








-
Traditionally, small molecules have exerted their pharmacological effects by binding to specific protein pockets or amino acid residues. However, only approximately 1.5% of the human genome encodes proteins[1,2], and fewer than 15% of the disease-associated proteins are considered druggable. Many proteins are considered "undruggable" due to the absence of suitable binding sites[3]. In contrast, RNA has emerged as a promising therapeutic target because its diverse structural motifs providing accessible recognition sites for small molecules and regulatory proteins. RNA-targeted therapeutics provide abundant druggable targets with potentially shortened preclinical research cycles and promising therapeutic potential in undruggable disease treatment. As a result, RNA-targeted therapies constitute a significant advancement in drug development for diseases that were previously considered "untargetable" and "undruggable," and serve as an emerging therapeutic paradigm for refractory diseases alongside small-molecule inhibitors (SMIs) and antibody-based drugs.
In recent years, RNA-targeting therapeutics have rapidly expanded beyond traditional antisense oligonucleotides (ASO). More than 100 RNA-based therapeutics are currently approved or undergoing clinical evaluation, including ASO, small interfering RNAs (siRNAs), messenger RNA (mRNA) therapeutics, aptamers, and emerging RNA-targeting small molecules, with ASO and siRNAs representing the most clinically advanced modalities[4]. However, the clinical translation of ASO is limited by its poor stability, susceptibility to nuclease degradation, and delivery challenges[5]. Notably, the internal loops and functional regions of RNAs facilitate the direct development of small molecules to modulate RNA's biological activity[6]. For example, Risdiplam and Branaplam directly splice the pre-mRNA of survival motor neuron-2 (SMN2), while bleomycin A5 cleaves the pyrimidine-rich RNA region. In addition, the world's first RNA-targeted SMI approved by the FDA, Evrysdi (Risdiplam), has demonstrated superior clinical efficacy and improved patient compliance than the ASO drug Spinraza[7−10]. Nevertheless, most currently available RNA-targeting small molecules still function through occupancy-driven mechanisms that require sustained target engagement, potentially limiting the degradation efficiency, selectivity, and durability of pharmacological responses[11,12].
Importantly, unlike proteins, RNAs possess unique secondary and tertiary structural motifs, including bulges, internal loops, hairpins, and G-quadruplexes (G4s), which provide accessible recognition pockets for small molecules[13−15]. In addition, both coding and non-coding RNAs participate in diverse biological processes, thereby greatly expanding the therapeutic target landscape beyond conventional protein-centric approaches. These structural and functional characteristics establish RNAs as promising druggable substrates and provide the conceptual foundation for the development of RNA-targeted degradation technologies[11].
In recent years, various therapeutic approaches based on targeted protein degradation (TPD) have generated tremendous excitement in the field of drug discovery; most of these approaches selectively degrade specific biomolecules by "hijacking" the cellular degradation machinery, including the ubiquitin-proteasome system (UPS) and the autophagy–lysosome system[16]. TPD strategies can effectively degrade drugs previously considered undruggable, such as scaffold proteins, transcription factors, and non-enzymatic proteins, etc., through an event-driven pharmacological approach to elicit the desired biological effects even at relatively low concentrations, thus reducing the risk of off-target side effects and drug resistance[17]. Since the first protein degrader was reported in 2001, PROTACs have been studied extensively (Fig. 1)[18]. Later on, molecular glues were also developed with simpler chemical structures[16,19]. As many pathogenic proteins are also located on the cell membranes, further investigations led to degraders targeting lysosomes and autophagosomes, such as autophagosome-tethering compound (ATTEC), lysosome-targeting chimaera (LYTAC)[16,20], ASGPR-targeting chimera (ATAC)[21], antibody-based PROTACs (AbTAC), and others (Fig. 1)[17,21,22]. Although RNA-targeting small molecules have demonstrated considerable therapeutic potential, their biological activity is often limited by transient target occupancy and incomplete suppression of pathogenic transcripts. Inspired by the success of TPD technologies, researchers have begun to explore whether event-driven pharmacology can also be extended from proteins to RNA substrates[17].
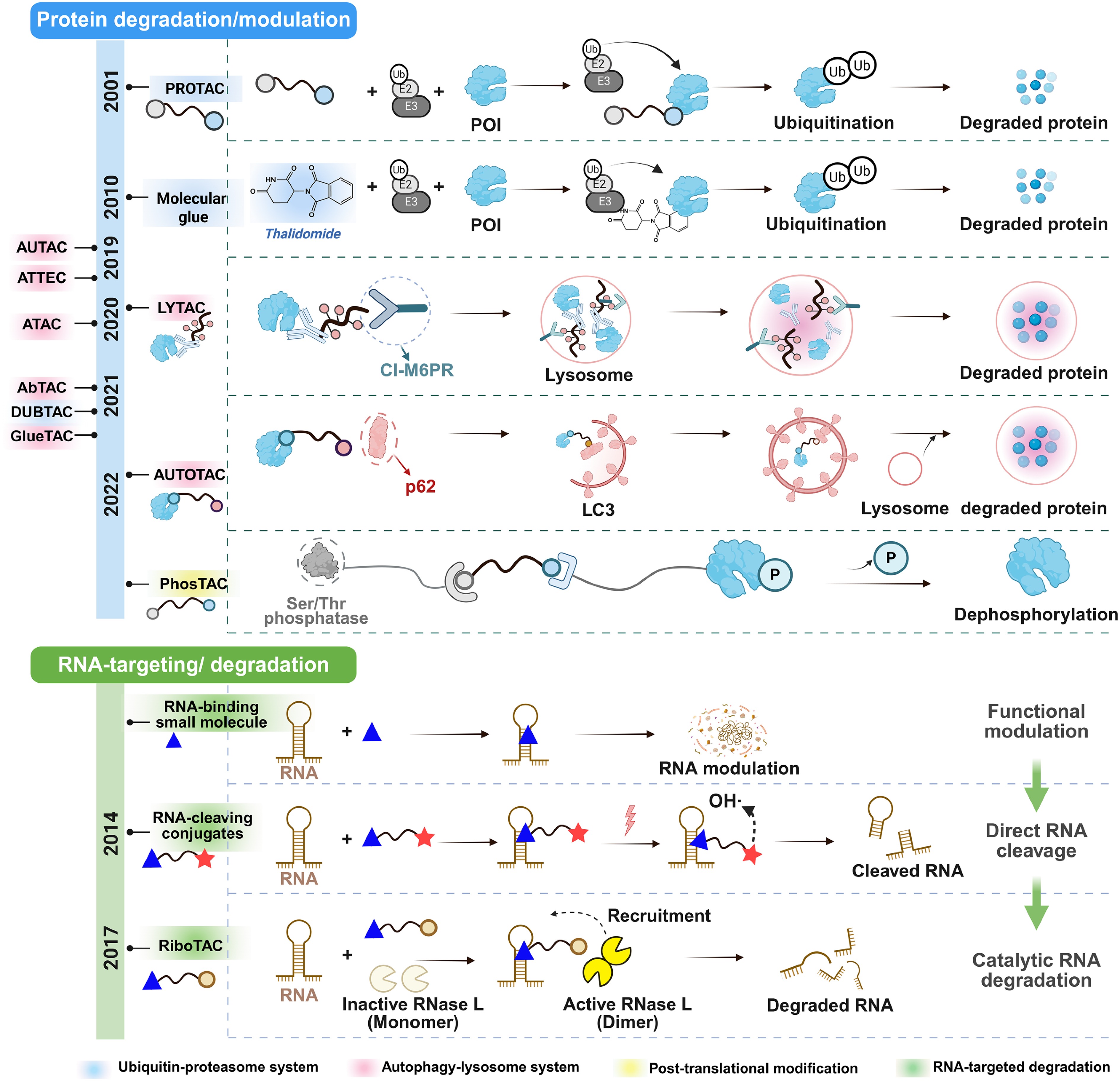
Figure 1.
Development history of targeted degradation and modulation technologies: from proteins to RNAs. PROTACs induce intracellular protein degradation by recruiting target proteins to E3 ubiquitin ligases, thereby triggering ubiquitination and subsequent proteasomal degradation. Molecular glues promote the interaction between target proteins and E3 ligases, leading to degradation mediated by the ubiquitin–proteasome system. LYTACs internalize extracellular or membrane-associated proteins and deliver them to lysosomes for degradation. Autophagy-targeting chimeras, represented by AUTOTACs, mediate target protein degradation through the autophagy–lysosome pathway. Phosphorylation-targeting chimeras (PhosTACs) achieve targeted protein dephosphorylation by recruiting phosphatases. In parallel, RNA-targeting strategies have progressed from RNA-binding small molecules that modulate RNA function without inducing RNA degradation to RNA-cleaving conjugates capable of directly cleaving target RNAs, and further to RiboTACs, which recruit endogenous RNase L to achieve catalytic RNA degradation.
With ongoing advancements, the scope of chimeric molecules has been continuously expanded from the field of proteins to that of nucleic acids[12,23−25]. It should be noted that small-molecule-mediated RNA degradation predates the formal development of the ribonuclease-targeting chimera (RiboTAC) platform. Early studies demonstrated that small molecules conjugated to RNA-cleaving modules could induce selective RNA degradation in cells, as exemplified by the work of Guan et al.[26]. Later, the development of RiboTACs by Costales et al.[23] introduced a distinct strategy in which RNA-binding small molecules recruit endogenous RNase L to catalytically degrade the target RNAs in cells. Therefore, RiboTACs should be viewed as an important mechanistic evolution from earlier RNA-cleaving conjugates toward programmable, endogenous nuclease-mediated RNA degradation. RiboTACs are generally composed of an RNA-binding small molecule, a linker, and an RNase L-recruiting group[27], with the key innovation of converting RNA binding molecules into degradation molecules (Fig. 1). Unlike conventional occupancy-driven SMIs, RiboTACs exhibit catalytic and event-driven pharmacological properties, enabling durable degradation of pathogenic RNA transcripts at relatively low concentrations[28,29]. Compared with ASO, the relatively lower molecular weight of RiboTAC facilitates improved druggability and cellular uptake[30]. In addition, the modular structure of RiboTACs enables further optimization of RNA recognition, ternary complex formation, and degradation selectivity through rational engineering of RNA ligands, linkers, and RNase L recruiters[27]. Therefore, RiboTACs represent a conceptual evolution from conventional occupancy-driven RNA targeting toward programmable and catalytic RNA degradation. In this review, we systematically summarize the classification of targetable RNAs, recent advances in RNA-targeting small molecules, and the emerging design principles, applications, and translational challenges of RiboTACs.
