study using cobalt phthalocyanine (CoPc) molecules (Fig. 6c) and clearly elucidated the sulfur reduction pathway in the presence of molecular catalysts. The results show that, at the initial stage of discharge, sulfide species preferentially nucleate near CoPc/CNT, where the catalytic effect significantly reduces the nucleation overpotential from 0.8 to 0.02 V (Fig. 6d). As discharge proceeds, the deposited sulfide layer expands into a thin film that progressively covers the CoPc/CNT catalytic sites. Subsequently, the reaction is driven by the self-catalytic behavior of the deposited Li2S2/Li2S (Fig. 6e). Notably, regardless of the presence of CoPc/CNT, the final discharge product is consistently Li5S3 (Li2S2 : Li2S = 1:4), indicating that molecular catalysts primarily facilitate the early-stage conversion from Li2S4 to Li2S2/Li2S without altering the intrinsic activity limitation of the cathode. Therefore, future research should focus on overcoming these intrinsic limitations to achieve a complete reaction pathway from S8 to Li2S.
Homogeneous catalytic role in Li–S batteries
Redox mediation
-
Soluble redox mediators refer to a class of electron–hole transfer agents that mediate the conversion of liquid-phase LPSs[58,59]. To ensure that a redox mediator can function effectively, it is generally required to satisfy the following conditions: (1) appropriate solubility and diffusion capability in the electrolyte to ensure rapid access to the reaction zone while minimizing undesired migration and parasitic reactions at the lithium metal anode; (2) a redox potential that closely aligns with the thermodynamic equilibrium potential of Li–S batteries; and (3) stability under both electrochemical and chemical conditions.
Metallocenes are compounds formed by the sandwich structure of a metal atom coordinated with two cyclopentadienyl ligands, exhibiting stable, reversible redox behavior. Cobaltocene (Co[Cp]2), as an electrolyte-soluble redox mediator, exhibits two key features in its cyclic voltammogram that demonstrate its catalytic mediation of LPSs redox reactions[60,61]: first, the reduction peak of Co(Cp)2 is shifted approximately 27 mV more negative than the Li2S formation peak, providing thermodynamic support for the chemical reaction Co(Cp)2+ + Li2Sn → Co(Cp)2 + Li2S; second, the Co(Cp)2/Co(Cp)2+ redox couple shows a Tafel slope of 67.6 mV dec−1, which is lower than the 177.8 mV dec−1 observed for LPSs, indicating faster electrochemical conversion kinetics for CoCp2/CoCp2+ than for intrinsic LPSs. This mediated reaction guides three-dimensional Li2S growth (Fig. 7a), resulting in an 8.1-fold increase in battery capacity under lean electrolyte conditions. In addition to cobaltocene, other metallocenes (e.g., titanocene[62], chromocene, and nickelocene[63]) have also been reported as redox mediators in Li–S batteries. Due to the fixed redox potential in a given electrolyte, a single redox mediator cannot simultaneously facilitate both discharge and charge reactions. Introducing dual-redox mediators provides an effective solution to this limitation. For example, Li et al.[63] constructed a Li–S redox flow battery using bis (pentamethylcyclopentadienyl) chromium (Cr[Cp]2) and bis (pentamethylcyclopentadienyl) nickel (Ni[Cp]2) (Fig. 7b). Because the electrode potentials satisfy E(Cr[III]Cr[Cp]2+/Cr[II][Cp]2) < E(S/Li2S) < E(Ni[III][Cp]2+/Ni[II][Cp]2), Cr(Cp)2 functions as the redox mediator during discharge, while Ni(Cp)2 serves as the mediator during charge. This novel operating mechanism provides an alternative strategy for Li–S battery systems.