-
Current RNA-targeting strategies primarily rely on recognizing defined secondary or tertiary structural RNA motifs or disrupting RNA–protein interactions[31]. These approaches primarily involve ASO and small molecules. However, ASOs are susceptible to enzymatic degradation due to their single-stranded nucleotide composition, resulting in limited pharmacokinetic stability and challenges for clinical translation[30]. Additionally, many RNA-binding proteins lack suitable binding pockets, which restricts the development of effective SMIs[31]. These limitations have stimulated increasing interest in directly targeting RNA structures with small molecules. In accordance with standardized classification criteria, targetable RNAs are divided into three major categories: coding RNA, non-coding RNA, and repeat-amplified RNA[11]. RNAs that can be effectively targeted by small molecules typically contain stem-loop structures suitable for binding. Representative examples include microRNA precursors, internal ribosome entry sites (IRESs) of mRNAs, and certain repeat regions within mRNAs (Fig. 2)[32,33]. Importantly, these RNAs frequently contain characteristic structural motifs, including bulges, internal loops, hairpins, and G4s, which provide accessible recognition pockets for small molecules and RNA degraders[11,15,34]. In this section, we mainly illustrate the binding modes and functional interference effects between small molecules and target RNAs, and further supplement the mainstream RNA-targeting strategies such as ASOs and RiboTACs besides small-molecule-based regulation.
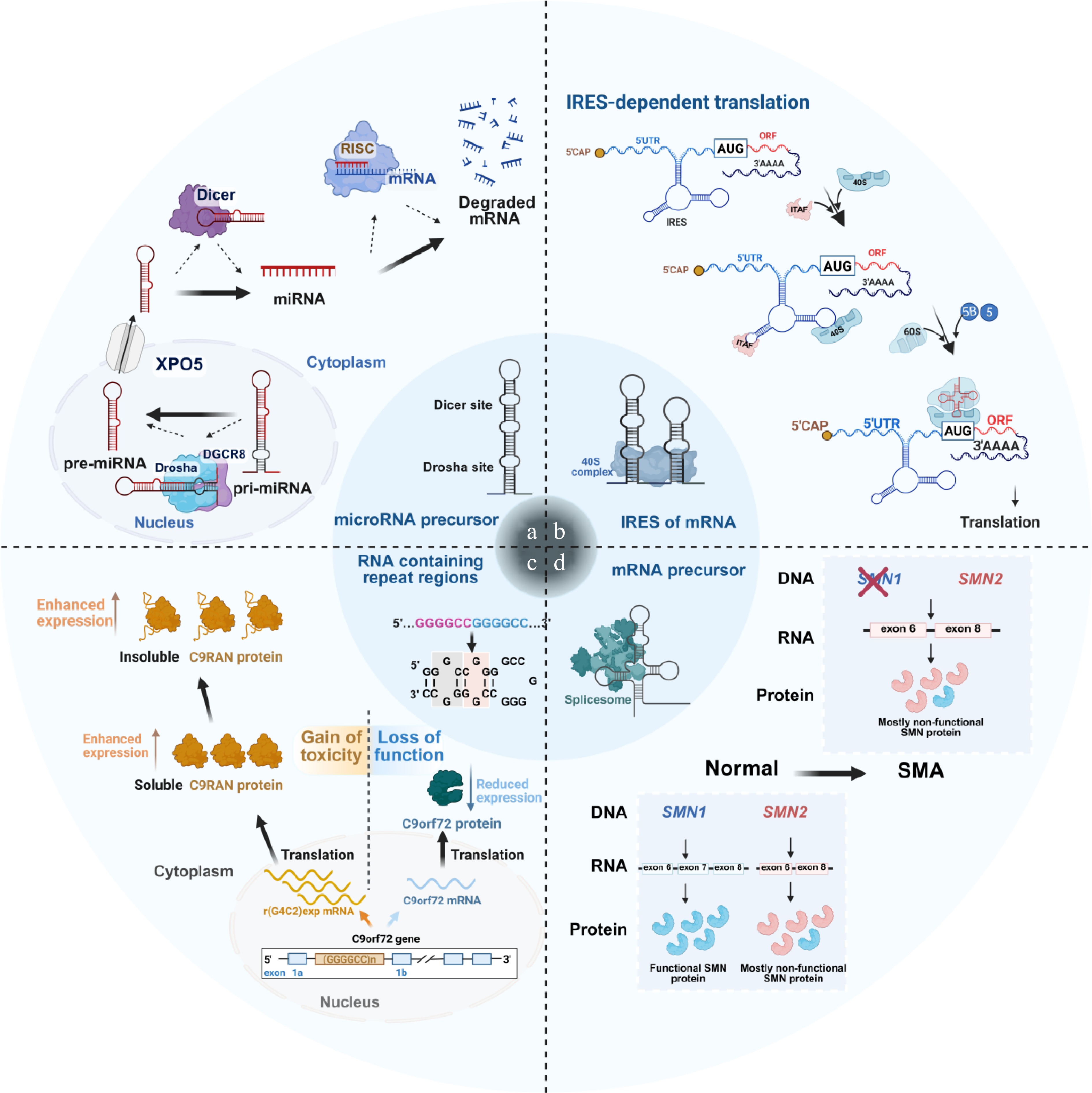
Figure 2.
Representative interaction patterns between small molecules and various target RNAs. (a) Small molecules bind to the Drosha or Dicer cleavage sites of miRNA precursors to block its maturation process. (b) Small molecules interact with the IRES of certain pathogenic mRNAs, inhibiting the translation process. (c) Small molecules attach to the repeat regions of certain mRNAs, causing nervous system-related diseases. (d) Small molecules influence the spatial structure of certain mRNA precursors to affect splicing and inhibit the expression of pathogenic proteins.
Non-coding RNA (microRNA precursors)
-
Most microRNA genes are transcribed by RNA polymerase II to generate primary miRNA transcripts (pri-miRNA) (Fig. 2a), which typically adopt extended stem-loop structures[35]. These pri-miRNAs are processed in the nucleus by the Drosha-DGCR8 microprocessor complex to produce precursor miRNAs (pre-miRNAs), approximately 60−70 nucleotides in length. Pre-miRNAs are then exported to the cytoplasm by exportin-5 (XPO5) and further cleaved by Dicer to yield mature miRNAs[36], which subsequently bind complementary sequences in target mRNAs, thereby repressing translation or promoting transcript degradation[37,38]. The defined secondary and tertiary structures of pri-miRNAs and pre-miRNAs make them attractive targets for small-molecule intervention. Small molecules that bind near Drosha or Dicer processing sites can block miRNA maturation and thereby alter downstream gene expression (Fig. 3a)[39,40]. These processing regions frequently contain stem loops, asymmetric bulges, and internal loop motifs that facilitate selective small-molecule recognition and targeted degradation[39]. This strategy is especially relevant for oncogenic miRNAs, whose aberrant expression contributes to tumor progression. A representative example is pri-miR-96, the precursor of the oncogenic miRNA-96, which is implicated in breast cancer. Specific small molecules, including compounds 1 and 2, have been shown to bind pri-miR-96 and interfere with its processing, thereby promoting apoptosis (Fig. 3a)[41,42]. Notably, dimerization or modular assembly of these ligands can markedly enhance their binding affinity for pri-miR-96. Consistent with this, the combined or dimeric optimization of compounds 1 and 2 more effectively suppressed pri-miR-96 and mature miR-96 expression, leading to restoration of FOXO1 signaling and induction of apoptosis in triple-negative breast cancer (TNBC) cells (Fig. 3b)[23,42−44].
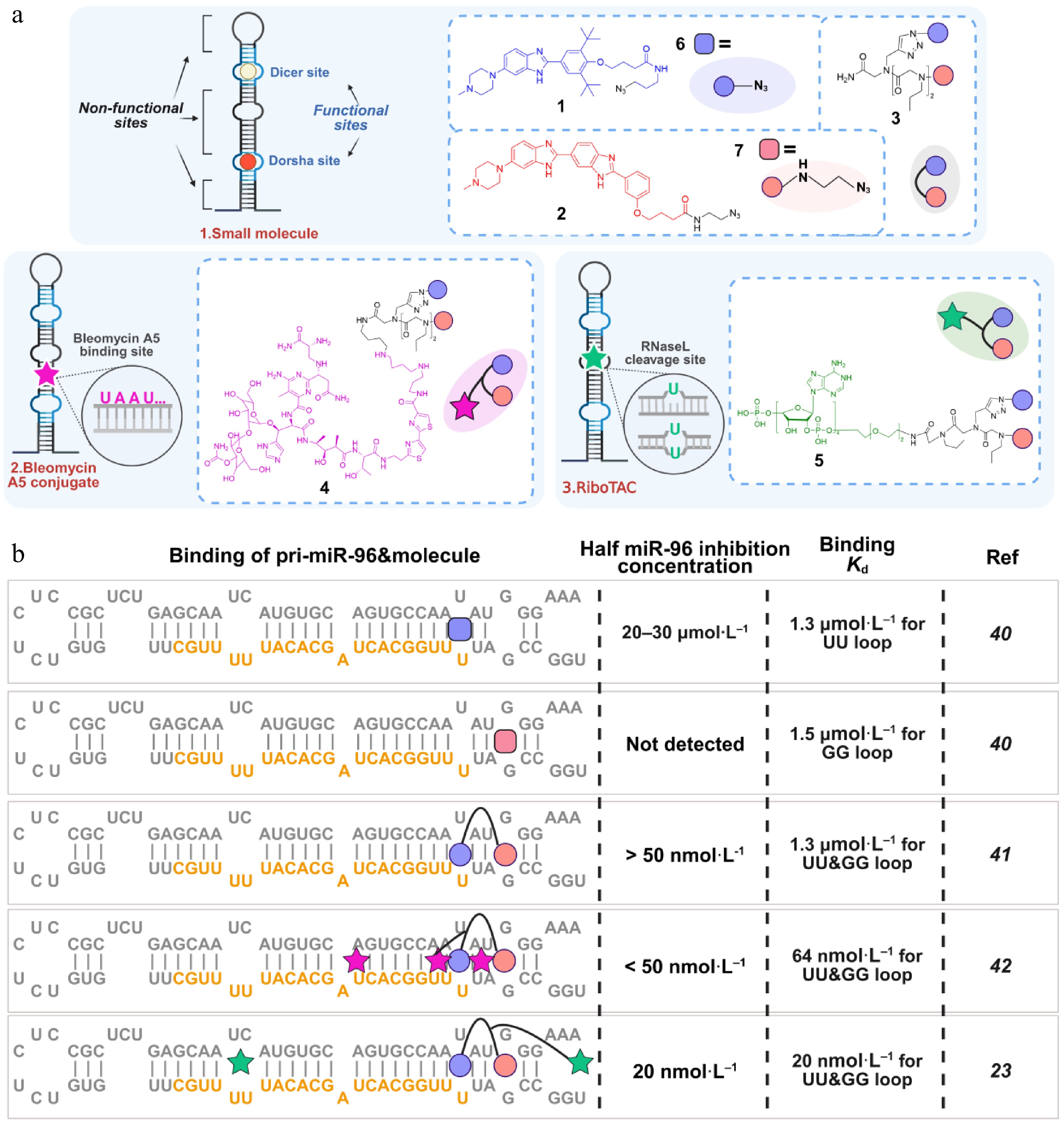
Figure 3.
Small molecule-RNA binding patterns and interaction modes. (a) Three classes of small molecules interacting with pri-miR-96. (b) Binding methods, affinity, and inhibitory effects of different compounds toward pri-miR-96. The orange sequences in (b) present mature miRNA-96.
In addition, G4s are distinctive secondary structural motifs that are widely distributed in non-coding RNAs and oncogenic transcripts[15]. These special stacked guanine-rich structures participate in the regulation of tumor-related gene expression and have become promising druggable targets for cancer therapy. Several G4-targeting small molecules, including the DNA G4 ligand CX5461 and RNA G4-directed compounds RC220 and AROG4-01, have advanced into clinical or preclinical development, highlighting the therapeutic potential of G4-targeted strategies in oncology[15,45].
Coding RNA (mRNA and mRNA precursors)
-
Most eukaryotic protein synthesis methods are based on a cap-dependent translation that relies on the m7G cap structure at the 5' end of the mRNA (Fig. 2b)[46,47]. This process involves the recognition of the 5' cap by the 43S translation initiation complex, followed by the binding of various eIF4 factors, which together facilitate codon recognition and the initiation of translation. In contrast, viral RNAs, such as those from the hepatitis C virus, often lack a cap structure and rely on their 5' untranslated region (5' UTR) containing an IRES to initiate translation (Fig. 2b). The IRES is situated within the 5' UTR and can directly recruit the 40S ribosomal subunit, allowing translation to commence independently of the 5' cap. These highly structured IRES motifs frequently contain characteristic bulges and internal loops, making them particularly suitable for selective small-molecule recognition[47]. This mechanism promotes tissue-specific translation, particularly under stress conditions when cap-dependent translation is inhibited[48,49]. IRESs can initiate translation from within the mRNA, enabling the co-expression of multiple genes regulated by the same promoter[48,49]. Given that many mRNAs containing IRES elements are implicated in human diseases, IRESs have emerged as promising therapeutic targets for conditions such as cancer and neurodegenerative diseases. Recent research utilizing the INFORNA tool has identified IRESs in the mRNAs of JUN and MYC, two critical oncogenic proteins that are typically considered undruggable. Small molecules have been shown to target these IRESs and possess RNase L cleavage sites, offering new avenues for therapeutic intervention[12,50−53].
Pre-cleaved pre-mRNA can undergo alternative splicing, allowing the inclusion or exclusion of exons to produce different protein isoforms. However, mutations at splice sites may alter protein expression and lead to various diseases[54,55]. Given the spatial structure of pre-mRNA, small molecules can effectively bind to it and guide the splicing process[56,57]. Importantly, splice-regulatory regions often adopt defined RNA conformations that provide structurally accessible motifs for selective small-molecule targeting[57]. A significant milestone was achieved in 2020 with the FDA approval of Risdiplam, the world's first RNA-targeting small-molecule drug[7−9]. This breakthrough is particularly relevant for spinal muscular atrophy (SMA), an autosomal recessive neurological disorder characterized by the degeneration of spinal motor neurons, resulting in muscle weakness[58]. SMA is primarily caused by deletions or mutations in the SMN protein. Errors in the splicing of SMN2 pre-mRNA can lead to insufficient levels of functional SMN protein (Fig. 2d). Risdiplam exerts its therapeutic effects by regulating the splicing of SMN2 pre-mRNA, promoting the production of functional SMN protein (Fig. 2d). Specifically, Risdiplam facilitates the inclusion of exon 7 in SMN2, effectively mimicking the function of SMN1 and alleviating the symptoms of SMA[7,8].
Repeat-amplified RNA
-
Numerous neurological diseases are associated with the expansion of repetitive RNA sequences. For example, the expanded r(CUG) sequence is implicated in myotonic dystrophy type 1 (DM1), while the G4C2 repeat is linked to chromosome 9-caused amyotrophic lateral sclerosis and frontotemporal dementia (c9ALS/FTD)[59−62]. r(CUG)exp in DM1 sequesters regulates proteins such as muscle blind-like protein 1 (MBNL1) and inhibits pre-mRNA splicing[63]. Cugamycin has been shown to selectively recognize the structure of r(CUG)exp, providing therapeutic benefits in DM1 mouse models. In contrast, oligonucleotides that target r(CUG) sequences have less selectivity and can cleave both long and short r(CUG)-containing transcripts[64]. c9ALS/FTD is characterized by progressive neurodegeneration, movement disorders, and cognitive, behavioral, and language deficits. The most common genetic cause of this condition is the G4C2 repeat expansion found in intron 1 of the chromosome 9 open reading frame 72 (C9orf72) gene (Fig. 2c)[65]. Healthy individuals typically have 2 to 30 G4C2 repeats, while patients with c9ALS/FTD may have hundreds to thousands. These repetitive RNA sequences can fold into stable hairpins, internal loops, and G-quadruplex-like conformations, thereby creating multiple accessible binding motifs for small molecules and RNA degraders[65]. The transcription of G4C2 repeat produces RNA, which reduces the expression of C9orf72 mRNA[61]. Additionally, the translation of repeat-associated non-AUG (RAN) proteins from G4C2 repeat produces a lot of toxic C9RAN protein, which can lead to neurodegeneration[61,66]. Targeted degradation of G4C2 repeat is expected to bring therapeutic hope to c9ALS/FTD[60].
Others
-
Bleomycin A5 has been shown to cleave purine-rich RNA base pairs, making it a valuable tool in RNA-targeting strategies[67]. If UA-rich bases are present near the RNA sequence that binds the small molecule, bleomycin A5 can achieve targeted cleavage. This conjugation strategy has been employed to cleave RNA repeats both intracellularly and in vivo, as well as various miRNAs within cells[44,68]. For instance, linking the bleomycin A5 conjugate to the Targapremir-96 dimer enhances the tumor suppressor effects of compound 3 (Fig. 3)[23,40−42]. Furthermore, the cleavage of purine-rich RNA base pairs helps clarify the binding site between the compound and the RNA target[31,44].
-
Small molecules can interact with RNA at either functional or non-functional sites, with the former directly inhibiting RNA biological function and the latter exhibiting limited functional effects. The binding selectivity of small molecules is largely determined by the intrinsic structural druggability of RNA, particularly the presence of accessible stem loops, bulges, internal loops, and other tertiary structural motifs. For example, when compounds directly bind to the Drosha or Dicer cleavage sites of miRNA precursors, the maturation of pathogenic miRNA is significantly inhibited[69−72]. Similarly, if the compound binds to the IRES of mRNA, it can disrupt RNA translation and inhibit protein production[47]. As shown in Fig. 3a, compounds 6 and 7 have low binding ability with the Drosha cleavage site of pri-miR-96 and weak inhibitory effect on miR-96. However, the affinity and inhibitory action of compound 3, which is obtained by linking compounds 6 and 7, are greatly enhanced[42,43]. A variety of RNA-targeting small molecules have been identified, such as benzimidazoles, aminoglycosides, and polyamines (Table 1). These compounds not only demonstrate the feasibility of occupancy-driven RNA targeting but also provide important RNA-binding scaffolds for the development of RNA degradation strategies such as RiboTACs. In the following sections, we mainly summarize the mechanisms of action for these small molecules and their derivatives, emphasizing their advantages and disadvantages.
Table 1. Targeted binding information for small molecules and RNA.
Type Name CAS. RNA Binding site Affinity Disease Validation status Ref. Benzimidazoles Hoechst 33258 207792-72-18 Introns of Candida albicans rRNA / / / Preclinical [73] 8 1174302-28-3 5'UAUAU/