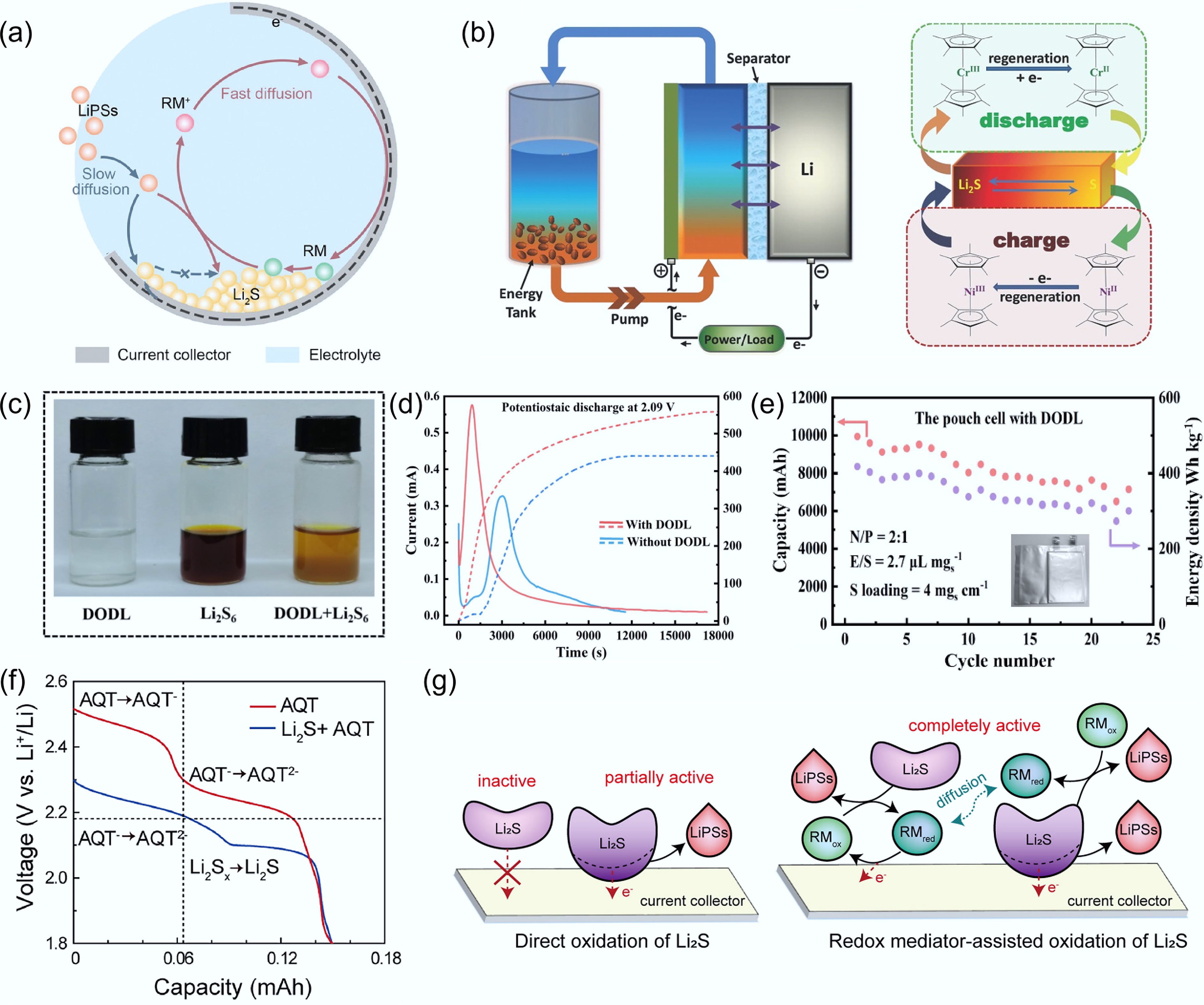
Figure 7.
(a) Schematic illustration of the Li2S nucleation pathway mediated by CoCp2. Reproduced with permission[60]. Copyright 2019, John Wiley & Sons. (b) Schematic illustration of redox reactions in a flow Li–S battery mediated by two metallocenes. Reproduced with permission[63]. Copyright 2015, John Wiley & Sons. (c) Color evolution of the Li2S6 solution after the addition of DODL. (d) Li2S nucleation chronoamperometry curves with, and without DODL addition. (e) Cycling performance of pouch cells with DODL at 0.2 C. Reproduced with permission[64]. Copyright 2022, Elsevier. (f) Discharge voltage profiles of a half-cell with a Li2S electrode and an AQT-containing electrolyte, and a half-cell containing only AQT without Li2S. (g) Schematic illustration of Li2S activation mediated by AQT as a redox mediator. Reproduced with permission[65]. Copyright 2019, Elsevier.