3'AGUCA/ / / Preclinical [74] 9 1311982-86-1 5'UUAU/3'AUGU / / / Preclinical [74] 11 1311982-88-3 Pri-miR-96 Drosha / / Preclinical [42,74] HL-1 263022-88-4 5'CUAA/3'GAUU / / / Preclinical [75] H1 4402-18-0 r(CUG)exp / / DM1 Preclinical [76] Ht-N3 1049722-30-6 r(5'CUG/3'GUC)exp / / DM1 Preclinical [26,77] 1a 70173-34-1 r(5'CGG /3'GGC)exp / 76 ± 4 nmol·L−1 FXTAS Preclinical [78,79] 1a 70173-34-1 r(5'GGG /3'GCC)exp / 9.7 ± 1 μmol·L−1 c9FTD/ALS Preclinical [79] 1 51877-67-9 r(5'CUG /3'GUC)exp / / DM1 Preclinical [80] Targapremir-210 1049722-30-6 Pre-miR-210 Dicer 165 nmol·L−1 TNBC Preclinical [40] TGP-210-RL / Pre-miR-210 Dicer 190 nmol·L−1 TNVBC Preclinical [24] Targaprimir-96 1655508-14-7 Pri-miR-96 Drosha 1.3 ± 0.1 μmol·L−1 TNBC Preclinical [43] TGP-96 Bleo / Pri-miR-96 Drosha 64 ± 11 nmol·L−1 TNBC Preclinical [24] TGP-96 RiboTAC / Pri-miR-96 Drosha 20 nmol·L−1 TNBC Preclinical [23] Targaprimir-515/885 1311982-86-1 Pri-miR-515 Drosha 15 ± 1.9 μmol·L−1 TNBC Preclinical [81] Targaprimir-515/885 1311982-86-1 Pri-miR-885 Drosha 9.0 ± 1.0 μmol·L−1 TNBC Preclinical [81] Targaprimir-515 2290585-84-9 Pri-miR-515 Drosha 940 ± 260 nmol·L−1 TNBC Preclinical [81] TGP-21 2769732-95-6 Pre-miR-21 Dicer 980 ± 97 nmol·L−1 TNBC Preclinical [25,82] TGP-21-C1-3 / Pre-miR-21 Dicer / TNBC Preclinical [25,82] TGP-377 2665700-67-2 Pre-miR-377 Dicer 5 ± 1 nmol·L−1 Coronary artery disease Preclinical [83] 63 2864325-32-4 r(CCUG)exp / 192 ± 20 nmol·L−1 DM2 Preclinical [84] 2b 2332612-58-3 r(5'CUG/3'GUC)exp / 40 ± 6 nmol·L−1 DM1 Preclinical [85] Pre-miR-155-RiboTAC 2763059-27-2 Pre-miR-155 5′GAU/3′C_A bulge 0.8 ± 0.1 μmol·L−1 TNBC Preclinical [12] Syn-RiboTAC / SNCA mRNA SNCA IRE 1.8 ± 0.3 μmol·L−1 PD Preclinical [86] RiboTAC 7 / Pre-miR-21 Dicer 4.6 ± 2.1 μmol·L−1 TNBC Preclinical [87] TGP-17-92 / Pre-miR-17,18a,20a / 120 ± 20 μmol·L−1 for pre-miR-17 and pre-miR-20a; 124 ±
22 μmol·L−1 for pre-miR-18aPC Preclinical [88] Aminoglycosides Kanamycin A 59-01-8 5'UUUU/3'AUCA / / / Preclinical [89] 3K-4 / r(CCUG)exp / / DM2 Preclinical [90,91] Neomycin 119-04-0 Pri-miR-525 Drosha 355 ± 13 nmol·L−1 HCC Preclinical [39] Polyamines D6 94345-49-0 r(5'CAG/3'GAC)exp / 0.06 ± 0.03 μmol·L−1 DM1 Preclinical [92] 2AU-2 / r(5'AUUCU)exp / 185 ± 1.0 nmol·L−1 SCA10 Preclinical [93] Others 1 51877-67-9 Pri-miR-544 Dicer 470 ± 20 nmol·L−1 Hypoxic BC Preclinical [94,95] Lomofungin 26786-84-5 r(5'CUG/3'GUC)exp / 998 ± 180 nmol·L−1 DM1 Preclinical [96] Dilomofungin 1383975-68-5 r(5'CUG/3'GUC)exp / 606 ± 300 nmol·L−1 DM1 Preclinical [96] Mitoxantrone 70476-74-3 Pre-miR-21 Dicer 22 ± 8.3 nmol·L−1 / Preclinical [97] C5 1225177-95-6 SARS-CoV-2 attenuator hairpin 5'CUU/3'GUA internal loop 11 ± 3.8 nmol·L−1 COVID19 Preclinical [98] CB253 1225169-17-4 r(GGGGCC)exp / 18 ± 4 μmol·L−1 c9FTD/ALS Preclinical [99] G4C2 RiboTAC / r(GGGGCC)exp / / c9FTD/ALS Preclinical [60] 3 950276-57-0 Pre-miR-372 Dicer 1.7 ± 0.6 μmol·L−1 Stomach cancer Preclinical [100] F1-RiboTAC / QSOX1-a mRNA / 11 ± 5 μmol·L−1 TNBC Preclinical [13,101] MYC-RiboTAC / MYC mRNA IRES 1.1 ± 0.1 μmol·L−1 Pan-cancer Preclinical [12] JUN-RiboTAC / JUN mRNA IRES 0.5 ± 0.2 μmol·L−1 Pan-cancer Preclinical [12] Pre-miR-372 degrader / Pre-miR-372 Dicer 0.03 ± 0.1 μmol·L−1 Gastric cancer Preclinical [102] Risdiplam 1825352-65-5 SMN2 pre-mRNA Exon 7–intron junction / SMA Approved [7−9,58,103] Branaplam 1562338-42-4 SMN2 pre-mRNA Exon 7–intron junction / SMA Clinical-stage [104] BC: breast cancer, c9FTD/ALS: chromosome 9-caused frontotemporal dementia or amyotrophic lateral sclerosis, COVID19: Coronavirus disease 2019, DM1: Myotonic dystrophy type 1, DM2: myotonic dystrophy type 2, FXTAS: fragile X-associated tremor ataxia syndrome, HCC: hepatocellular carcinoma, PC: prostate cancer, PD: Parkinson's disease, SCA10: spinocerebellar ataxia type 10, SMA: spinal muscular atrophy, TNBC: triple-negative breast cancer. Benzimidazoles
-
Benzimidazoles are one of the earliest and most extensively studied classes of compounds capable of binding to RNA. The first RNA-targeting compound was identified in bacteria[73], among which approximately 40% of the strains possess group I self-splicing introns in their large subunit rRNA precursors[105]. These introns play a critical role in regulating protein expression, making rRNA precursors viable drug targets. In 2004, Disney et al.[73] demonstrated that Hoechst 33,258 could inhibit the self-splicing of group I introns in Candida albicans, thereby affecting ribosome assembly and ultimately inhibiting the activity of the organism. Since this foundational work, numerous Hoechst 33258 derivatives have been shown to bind to RNA and exhibit various therapeutic effects, including anti-cancer properties and treatment of neurodegenerative diseases (Fig. 4)[23,40,44,53,73−77]. For instance, Targapremir-210 binds to the Dicer cleavage site of pre-miR-210, inhibiting the production of mature miRNA and subsequently activating glycerol-3-phosphate dehydrogenase 1-like enzyme (GPD1L), which triggers apoptosis in TNBC cells under hypoxic conditions[40]. Additionally, compound 1a can bind to the 5'CGG/3'GGC loop in an mRNA precursor, alleviating symptoms of fragile X-associated tremor/ataxia syndrome (FXTAS), or target the G4C2 repeat expansion to potentially treat FTD or amyotrophic lateral sclerosis (ALS)[78,106]. Despite their potential, benzimidazoles exhibit low selectivity and a risk of off-target effects. To address these limitations, researchers have begun to link two or more compounds into a single entity to enhance both the affinity and selectivity for RNA. For example, certain benzimidazoles can bind simultaneously to the Dicer site and an adjacent U bulge of pre-miRNA[25]. The dimeric form, Targapremir-96, demonstrates greater Drosha enzyme cleavage capability for pri-miR-96 in TNBC cells compared to its monomeric counterparts, with its efficacy largely attributed to increased expression of the pro-apoptotic protein FOXO1[42,43,82]. The accumulated evidence on benzimidazole-based ligands shows that rational recognition of RNA structural motifs can achieve selective functional inhibition of pathogenic RNAs while providing versatile RNA-binding modules for RiboTAC construction.
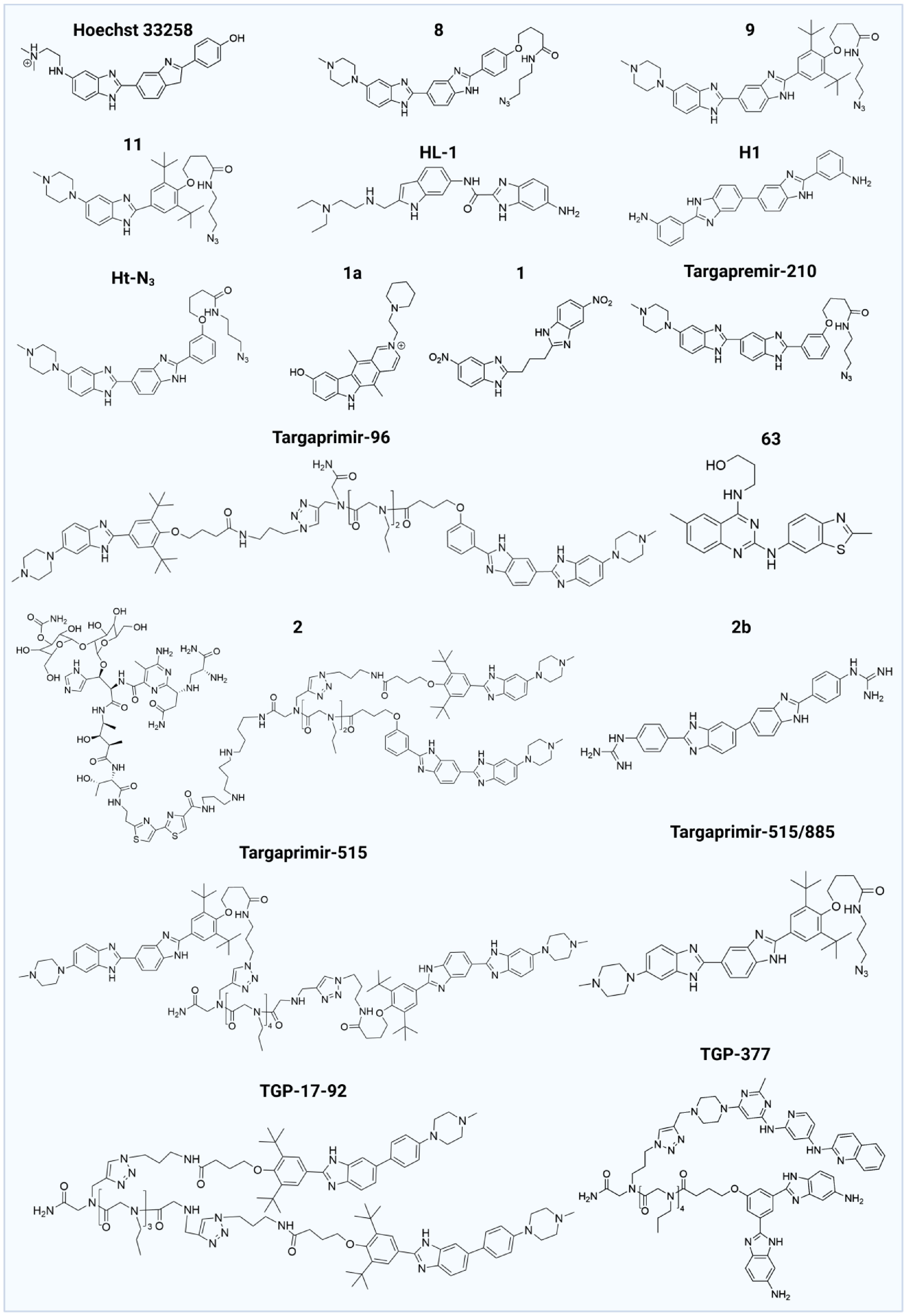
Figure 4.
Structures of RNA-bound benzimidazoles.
Aminoglycosides
-
Aminoglycosides are the second largest class of compounds capable of binding to RNA (Fig. 5). These compounds, particularly the antibiotic kanamycin A, are known to inhibit protein synthesis by binding to specific sites on bacterial ribosomes[107−109]. In a pivotal study in 2008, Disney's team utilized a microarray approach to investigate the binding properties of kanamycin by fixing it on agarose resin and screening an RNA 3*3 internal loop library. They found that kanamycin exhibits high affinity for a 5'-UU/3'-UC 2*2 internal ring closed by an AU pair, demonstrating its potential as a therapeutic agent for RNA-related diseases[89]. Subsequent research revealed that kanamycin could also bind to RNA containing repeated 5'CUG loops, which has implications for treating myotonic dystrophy type DM1[110]. Furthermore, kanamycin A was shown to interact with pyrimidine-rich nucleotide loops (5'UU/3'UC or 5'CU/3'UU), specifically targeting the cCCUG repeat expansion associated with DM2[90,91]. When kanamycin A was assembled on a peptide scaffold, the resulting compound 3K-4 showed a significantly enhanced binding ability to DM2 RNA, 15 times stronger than its interaction with the downstream MBNL1, making it a promising candidate for DM2 therapy (Fig. 5)[91]. Other aminoglycosides, such as neomycin A and neomycin, have demonstrated the ability to bind to specific RNA motifs, such as G-A internal loops and the Drosha restriction site of pri-miR-525, respectively, highlighting their potential applications in cancer treatment[39,90]. Importantly, the strong RNA-binding capability, structural modularity, and accessible derivatization sites of aminoglycosides make them attractive scaffolds for RNA-targeted degradation strategies, including RiboTAC design. However, the clinical utility of aminoglycosides is hampered by their side effects, including ototoxicity and renal toxicity[74,111].
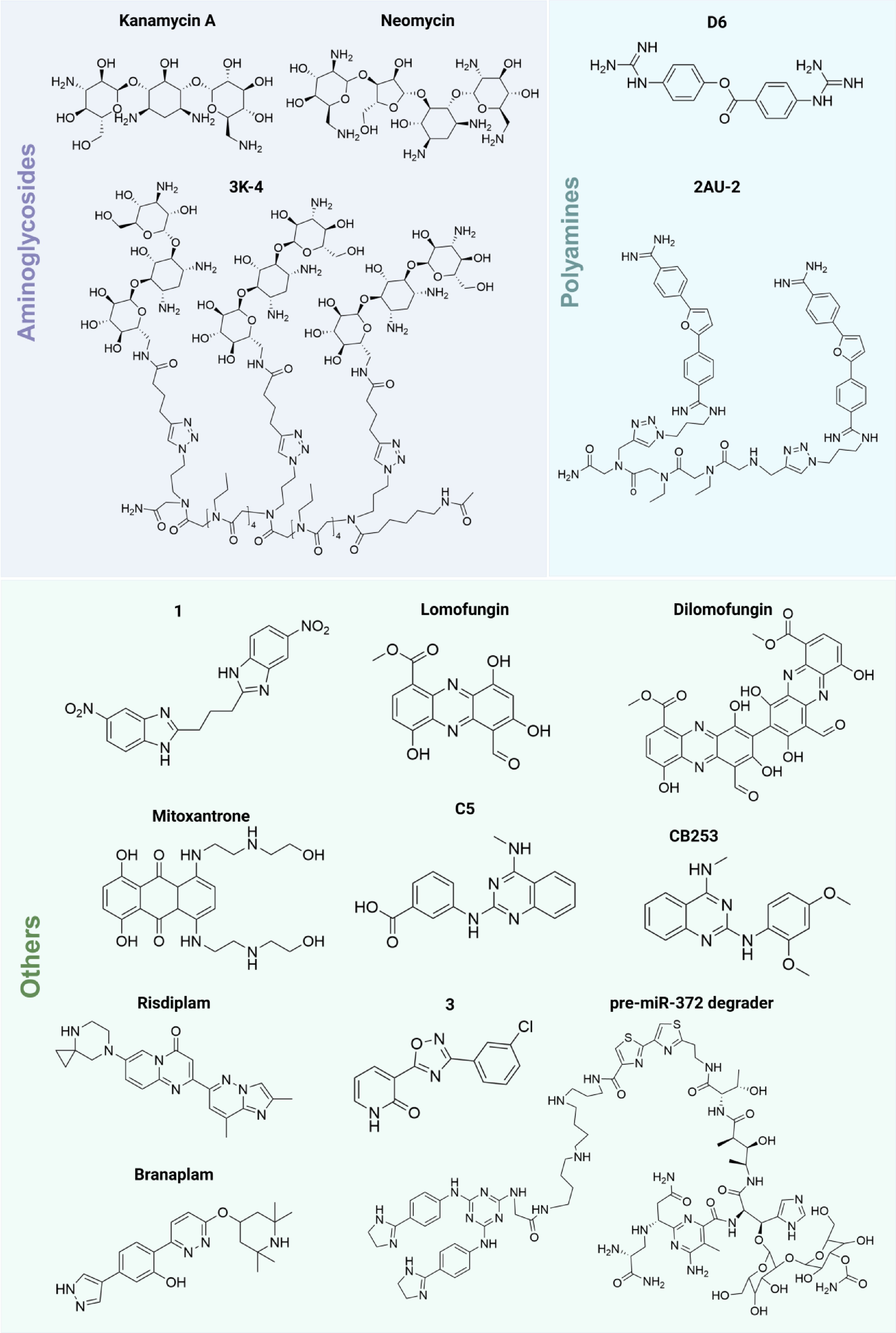
Figure 5.
Structures of aminoglycosides, polyamines, and other compounds.
Polyamines
-
In 2009, Disney's team[112] developed a peptide library to identify small molecules that bind RNA, leading to the discovery of second-generation polyamine/peptide inhibitors for rRNA introns. Fluorescence resonance energy transfer (FRET) assays demonstrated that the polyamine compound D6 binds to r(CAG) triplet repeats, releasing sequestered MBNL1 and improving splicing defects, thereby inhibiting the progression of neurodegenerative diseases[92]. Additionally, the bis-benzamidine compound AU-2 targets r(AUUCU) amplifications associated with spinocerebellar ataxia type 10 (SCA10). The dimeric form, 2AU-2, has shown promise in improving mitochondrial dysfunction and reducing caspase 3 activation in SCA10 fibroblasts, indicating good target selectivity (Fig. 5)[93]. These studies further suggest that polyamine-derived compounds can selectively recognize structurally complex and negatively charged RNA motifs, highlighting their potential utility in programmable RNA-targeting platforms.
Others
-
Through FRET screening, lomofungin and its dimer, dilomofungin, were identified as inhibitors of MBNL1-CUG RNA binding, providing a potential therapeutic approach for DM1[96]. The topoisomerase inhibitor, mitoxantrone, targets the Dicer cleavage site of pre-miR-21, selectively inhibiting the biosynthesis of miR-21 and the invasion ability of MDA-MB-231 cells[97]. Other compounds of interest include those that bind to the Drosha cleavage sites of oncogenic miRNAs, such as miR-544 and miR-372, which are activated in various cancers (Fig. 5). For example, compound 1 inhibits the biosynthesis of miR-544 by binding to the AUU/UUA of its Drosha cleavage site, regulating the ATM/mTOR signaling pathway and inducing apoptosis[95]. Using the two-dimensional combinatorial screening (2DCS) method, another compound 3 was discovered to selectively inhibit miR-372 biosynthesis, reversing proliferation and invasive phenotypes in gastric cancer[100]. Notably, Risdiplam and Branaplam, small molecules identified for their splicing regulatory capabilities, have progressed through clinical trials for SMA[7−9,58,103,104]. The ongoing exploration of small molecules that bind to RNA holds significant promise for advancing our understanding and treatment of RNA-related diseases, particularly as selective binding and degradation strategies are developed. Collectively, these occupancy-driven RNA-targeting small molecules demonstrate the feasibility of modulating pathogenic RNA function through structure-guided recognition. However, limitations such as transient target engagement, incomplete suppression of pathogenic transcripts, and suboptimal selectivity remain important challenges for clinical translation[11,12].
These limitations have stimulated increasing interest in event-driven RNA degradation strategies capable of converting RNA-binding ligands into catalytic degraders. Inspired by TPD, emerging RNA-targeting chimeras such as RiboTACs recruit endogenous ribonucleases to achieve selective degradation of pathogenic RNAs, thereby extending RNA-targeting pharmacology from functional inhibition to programmable RNA elimination[1,11,12].
-
RiboTACs represent a promising RNA degradation strategy that mainly consists of three components: an RNase L activator, a linker, and an RNA binding moiety[60]. Some tags will also be used in the study of RiboTAC-related mRNA and proteins involved in chemical pull-down cross-linking and separation (Chem-CLIP) experiments[113]. This strategy operates by binding to the target RNA and subsequently recruiting RNase L, thereby promoting the degradation of the RNA molecule. One notable advantage of RiboTACs over conventional therapies, such as ASO, siRNAs, and SMIs, is their catalytic and event-driven pharmacological mechanism[113]. Unlike occupancy-driven RNA inhibition, RiboTACs can repeatedly recruit endogenous RNase L to achieve sustained degradation of pathogenic RNAs at relatively low concentrations, thereby enhancing the therapeutic efficiency and selectivity[27]. For instance, TGP-96-RiboTAC (compound 5) has been shown to enhance the RNA-binding capability and miR-96 inhibition ability of TGP-96 (compound 3), demonstrating its utility in therapeutic applications (Fig. 3)[23,42−44]. RiboTACs targeting the IRES of mRNAs, such as MYC-RiboTAC and JUN-RiboTAC, convert inactive binding interactions into effective RNA degradation mechanisms[12]. This conversion leads to significant anti-cancer effects by reducing the levels of oncogenic mRNAs associated with these targets. To date, a variety of RiboTACs have been rationally designed and synthesized, reflecting ongoing advancements in this field (Fig. 6). In this section, we summarize the design and optimization principles of the three RiboTAC components, as well as their recent applications, providing a theoretical foundation for future development.
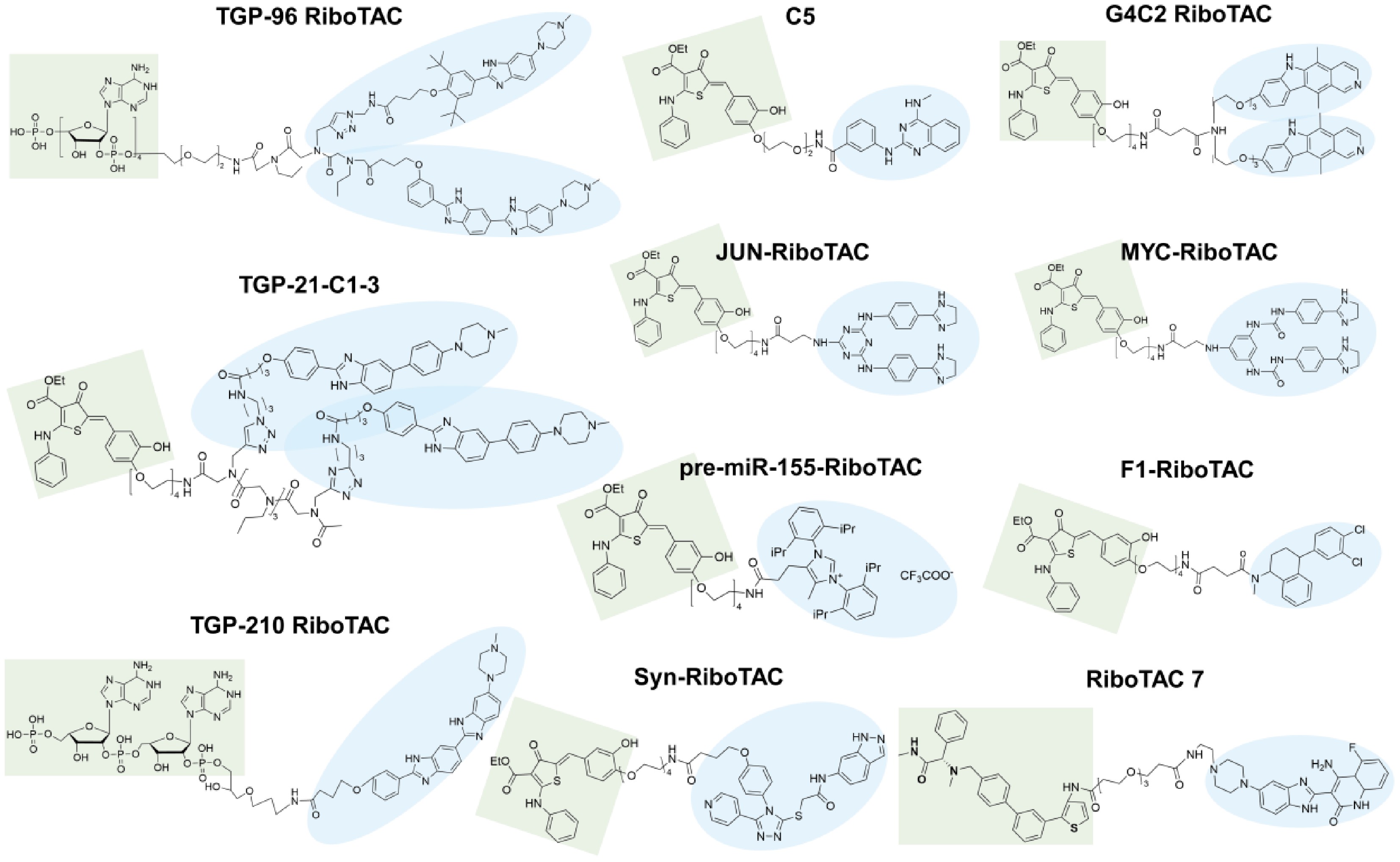
Figure 6.