Beyond metallocenes containing metal centers, certain metal-free soluble organic molecules have also been reported to homogeneously mediate LPSs conversion. For instance, Gu et al.[64] synthesized lithiated 3, 6-dioxa-1, 8-octane dithiol (DODL) via a lithiation reaction. DODL can react with LPSs (e.g., Li2S6) to form Li2S4 and Li2S2 precipitates (Fig. 7c), demonstrating its chemical reduction capability toward polysulfides, which is a key prerequisite for its function as a redox mediator. Moreover, S8 was found to be soluble in the DODL-based electrolyte, which helps activate 'dead sulfur' and improve sulfur utilization. Consequently, the introduction of DODL leads to enhanced Li2S nucleation capability (Fig. 7d). Benefiting from these advantages, a pouch cell with a high sulfur loading of 4 mgS cm−2 and a low electrolyte-to-sulfur ratio of 2.7 μL mgS−1 delivered an energy density of 397.9 Wh kg−1 at a discharge rate of 0.2 C (Fig. 7e). The mechanism of redox mediators is often complex and accompanied by multiple side reactions. In another study, Tsao et al.[65] designed a well-controlled experiment to exclude the influence of other chemical/electrochemical reactions. Specifically, 1,5-bis(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)anthra-9,10-quinone (AQT) was introduced into the electrolyte. A half-cell with a Li2S cathode and AQT-containing electrolyte was then compared with a control half-cell containing AQT, but no Li2S (Fig. 7f). The discharge profiles reveal two distinct reduction plateaus: 2.25 V corresponding to AQT, and 2.1 V corresponding to Li2Sx. Notably, both cells exhibit identical discharge capacities, indicating that no additional side reactions occur and that the total charge remains unchanged. This mechanism effectively activates the Li2S cathode (Fig. 7g) and preserves its original morphology after 250 cycles. Overall, soluble redox mediators provide an effective strategy to regulate the conversion kinetics of LPSs and improve sulfur utilization. However, their practical application still requires precise control over redox potential matching and the suppression of parasitic side reactions.
Polysulfide intermediate modification
-
Another mode of action for molecular catalysts in electrolytes involves reacting with LPSs to generate transition-state intermediates, which participate in subsequent transformations rather than operating through a conventional reversible redox couple. Rao et al.[66] conducted a detailed characterization of the structure of molecularly modified polysulfides. The results reveal that Li in LPS coordinates with S in 4-(trifluoromethyl)thiobenzamide (TFBCA) molecules, leading to the cleavage of the C=S double bond. Meanwhile, S species in LPS react with N atoms in TFBCA to form reactive polysulfide intermediates, significantly reducing the nucleation overpotential and delivering a high-rate performance of 888 mAh g−1 at 5 C (Fig. 8a). In another study, tert-butyl disulfide (DtbDS) was employed to chemically react with LPSs to form a new transition-state species, lithium tert-butyl polysulfides (LitbPSs)[67]. These intermediates exhibit enhanced redox mediation capability, accelerating liquid–liquid conversion kinetics and inducing 3D nucleation and growth of Li2S (Fig. 8b). As a result, a pouch cell with an electrolyte-to-sulfur ratio of 4.0 μL mg−1 and a sulfur loading of 4.9 mg cm−2 delivers stable discharge at 0.05 C (Fig. 8c). However, transition-state polysulfide species suffer from severe shuttle effects and typically induce parasitic corrosion of the lithium anode. The combination of weakly solvating electrolytes with effective molecular catalysts provides an effective strategy to enhance redox kinetics under lean-electrolyte conditions. For example, Liu et al.[68] leveraged this strategy by employing 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE) as a solvation sheath to encapsulate dimethyl diselenide (DMDSe)-modified polysulfide species. This design enhances reaction activity while mitigating parasitic reactions on the lithium metal anode (Fig. 8d).