Structures of currently reported RiboTAC molecules. The green moieties represent RNase L-recruiting groups, whereas the blue moieties represent RNA-binding groups.
Beyond classical RiboTAC technology, several emerging small molecule-mediated targeted RNA degradation approaches have been developed in recent years. Among them, the proximity-induced nucleic acid degrader (PINAD) platform, proposed by Mikutis et al.[114], achieves efficient and selective RNA cleavage through proximity-induced effects, further expanding the toolbox of targeted RNA degradation strategies. In addition, in a recent review, Bonet-Aleta et al.[115] have systematically summarized and classified existing small-molecule RNA degraders. In contrast to these broader classification-oriented reviews, in this section we mainly focus on RiboTACs, emphasizing their structural design principles, mechanistic advantages, optimization strategies, and preclinical therapeutic potential, and particularly on their emerging applications in refractory cancers to underscore the unique characteristics and translational significance of this emerging RNA degradation modality.
RNA binding part
-
The RNA binding component of RiboTACs primarily utilizes compounds such as benzimidazoles, aminoglycosides, and polyamines, among others (Fig. 7a). To facilitate the discovery and optimization of such RNA-targeting ligands, multiple mainstream strategies have been established for mining and optimizing RNA-binding ligands, including 2DCS-Inforna screening, structure-based rational design, and single-motif high-throughput screening (Fig. 7a)[116]. Combining microarray technology and high-throughput sequencing, the 2DCS-Inforna platform efficiently profiles intermolecular interactions between small molecules and RNA motifs, and further establishes the Inforna database for the rapid mining of pathogenic RNA-targeted compounds[116−118]. In comparison, the structure-based design optimizes the binding affinity by resolving RNA-ligand three-dimensional (3D) conformations using nuclear magnetic resonance (NMR) or X-ray crystallography, coupled with virtual screening techniques. Additionally, single-RNA-motif screening using by the automated ligand identification system (ALIS) enables target-oriented small-molecule discovery and is particularly suitable for the precise identification of miRNA-targeted inhibitors[119]. Collectively, these complementary screening strategies substantially expand the repertoire of RNA-binding small molecules available for the development of RiboTACs. Importantly, efficient RNA-binding moieties must not only recognize pathogenic RNAs with high affinity but also position the RNase L recruitment module in an orientation compatible with ternary complex formation and catalytic RNA cleavage[120,121].
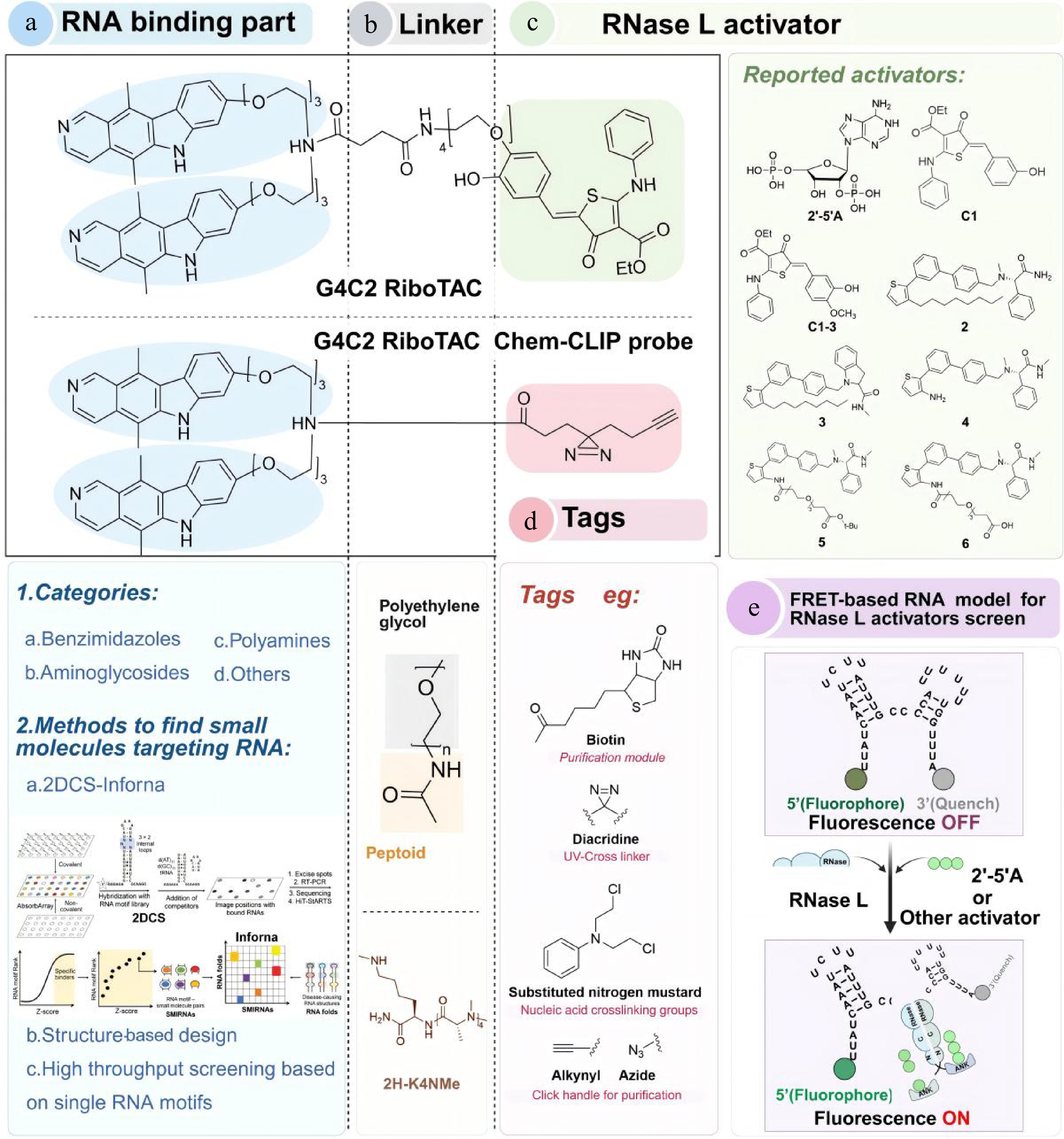
Figure 7.
Optimization of RiboTACs. (a) Classification and screening strategies for the RNA binding part. (b) The linker of RiboTACs is generally a combination of PEG and peptides. (c) Reported RNase L activators. (d) Tags used in RiboTACs. (e) FRET-based RNA model for the RNase L activators screen. When RNase L is dimerized and activated by 2'-5'A or other small molecules, it cleaves the RNA and displays fluorophores.
Nevertheless, despite these advances, several intrinsic challenges remain. The highly dynamic and conformationally flexible nature of RNA structures complicates reliable target validation, while sequence homology among RNA motifs frequently leads to off-target interactions and limited molecular selectivity[11,117,122]. Moreover, inadequate pharmacokinetic properties and the poor in vivo stability of current RNA binders continue to impede the clinical translation of RiboTAC-based therapeutics[123]. These bottlenecks are primarily derived from the imperfect druggability of RNA targets that fail to satisfy the four unified evaluation criteria.
Linker optimization
-
Linkers play a crucial role in the design of effective and specific RiboTACs, yet a universal linker has not yet been established. In 2013, Professor Disney investigated small molecules targeting the DM1 repeat region r(CUG)exp in a modular assembly manner[124]. By testing different linkers, such as polyamines, α-peptides, β-peptides, and peptide tertiary amides (PTAs), the researchers found that PTA was the best ligand for improving DM1-related defects and assessing protease stability, cell permeability, and toxicity. The most efficient connectome structure 2H-K4NMe is shown in Fig. 7b[124].
In addition to PTA, researchers have used a combination of polyethylene glycol (PEG) with peptides, including α-peptides and β-peptides, as linkers for RiboTACs (Fig. 7b). Rather than simply serving as passive connectors, linker composition and geometry critically govern ternary complex formation, spatial accessibility of RNase L, conformational flexibility, and overall degradation efficiency[117]. PEG increased the hydrophobicity of the RNase L recruitment group, thereby improving the permeability and bioavailability of RiboTACs[25]. Importantly, linker length should be carefully optimized to balance RNA binding, RNase L recruitment, and catalytic degradation efficiency, while minimizing steric hindrance and off-target effects[27].