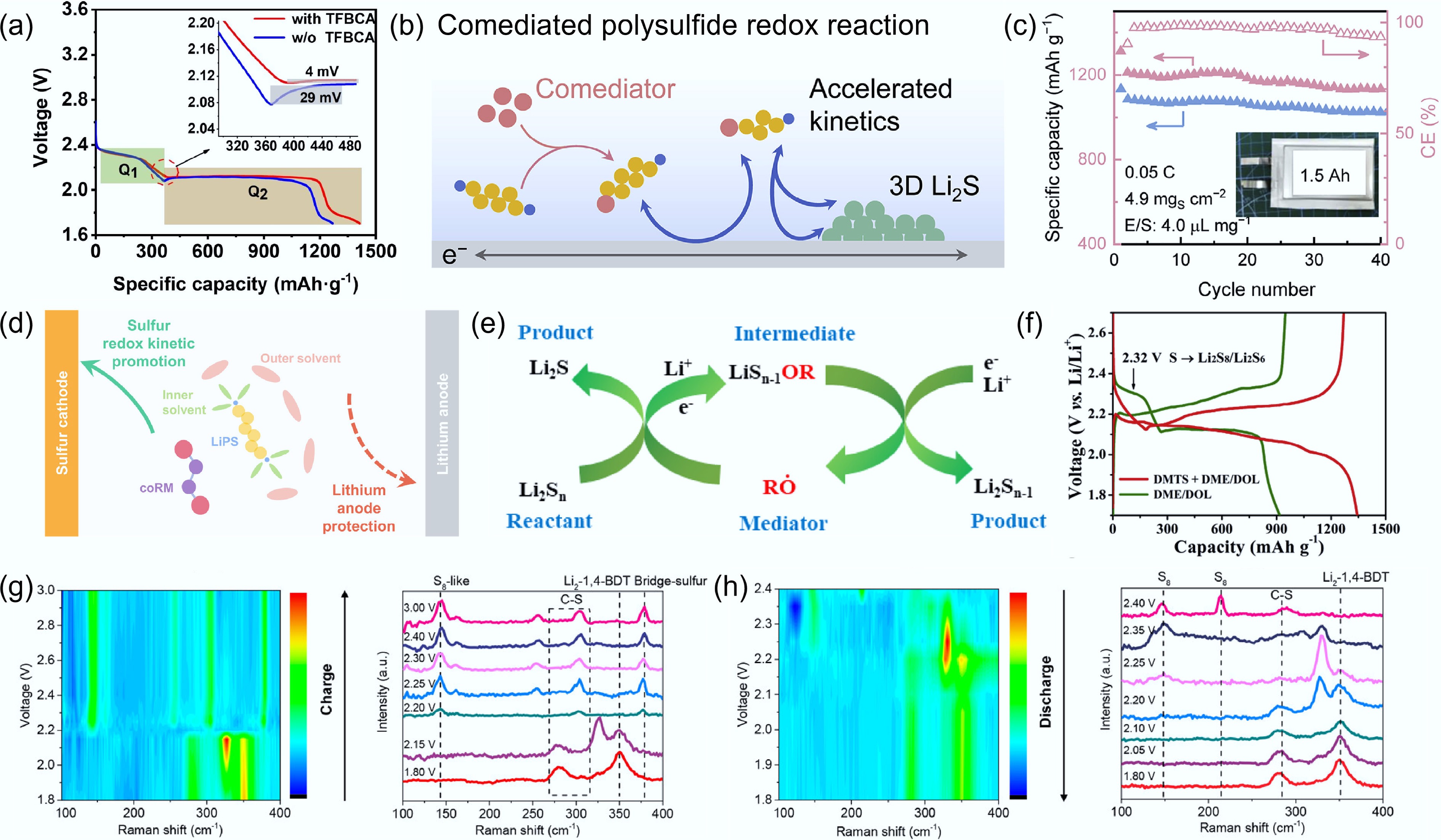
Figure 8.
(a) Charge–discharge curves at a rate of 0.2 C with, and without the incorporation of TFBCA. Reproduced with permission[66]. Copyright 2024, Elsevier. (b) Reaction equation between DtbDS and LPSs. (c) Cycling performance of Li–S pouch cells with the addition of DtbDS. Reproduced with permission[67]. Copyright 2020, Elsevier. (d) Schematic illustration of the mechanism of molecularly modified LPSs under weak solvation conditions. Reproduced with permission[68]. Copyright 2023, John Wiley & Sons. (e) Oxygen radical-mediated cyclic catalytic process. Reproduced with permission[69]. Copyright 2022, American Chemical Society. (f) Charge–discharge curves of Li–S batteries with DMTS-modified and conventional electrolytes. Reproduced with permission[70]. Copyright 2020, Elsevier. (g) In-situ Raman spectra during the initial charge, and (h) discharge processes of the Li–S battery containing 1,4-benzenedithiol (1,4-BDT). Reproduced with permission[71]. Copyright 2021, American Chemical Society.
Most organic molecules exhibit good solubility in non-aqueous electrolytes, enabling their uniform distribution throughout the electrolyte and facilitating the formation of a stable solid electrolyte interphase (SEI) on the lithium anode surface[72]. More importantly, the S–S bonds in LPSs undergo continuous cleavage and recombination during cycling, allowing facile grafting of organic moieties and thereby mediating efficient sulfur conversion[73,74]. For example, Zhang et al.[69] reported that 1,4-difluoroanthraquinone (DFAQ), as an electrolyte additive, reacts with Li to generate oxygen-centered radicals (RO•). These radicals selectively attack S–S bonds in Li2S4 to form LiS2OR intermediates. Owing to their low dissociation energy, these intermediates readily release RO• and generate Li2S2, which is further converted into LiSOR via continued radical attack. This sequence establishes an oxygen radical-mediated catalytic cycle (Fig. 8e). Such modified polysulfide intermediates play a critical role in determining electrochemical reaction pathways. In particular, low-solubility intermediates can enable a single-plateau 'solid–solid' conversion pathway. For instance, Liu et al.[70] demonstrated that dimethyl trisulfide (DMTS) generates radicals (CH3SS• and CH3S•) during electrochemical reactions, which react with LPSs to form dimethyl polysulfide intermediates. These intermediates are subsequently reduced directly to Li2S, effectively bypassing the formation of soluble LPS species. As a result, a single discharge plateau at 2.06 V is observed, delivering a high initial capacity of 1,497.3 mAh g−1 (Fig. 8f). In addition, diphenyl ditelluride (DPDTe) and diphenyl diselenide (DPDSe) have been reported to establish chalcogen radical-mediated catalytic cycles[75]. Similarly, para-dichlorobenzene (1,4-BzCl2) enables a LiBzMe• (Me = methyl)-mediated system with enhanced cycling stability[76].
It is worth noting that the proposed mechanisms largely rely on the assumption and identification of intermediates capable of attacking S–S bonds. Therefore, the effective characterization of such intermediates is crucial for establishing reliable reaction mechanisms. For example, Deng et al.[77] identified the key intermediate Li-pyS, generated from the reaction between 4-mercaptopyridine (4Mpy) and LPSs, using liquid chromatography–mass spectrometry (LC–MS). This finding provides critical evidence for a regeneration-mediated catalytic mechanism involving Li-pyS. However, such ex situ characterization techniques mainly capture static information after the reaction and fail to reflect the dynamic evolution of intermediates during operation. Therefore, in situ characterization techniques have become essential tools for elucidating molecular mechanisms. For instance, Lian et al.[71] employed in situ Raman spectroscopy to investigate the interaction between 1,4-benzenedithiol (1,4-BDT) and sulfur during the first charge–discharge cycle (Fig. 8g, h). The disappearance of the S–H stretching peak and the emergence of Li2-1,4-BDT signals indicate that Li extracts hydrogen from the S–H groups to form Li2-1,4-BDT. During the subsequent charging process, the appearance of bridging sulfur bond peaks suggests the formation of polysulfide oligomers after delithiation. These oligomers further undergo S–S bond cleavage during the following discharge, leading to the formation of Li2S and regeneration of Li2-1,4-BDT. The structural reconstruction induced by sulfur incorporation into oligomers suppresses the formation of soluble polysulfides, enabling the 1,4-BDT-based Li–S battery to achieve stable cycling over 500 cycles at 0.5 C. In summary, molecular catalysts in the electrolyte can significantly regulate the conversion kinetics of polysulfides by constructing transition-state intermediates. However, these highly reactive species exacerbate the shuttle effect, indicating that a delicate balance between catalytic activity and electrochemical stability is required.
-
This article systematically reviews the recent advances and underlying mechanisms of molecular catalysts in Li–S batteries. Owing to their highly tunable structures, low molecular weights, and minimal loading requirements, molecular catalysts can be flexibly integrated into various battery components and exert synergistic catalytic effects at multiple interfaces. As shown in Table 1, among these numerous and structurally diverse molecular catalysts, we summarize several representative molecular catalysts that function in different components, thereby providing readers with a macroscopic comparison of key data.