RNase L activator optimization
-
As a core component of the innate immune response, RNase L helps cells resist the viral invasion[121]. Upon infection, double-stranded RNA activates oligoadenylate synthetase (OAS), leading to the production of 2'-5' linking oligoadenylate (2'-5'A). This activates RNase L, enabling it to cleave RNA containing unpaired uridines (UNN pattern)[120,125−127]. The initial RiboTACs were formed by linking small molecules with 2'-5'A; however, sustained RNase L activation by 2'-5'A can induce apoptosis[121]. Moreover, the rapid degradation of 2'-5'A and its limited cell membrane permeability hindered further development[28]. Fortunately, recent studies have identified small molecules that activate RNase L similarly to 2'-5'A, making them suitable for RiboTAC applications (Fig. 7c)[12,31,128]. High-throughput screening using FRET was used to identify structural alternatives to 2'-5'A. This approach monitored the effect of compounds and RNase L on the fluorescence signal of a FRET probe corresponding to a segment of genomic RNA from paramyxovirus and respiratory syncytial virus, which contains multiple RNase L cleavage sites (UU or UA) in an optimal cleavage context (Fig. 7e)[25,129−131]. The first series of compounds selected, C1 and C2, significantly enhanced the fluorescence signal of the FRET probe but were less effective than 2'-5'A[129]. Further screening for structural similarities led to the identification of C1-3 as more active RNase L activators[25,120]. Two subsequent compounds, 2 and 3, demonstrated dose-dependent enhancements in RNA cleavage, with compound 2 exhibiting a stronger effect than 3. Further structural modifications to compound 2 yielded compounds 4, 5, and 6, with compound 6 showing the best activation effect[87].
Collectively, optimization of RNase L activators primarily aims to balance the activation potency, metabolic stability, membrane permeability, and controlled catalytic RNA degradation while minimizing excessive immune activation and cytotoxicity[25]. It is essential to note that the targeted degradation capability of RiboTACs largely depends on the content of RNase L, predominantly found in the cytoplasm, limiting RiboTACs' efficacy in degrading nuclear RNAs[25].
Tags
-
The predominant method for confirming the targets in RiboTAC research is Chem-CLIP[60,117,132,133]. Its probes typically consist of three components: a photocrosslinking unit (e.g., diazirine), an enrichment and purification moiety (e.g., biotin), and a click chemistry element (e.g., azide or alkyne) (Fig. 6d). This setup facilitates a crosslinking reaction upon binding to the RNA motif[132,133]. The Chem-CLIP process generally involves extracting total RNA, purifying and enriching the crosslinked RNA using a purification module, followed by RNA sequencing or quantification through quantitative reverse transcription-polymerase chain reaction (qRT-PCR)[133]. Importantly, tags in RiboTAC systems primarily function as mechanistic validation tools for confirming RNA engagement and target selectivity, rather than directly enhancing the degradation activity.
Construction strategy of RiboTACs
-
The synthesis of RiboTACs follows a modular design in which three functional elements (RNA binding moiety, linker, and RNase L activator) are covalently assembled to enable selective RNA degradation[12,13,87]. The process typically begins with functionalization of the RNase L activator to introduce a reactive amino group (-NH2), allowing subsequent conjugation. Common activators, including 2'-5'A derivatives and C1-3 analogues, are chosen for their stability and efficient activation of RNase L. Their amino modification is typically achieved through N-hydroxysuccinimide (NHS) ester-mediated amidation or reductive amination[86,129].
The linker is then installed to define the spatial relationship between the RNA-binding module and the RNase L recruiter. Importantly, linker composition and geometry play critical roles in regulating ternary complex formation, RNase L accessibility, conformational flexibility, and overall degradation efficiency[134]. Short (e.g., α-peptides with 2–4 amino acid residues) or rigid linkers (e.g., PEG4–PEG6) are generally suitable for simple RNA motifs, whereas flexible peptide or hybrid linkers improve the adaptability for structurally complex RNAs[25,124]. Systematic optimization has identified intermediate-length PEG linkers (PEG4–PEG8) as optimal, as they balance the solubility, cellular uptake, and conformational flexibility. In contrast, excessively short (PEG2) or long (PEG10–PEG12) linkers, as well as rigid scaffolds, often impair activity due to suboptimal spatial alignment[134]. These coupling steps are typically carried out under anhydrous dimethylformamide (DMF) or dichloromethane (DCM) using condensation reagents such as N,N'-diisopropylcarbodiimide (DIC), exafluorophos-phate azabenzotriazole tetramethyl uronium (HATU), and Oxyma to ensure high yields[13,25,134].
The RNA-binding ligand (equipped with a carboxyl group, -COOH) is subsequently conjugated to the linker-activator intermediate. The RNA binding moiety is pre-optimized through high-throughput screening or structure-based design to ensure specific recognition of target RNA motifs (e.g., Drosha/Dicer cleavage sites, IRES regions, G4s, or repeat sequences)[12,13]. For example, benzimidazole-based ligands have been successfully employed to target IRES-containing transcripts such as MYC and JUN. Their carboxyl groups are activated by 1-hydroxybenzotriazole (HOBt) to facilitate efficient coupling with the amino-terminated linker[12]. Final products are validated by chromatography and mass spectrometry analyses to confirm purity and structural integrity[87,102].
Recent advances have expanded RiboTAC's construction strategies beyond conventional stepwise synthesis. Fragment-based approaches, including click chemistry, enable independent optimization of RNA-binding and RNase L-recruiting modules prior to conjugation, thereby improving the synthetic efficiency[102]. This approach has been successfully applied to the synthesis of pre-miR-372 degrader, achieving a 3-fold increase in yield compared to classical amidation. Another innovative strategy involves the solid-phase synthesis, in which the RNA binding moiety is immobilized on a resin and the linker and RNase L activator are introduced sequentially. This method enables rapid screening of linker length and composition, facilitating structure-activity optimization[102]. In parallel, the incorporation of stimulus-responsive linkers, such as hydrazone bonds, allows controlled release of RiboTACs in acidic tumor microenvironments, thereby enhancing the target specificity and minimizing the off-target effects[115]. Emerging assembly-based platforms have been developed to overcome the complexity of conventional RiboTAC synthesis. For example, LipoSM-RiboTAC employs liposome-mediated self-assembly to co-deliver DSPE-PEG2000-modified RNase L recruiters and RNA-binding ligands, forming stable nanoparticles (~150–160 nm) that are internalized through clathrin-mediated endocytosis. Optimal activity is achieved at a 1:5 molar ratio of the RNase L recruiter to RNA binder, enabling efficient degradation of MYC mRNA, JUN mRNA, and pre-miR-155[135]. Similarly, aptamer-based RiboTAC (ARiboTAC) incorporates the nucleolin-targeting aptamer AS1411 with 4A-ASO chimeras to enable tumor-selective delivery and degradation of miR-210-3p and miR-155-5p, with minimal off-target effects[136]. Collectively, these advances demonstrate that RiboTAC construction strategies are evolving from conventional modular conjugation toward programmable, delivery-integrated, and stimulus-responsive degradation platforms.
Application of RiboTACs
-
To date, RiboTACs have been designed and synthesized to degrade various RNAs, employing four primary binding modes (Fig. 7). The first mode focuses on the Drosha and Dicer enzyme cleavage sites of cancer-related microRNAs, such as pri-miR-96[23], pre-miR-210[24], pre-miR-21[25,87], and pre-miR-155[12], and is the most extensively studied. The second mode targets viral RNA, such as the SARS-CoV-2 RNA genome[137]. The third mode addresses the repeat region of disease-causing RNAs, such as the G4C2 repeat region of c9ALS/FTD[60]. The fourth mode targets IRESs for pathogenic RNA, such as MYC and JUN[12]. These applications collectively demonstrate the broad adaptability of RiboTACs to structurally diverse RNA substrates.
An example of RiboTAC's application is the connection of 2'-5'A to the Targapremir-96 dimer, which enables selective RNA cleavage by locally activating RNase L, resulting in enhanced anti-cancer activity[23]. Additionally, C1 has been combined with two small molecules to create TGP-21-C1-3 (Fig. 3), an active miR-21 RiboTAC that outperforms the bleomycin A5 conjugate[25]. TGP-21-C1-3 promotes downstream PDCD4 expression, thereby inhibiting breast cancer cell invasion in vivo and lung metastasis in mice[25]. Notably, in this context, RiboTACs demonstrate greater selectivity compared to the bleomycin A5 conjugate.
In general, distinct RiboTAC design strategies display different performance characteristics in degradation efficiency, selectivity, and intracellular delivery[1,117]. Benzimidazole-based ligands preferentially recognize bulged miRNA precursors, whereas IRES-targeted binders are more suitable for oncogenic mRNAs[74]. Moderate-length PEG-peptide hybrid linkers help balance conformational flexibility and cellular permeability, thereby facilitating ternary complex formation and catalytic RNA degradation[134]. Compared with traditional 2'-5'A, synthetic RNase L activators exhibit improved metabolic stability and pharmacokinetic properties[25]. In contrast, Chem-CLIP tags primarily function in target validation rather than degradation enhancement[133]. Collectively, the efficiency of RiboTAC design generally depends on the coordinated optimization of RNA-binding ligands, linker architecture, and RNase L activators, providing a conceptual framework for the rational development of next-generation RNA degraders.
-
Small molecules targeting RNA have emerged as a transformative therapeutic paradigm, extending drug discovery beyond the limitations of conventional protein-based approaches to access the largely "undruggable" transcriptome. Among these strategies, RiboTACs stand out owing to their event-driven degradation mechanism, combining the advantages of small-molecule drugs (good membrane permeability and structural tunability) and oligonucleotide therapies (specific RNA recognition) to address pathogenic RNA-related diseases[27,31]. Despite these advances, key challenges persist: RNA's structural flexibility hinders small-molecule binding, RiboTAC's large molecular weight (> 800 Da) impairs cellular uptake, and heterogeneous RNase L expression limits therapeutic efficacy[12,25]. Addressing these limitations will require interdisciplinary integration across chemistry, biology, materials science, and computational modeling.
Expanding RNA recognition modalities
-
Peptides have emerged as promising RNA-targeting candidates that complement small molecules and oligonucleotides because of their distinct physicochemical properties and binding modes. Unlike small molecules, which typically engage shallow RNA pockets, peptides can adopt defined secondary structures (e.g., amphipathic α-helices or β-sheets) that enable their insertion into deeper grooves or recognition of complex tertiary architectures through sequence-specific hydrogen bonding and electrostatic interactions[137]. For instance, Pomplun et al.[71] developed biohybrid nucleobase peptide libraries with nanomolar affinity (Kd = 10–50 nmol·L−1) for pre-miRNA hairpins, achieving 10-fold higher selectivity than traditional small molecules.
Peptide-based strategies offer several key advantages. (1) Their structural adaptability allows them to accommodate dynamic RNA conformations, overcoming the limitations associated with rigid small-molecule binding[137]. (2) Peptides derived from natural amino acids generally exhibit favorable biocompatibility and reduced off-target toxicity compared with aminoglycosides[112]. (3) Their modular architecture enables straightforward integration with functional elements such as cell-penetrating peptides (e.g., TAT, RGD) or RNase L recruiters, facilitating the construction of peptide-based RiboTAC systems[17]. More importantly, peptide-based recognition strategies further expand the accessible RNA target space by enabling engagement of structurally disordered or highly dynamic RNA regions that are difficult to target with conventional small molecules[71].