Table 1. Electrochemical performance of molecular catalysts in Li–S batteries
Molecular catalyst Incorporation
locationSulfur loading
(mgS/cm2)Rate performance Cycle performance Ref. Rate (C) Capacity (mAh g−1) Cycle rate Cycle number Retention CoPc Cathode 4 0.12 About 800 − − [17] CoTnPc Cathode 5 0.5 940 0.5 100 84.8 [18] CoPc Cathode 6.6 5 667.9 0.2 200 81.5 [38] F-Co(II)Pc Cathode 12 5 302.2 0.5 700 70 [54] FePc Cathode 4 1.5 915 1 1,000 45 [78] FePc Cathode 9.2 4 600 1 1,500 47.5 [46] InPc Electrolyte 8 1 585.2 0.2 260 75.8 [79] Ni-PCTs Cathode 4.7 − − 0.02 400 48.6 [48] InPc Separator 10 2 854 0.5 450 50 [34] CuPc Electrolyte 3 4 560 0.5 150 82.1 [80] FePc@WS2 Separator 4 2 833.9 2 1,000 74 [81] Li2TaPc Separator 5 3 About 600 0.5 350 88.2 [82] CoPc Electrolyte 5 4 636 0.5 500 81.1 [83] CoTAP Separator 1 5 672.5 2 500 71.5 [51] FeTNPc@ACNT Separator 5.04 3 861.9 2 1,000 62.4 [32] Fe2-Pc Cathode − − − 1 400 62.8 [84] PPc-CE6 Cathode 6.63 2 688 1 100 85.2 [39] NiTnPc Separator 0.9 2 534 1 500 38.9 [85] FePcF16 Separator 1 5 624.9 2 300 87.63 [86] ZnPc@MXene Separator 1.2 1 806.9 1 500 70 [87] NiCuTnPc Separator 0.9 2 564.4 1 500 60.6 [88] TaPcNiCu Cathode 5.3 2 603.65 1 500 75.4 [89] CoPTpz/rGO Cathode 2.75 4 About 800 5 500 45 [90] G@ppy-por Cathode 5 4 649 2 50 95.2 [91] PCN-222(Cu)-NS Separator 5.7 3 718.3 1 500 77 [47] Al-CPP Separator 1 4 812 1 500 61 [92] Por(Co)-POP/GN Separator 4 2 About 830 1 1,000 54 [93] Mn-COP Cathode 8.6 2 748.6 4 1,000 46 [94] NiTPP Electrolyte 3.9 4 634.9 0.5 200 76 [95] CoTPyP-Mn Separator 6.2 4 712.5 2 1,000 65.8 [55] NUST-66-Ni Cathode 4.2 3 728.8 1 500 74 [96] Fe-TCPP@Cu-BTC Cathode 5.6 2 970 1 600 73.9 [57] FeTPP Cathode 5.7 3 616 2 950 55.7 [36] TaTp-COF Separator 1 2 382.1 1 500 48.8 [97] Porphyrin(Cu/Fe)-S-rGO Cathode 7.79 2 502.4 0.5 300 64.9 [98] FeTPP Electrolyte 6.6 2 543.6 1 400 74.5 [99] Li2S-T4PP-GO-CNT Cathode 2 2 307.7 2 200 66 [100] COF-366-Co Cathode − 2 497 0.5 1,000 65.5 [101] EDOT-Por-COF Separator 8.74 5 763.9 1 2,000 36 [50] Mn-TAPP Separator 2 1 850.36 1 500 61.8 [102] Zr-TCPP(Ni) Separator − 5 986.17 3 600 69.7 [103] S/G-AT Cathode 5.7 0.1 1140 1 450 58.9 [104] THPP Separator − 5 734.2 1 200 83.6 [105] DMAQ Electrolyte 5 0.5 708 1 600 80 [37] DHAQ Cathode 5 0.2 643 0.2 120 94.3 [49] AQ/Fe-NC Cathode 0.63 1 715 1 600 43 [56] DFAQ Electrolyte 5.8 0.1 1,656.4 0.1 80 61.2 [69] AQ/Co-N-C Cathode 1.2 0.1 1,290 0.2 120 62 [106] PG-DAAQ Cathode − 2 870 1 200 71.4 [107] DHAQ/Co-C Cathode 1.19 0.1 1,385 1 600 63 [108] Li+TBAQ·− Electrolyte 5 0.1 899.9 0.2 100 80 [109] Overall, molecular catalysts possess multiple significant advantages, including facile integration into electrochemical systems and high catalytic efficiency. These characteristics enable their promising applications in both homogeneous and heterogeneous catalytic modes. However, each mode has its inherent limitations: heterogeneous molecular catalysts are prone to deactivation due to active site blockage by deposited species, whereas homogeneous molecular catalysts may induce irreversible shuttle effects, thereby shortening battery cycle life. In recent years, semi-confined catalytic architectures have been proposed to address this issue. Zhao et al.[91] proposed a covalent grafting strategy to immobilize porphyrins on electrode surfaces. The synthesis strategy of the material can be summarized in two steps: polypyrrole (PPy) was first integrated onto a graphene (G) conductive substrate, followed by covalent grafting of porphyrin units onto PPy via an amidation reaction between the carboxyl groups of porphyrins and the pyrrole rings of PPy, thereby forming a semi-immobilized heterogeneous catalyst. The porphyrin active sites retain certain characteristics of homogeneous catalysis, leading to faster redox kinetics. This approach provides a promising route toward advanced Li–S batteries. On the other hand, current research remains largely focused on material screening and phenomenological observations. A key challenge lies in establishing descriptors that can accurately capture the effectiveness of molecular catalysts. The performance improvements induced by molecular catalysts in Li–S batteries typically involve multiple physicochemical processes, making it difficult for a single experimental metric to comprehensively reflect their catalytic roles. Moreover, traditional DFT calculations for screening adsorption–catalysis descriptors are computationally expensive and lack systematic efficiency. In contrast, database training based on machine learning and artificial intelligence enables correlation analysis between structural features and chemical or electrochemical signals, as well as efficient processing of large diffraction and spectroscopic datasets to uncover trends obscured by noise. Screening optimal adsorption–catalysis descriptors through data-driven approaches is therefore beneficial for elucidating the intrinsic mechanisms of sulfur conversion. Meanwhile, it is necessary to integrate diverse in situ and ex situ characterization techniques with electrochemical measurements to systematically resolve the coordination environment, electronic structure, and dynamic evolution of molecular catalysts during reactions, thereby establishing clear structure–performance relationships.
In summary, molecular catalysts exhibit great potential for realizing practical Li–S batteries with high specific energy density. In particular, their ability to regulate key interfacial chemistry and reaction kinetics, together with their well-defined molecular structures and minimal mass contribution, effectively promotes the development of next-generation energy storage batteries.
-
The authors confirm their contributions to the paper as follows: Yi Ke: writing – original draft, investigation, methodology, formal analysis; Can Qian: writing – original draft, investigation; Zhuang ji: investigation, resources; Yuan Yang: investivation; Cai Qi: investigation; Xinwei Wang: investigation; Jinhai Zhang: investigation; Yupin Wu: investigation; Yiren Zhong: writing – review & editing, supervision, investigation, conceptualization. All authors reviewed the results and approved the final version of the manuscript.
-
This review article does not report new experimental data. All data discussed in this work are available from the cited literature and publicly accessible databases referenced within the article.
-
This work was supported by the National Natural Science Foundation of China (Grant No. 22372033), the Jiangsu Provincial Science and Technology Program (Major Project) (Grant No. BG2024020) and the Fundamental Research Funds for the Central Universities (Grant No. 2242024k30047 and No. 2242025K30015).
-
The authors declare that they have no conflict of interest.
-
Full list of author information is available at the end of the article.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Ke Y, Qian C, Ji Z, Yang Y, Qi C, et al. 2026. Heterogeneous/homogeneous catalytic role of molecular catalysts in lithium–sulfur batteries. Energy & Environment Nexus 2: e015 doi: 10.48130/een-0026-0009 

Heterogeneous/homogeneous catalytic role of molecular catalysts in lithium–sulfur batteries
- Received: 01 April 2026
- Revised: 19 April 2026
- Accepted: 30 April 2026
- Published online: 25 May 2026
Abstract: Lithium–sulfur (Li–S) batteries are regarded as one of the most promising advanced energy storage systems owing to their high theoretical specific capacity and environmental friendliness. However, the shuttle effect of soluble lithium polysulfides (LPSs) and the sluggish reaction kinetics severely limit further improvements in electrochemical performance. Therefore, there is an urgent need to address these issues through the rational design of catalysts. Among various strategies, molecular catalysts have been widely applied to different battery components owing to their well-defined and tunable structures. Nevertheless, systematic reviews focusing on the catalytic mechanisms and structure–performance relationships of molecular catalysts in Li–S batteries remain scarce. This article first outlines the unique advantages of molecular catalysts in Li–S batteries. Subsequently, the mechanistic principles and representative applications of homogeneous and heterogeneous molecular catalysis in Li–S electrochemistry are comprehensively analyzed. Finally, the current research status of molecular catalysts is discussed, and future research directions of molecular catalysts in Li–S batteries are proposed.