Nevertheless, peptide-based RNA targeting still faces inherent bottlenecks that hinder their further application. The limited high-resolution structural datasets of RNA–peptide complexes hinder the rational screening of high-affinity peptides[5]. Moreover, prospective biological validation remains challenging, as most candidates display inconsistent binding behavior between in vitro screening and intracellular environments.
AI-assisted drug design accelerating RiboTAC development
-
Artificial intelligence (AI) has emerged as a core driver in advancing RiboTAC development, addressing the key bottlenecks in traditional drug discovery, such as limited RNA structural data and inefficient modular optimization[138,139]. A critical breakthrough lies in AI-enabled RNA structure prediction: deep learning models trained on large sequence-structure datasets can generate reliable secondary and tertiary RNA structures (with 85%–90% secondary structure accuracy and 2–4 Å RMSD for tertiary structures) even in the absence of NMR or X-ray data[140]. Platforms such as INFORNA 2.0 further integrate structure prediction with binding affinity modeling, enabling de novo design of RNA-binding moieties with predicted Kd values that are closely correlated with the experimental results (R2 = 0.85)[118,141]. These capabilities provide a foundation for the precise targeting of RNA structural motifs, including G4s and IRES regions, in RiboTAC design[13,142].
Beyond target identification, AI facilitates the optimization of RiboTAC components. For linker optimization, AI-driven models incorporating 3D ternary complex predictions enable the rational selection of linker length and composition, minimizing steric hindrance while promoting productive complex formation. For example, an AI-optimized PEG6-peptide linker increased the pre-miR-21 RiboTAC degradation efficiency by 40%[87]. Moreover, machine learning approaches applied to high-throughput screening datasets reduce the false-positive rates by 40%–60%, thereby accelerating the discovery of specific RNA binders[138]. These advancements have the potential to accelerate the iterative optimization of RiboTAC candidates and further improve their druggability.
Despite these advances, AI-assisted RiboTAC design still faces major limitations. Insufficient high-quality RNA tertiary structure datasets restrict model generalization, and discrepancies between computational prediction and in vivo pharmacological performance remain significant challenges[142].
Improving druggability and delivery
-
Despite rapid progress, the clinical translation of RiboTACs requires further advances in delivery, selectivity, and mechanism research. Targeted delivery remains a central challenge. Ligand-functionalized nanoparticles (e.g., folate-directed systems) and cell-penetrating peptides enhance cellular uptake and tissue specificity[24], while tumor microenvironment (TME)-responsive designs enable spatially restricted RNase L activation with reduced off-target effects[115]. Linker engineering also plays a critical role. PEG4–PEG8 linkers provide a balance between solubility and degradation efficiency[134], and emerging platforms such as LipoSM-RiboTAC and ARiboTAC further improve delivery precision and biosafety through self-assembly or receptor-mediated targeting strategies[134,136].
Beyond delivery barriers, the intrinsic druggability of RNA-targeting small molecules and RiboTACs poses another major challenge to medicinal chemistry. Most RiboTAC chimeras possess relatively high molecular weights (> 800 Da), which compromise membrane permeability and oral bioavailability[143,144]. Their modular architectures also increase synthetic complexity and manufacturing cost. Although PEG-based linkers may partially improve hydrophilicity and pharmacokinetic properties, excessive molecular weight and conformational flexibility continue to limit in vivo translation[31].
Notably, the pharmacological behavior of RiboTACs differs fundamentally from that of conventional occupancy-driven inhibitors[27]. Unlike reversible binders that require sustained target engagement, RiboTACs act through an event-driven catalytic degradation mechanism, enabling a single molecule to repeatedly eliminate multiple RNA transcripts. This mechanism contributes to the sustained pharmacodynamic effects at relatively low systemic exposure. However, the therapeutic efficacy remains highly dependent on the expression of endogenous RNase L, which varies substantially across tissues and may contribute to heterogeneous degradation responses[12,25]. Furthermore, the flexible and dynamic tertiary structures of RNA complicate the establishment of intuitive structure-activity relationships, increasing the uncertainty in rational optimization and prediction of RNA-ligand interactions[31,134].
The selectivity of RiboTACs is determined by the coordinated contribution of multiple structural factors[27,144]. RNA-binding ligands that recognize specific secondary or tertiary RNA motifs provide the primary basis for target recognition, while linker geometry and conformational flexibility influence ternary complex assembly and spatial orientation[31]. In addition, localized recruitment of RNase L further contributes to selective RNA cleavage within defined intracellular contexts[12,17,25].
Beyond RNase L
-
Expanding the scope of RNA targets and enzymatic tools represents another key direction for next-generation RNA degraders. Recent studies have demonstrated RiboTAC-mediated degradation of diverse RNA classes, including lncRNAs such as TERRA via G4s recognition and viral RNAs such as SARS-CoV-2 genomes using bioorthogonally activated degraders[14,132]. Future efforts may focus on enabling nuclear RNA targeting through incorporation of nuclear localization signal (NLS)-modified linkers[27].
In parallel, the development of low-immunogenicity RNase L activators and exploration of alternative nucleases (e.g., EndoU) may help overcome the current reliance on a single enzymatic pathway[18]. Personalized approaches that account for RNase L genetic variability could further improve therapeutic consistency[19]. Overcoming heterogeneity in RNase L expression is also critical for maximizing efficacy. Strategies such as co-administration of RNase L agonists or gene-based modulation may enhance degradation capacity in low-expression contexts[12].
Beyond RNA degradation, next-generation RiboTAC systems are beginning to expand toward programmable regulation of RNA fate through recruitment of enzymes involved in RNA editing, uridylation, and decapping, including ADAR-related editing systems[20]. Advanced validation technologies, including fluorescence binding assays, NanoBRET, and live-cell imaging, are also facilitating mechanistic characterization of RiboTAC-mediated RNA degradation processes[87,145,146]. Collectively, these emerging strategies are extending RNA-targeted therapeutics beyond simple transcript degradation toward programmable manipulation of RNA function and metabolism.
Clinical translation challenges
-
Therapeutically, RiboTACs show broad potential across oncology, neurodegenerative disorders (PD, c9ALS/FTD), and viral infections[60,137]. Preclinical data have demonstrated 10-fold enhanced antiviral potency against SARS-CoV-2, 74% reduction in c9ALS/FTD poly (GP) levels, and cytoprotective effects in PD models[9,10,12]. In oncology, platforms such as Aptamer-RiboTAC have shown improved RNA degradation efficiency and antitumor activity[136].
Nevertheless, several important translational challenges remain unresolved. The dynamic conformational behavior of RNA, together with sequence similarity among related RNA family members, may increase the risk of off-target interactions[147,148]. In addition, heterogeneous intracellular RNase L distribution, protein competition, and variable cellular microenvironments can compromise degradation consistency across tissues and disease contexts. Efficient RNase L recruitment also requires coordinated optimization of recruiter potency, linker flexibility, and intracellular spatial organization, all of which collectively influence the stability of the ternary complex and efficiency of RNA cleavage[149].
Overall, further advances in RNA-targeted therapeutics will require coordinated advances in RNA structural biology, medicinal chemistry, delivery technologies, and computational modeling. With continued improvements in molecular specificity, delivery efficiency, and pharmacological properties, RiboTACs and related RNA degraders are expected to emerge as a new class of therapeutics capable of targeting previously inaccessible disease-associated RNAs.
-
Not applicable.
-
The authors confirm their contributions to the work as follows: conception and design: Jin J, Luan X; draft manuscript preparation: Zhou W, Jin J; analysis and interpretation: Wu Y, Chen H, Zhang W. All authors reviewed and approved the final version of the manuscript.
-
Data sharing is not applicable to this review as no datasets were generated or analyzed.
-
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 82322073, 82173846, 82430119, 82304790, and 82304533), Shanghai Rising-Star Program (No. 22QA1409100), Oriental Scholars of Shanghai (TP2022081), Jiangxi Province Thousand Talents Program (jxsq2023102168), Shanghai Shuguang Program (No. 24SG40), the Organizational Key Research and Development Program of Shanghai University of Traditional Chinese Medicine (2023YZZ02), National Key R&D Program of China (Grant No. 2024YFC3506603), Shanghai Sailing Program (No. 23YF1442600), the grant from the State Key Laboratory of Discovery and Utilization of Functional Components in Traditional Chinese Medicine (SHUTCM-SKL-202509, SHUTCM-SKL-202520), CAMS Innovation Fund for Medical Sciences (CIFMS) (2023-I2M-3-009), Key project at central government level: The ability establishment of sustainable use for valuable Chinese medicine resources (2060302), Three-year Action Plan for Shanghai TCM Development and Inheritance Program [ZY(2025-2027)-2-2-1], High level Key Discipline of National Administration of Traditional Chinese Medicine (No. zyyzdxk-2023071), and Innovation team of high-level local universities in Shanghai: Strategic Innovation Team of TCM Chemical Biology, Shanghai Key Laboratory of Traditional Chinese Clinical Medicine (20DZ2272200). We acknowledge the online drawing software for supporting the figure creation (BioRender, https://biorender.com).
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Wei Zhou, Jinmei Jin
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhou W, Jin J, Wu Y, Chen H, Zhang W, et al. 2026. Small-molecule-mediated RNA targeting: emerging strategies and therapeutic potential of RiboTAC. Targetome 2(3): e025 doi: 10.48130/targetome-0026-0023 

Small-molecule-mediated RNA targeting: emerging strategies and therapeutic potential of RiboTAC
- Received: 03 April 2026
- Revised: 24 May 2026
- Accepted: 27 May 2026
- Published online: 24 June 2026
Abstract: Many disease-associated proteins are difficult to target using conventional small molecules or biologics. RNA offers a promising therapeutic strategy because its structural motifs can create recognition sites for regulatory proteins and small molecules, enabling intervention upstream of proteins that are otherwise considered "undruggable". Recent advances in RNA-targeting therapeutics, particularly RNA-binding small molecules, have demonstrated the feasibility of selectively modulating RNA maturation, splicing, translation, and degradation. However, their broader clinical application is still limited by non-specific toxicity, off-target effects, stability concerns, and delivery challenges. Targeted protein degradation is a remarkable therapeutic modality that has been widely used to target undruggable pathogenic proteins. In relation to this, a currently emerging research field is that of ribonuclease-targeting chimeras (RiboTACs). Unlike the "occupancy-based" mode of small-molecule inhibitors (SMIs), RiboTACs recruit endogenous ribonucleases to selectively degrade disease-associated RNA transcripts through an event-driven mechanism, which enables efficient removal of pathogenic RNAs while offering advantages with regard to medicinal chemistry optimization compared with oligonucleotide-based approaches. In this perspective, we systematically summarize the classification of targetable RNAs, recent advances in RNA-targeting small molecules, and the design principles, applications, and future challenges of RiboTACs.
-
Key words:
- Small molecules /
- RiboTACs /
- RNA-targeting therapeutics