









-
The rapid development of high-throughput sequencing technologies has led to an exponential increase in microbial genomic data[1−3]. However, functional annotation of the vast number of genes identified from these genomes remains a major bottleneck, with a considerable proportion of genes still labeled as 'hypothetical proteins' whose biological functions are unknown, severely hindering the translation of genomic data into biological insights and practical applications[4,5].
One of the factors affecting the efficiency of functional annotation is that while substantial valuable enzyme functional information has been obtained through experimental studies, this information is dispersed across vast amounts of scientific literature, making systematic utilization challenging. When conducting functional annotation, researchers often need to manually search and review relevant literature to obtain detailed information about specific enzymes, such as enzymatic activity, substrate specificity, optimal reaction conditions, and other key biochemical parameters[6,7]. This manual literature mining process is relatively time-consuming and susceptible to subjective factors, which may limit the effective translation of existing knowledge into genomic annotation[8,9].
The conventional pipeline for systematic literature review encompasses a series of pivotal steps: (1) defining research themes and formulating search strategies, followed by selecting pertinent databases such as PubMed for biomedical literature, Web of Science for multidisciplinary citations, Embase for pharmacology and life sciences, and the Chemical Abstracts Service (CAS) for chemical literature[10,11]; (2) pinpointing key search terms and utilizing controlled vocabularies like the Medical Subject Headings (MeSH) and the CAS registry to expand topics and standardize terminology[12]; (3) meticulously amalgamating search terms via Boolean logic, stemming analysis, synonym expansion, and term relevance weighting, employing natural language processing techniques to construct sophisticated and precise queries[12]; (4) conducting searches within chosen databases to gather an initial collection of potentially related literature[13]; (5) manually screening the literature by researchers for duplication and thematic relevance, implementing predetermined inclusion and exclusion criteria to curate a core collection of applicable literature[14]. The traditional information extraction stage (6) is typically a process involving two or more researchers repetitively reading through each article within the core literature set, manually identifying, and distilling the necessary data to form a standardized, structured dataset. This manual operation usually necessitates a certain level of redundant work, such as independent extractions followed by cross-verification between researchers, to ensure the accuracy and consistency of the extracted information[15,16]. This comprehensive manual process of literature retrieval and data extraction is laborious, protracted, and susceptible to the influence of subjective human factors, which can lead to variable quality and consistency in the outcomes.
The field of artificial intelligence, particularly through large language models (LLMs), offers new possibilities in this regard. Trained on massive textual datasets, LLMs acquire a broad range of knowledge, from simple facts to complex theories[17], enabling them to rapidly and accurately extract the information required by researchers from large-scale literature collections[18]. In tasks such as systematic reviews, which involve extensive literature screening and synthesis, LLMs demonstrate significant advantages in terms of time and cost efficiency[19,20]. More importantly, their algorithmic objectivity helps reduce biases arising from subjective judgments in traditional manual reviews, thereby enhancing the transparency and reproducibility of research[19,21]. Nevertheless, existing studies on LLM-assisted biological information extraction exhibit notable limitations. Most efforts have not systematically evaluated the accuracy of LLMs in extracting fine-grained functional information from the literature, such as enzyme activity parameters and sequence identifiers[22,23], nor have they established performance benchmarks tailored to specific application scenarios, such as enzyme function mining.
Furthermore, in the field of genomics, in addition to textual literature, there exists a substantial amount of structured and visual information derived from genomic data, such as phylogenetic trees, multiple sequence alignments, and functional domain architecture diagrams, that encodes evolutionary relationships and structural constraints, which are crucial for functional inference[24,25]. However, whether LLMs can effectively analyze such multimodal information and integrate it into functional annotation pipelines remains unexplored.
Mycotoxin-degrading enzymes serve as an ideal case for examining the aforementioned issues. Mycotoxins are toxic secondary metabolites produced by fungi, which are frequently found in a wide range of foodstuffs and pose a major threat to human and animal health[26]. Mycotoxins can be produced by a wide range of microorganisms, such as Aspergillus, Penicillium, Fusarium, Alternaria, and Claviceps[15]. Research in the food industry has been focused on characterizing enzymes capable of degrading these harmful compounds[27]. However, the vast and rapidly growing body of literature on mycotoxin-degrading enzymes presents a challenge for researchers seeking to stay up-to-date with the latest findings. Currently, there is no complete database storing information on mycotoxin-degrading enzymes. Therefore, we plan to collect published literature on this subject by manually extracting it from information on mycotoxin-degrading enzymes to construct the database. Unfortunately, in practice, we found that it is very time-consuming and inefficient to manually find the target information from the downloaded literature one by one. For example, for finding the conditions under which an enzyme degrades all substrates, it would require an author to repeatedly scrutinize the entire text before finally identifying the target information, which is likely to be scattered in the descriptions of different paragraphs in an article.
This study aims to evaluate the performance of large language models in literature information extraction within the microbiology domain. We systematically assessed six large language models, including ChatGLM-3-Turbo, ChatGPT-4o, two versions of the Claude series (Claude2 and Claude Sonnet 4), DeepSeek-V3, and Kimi Chat, for their accuracy and efficiency in extracting biologically relevant information from literature. The target information evaluated included experimental and functional attributes, as well as sequence-related information and enzyme-associated database identifiers. Additionally, we conducted a preliminary exploration of large language models' capability to analyze genome-derived visual information, using Claude Sonnet 4 to analyze phylogenetic trees, multiple sequence alignments, and functional domain architecture diagrams, and compared its outputs with expert manual analysis. Based on the evaluation results, we developed an assistive workflow tool named 'ptol' that supports literature retrieval, text preprocessing, and standardized input generation. We anticipate that this work may provide methodological reference for literature mining in the microbiology field and establish performance benchmarks for related information extraction tasks.
-
We initially surveyed foundational large language models accessible through various platforms and APIs during the study period. The selection was conducted according to three predefined criteria: (i) direct model accessibility during the study period, (ii) support for processing long document texts, and (iii) availability of documented model specifications and parameters. Based on these criteria, we selected six foundational LLMs for formal comparison: ChatGLM-3-Turbo, ChatGPT-4o, Claude2, Claude Sonnet 4, DeepSeek-V3, and Kimi Chat. Notably, we deliberately retained two versions of Anthropic's Claude series (Claude2 and Claude Sonnet 4) to evaluate performance changes within the same model family during technological iteration, providing valuable comparative data for understanding the developmental trajectory of large language models in biological information extraction tasks. Given the rapid updates and iterations of LLMs, and considering that our research timeframe might involve different model versions, this cross-version comparison helps assess the impact of technological advancement on practical application effectiveness.
We used keywords such as Mycotoxin, enzyme, and degrad* as indices for the collection of literature related to mycotoxin-degrading enzymes in PubMed, Web of Science, and CNKI databases, and randomly downloaded 86 documents. Thirteen papers containing more complete information on mycotoxin-degrading enzymes were hand-selected as targets for testing through careful reading and discrimination (Supplementary Table S1).
At the paper level, the 13 selected studies covered six major mycotoxin groups, including deoxynivalenol (DON; three papers), zearalenone (ZEN; two papers), aflatoxins (including AFB1, AFB2, AFG1, AFG2, and AFM1; four papers), fumonisins (including FB1, FB2, FB3, and hydrolyzed FB1; two papers), ochratoxin A (OTA; three papers), and patulin (PAT; one paper). Because some papers reported enzymes acting on more than one mycotoxin, these counts are not mutually exclusive. Therefore, the benchmark dataset spans multiple toxin classes rather than a single narrowly defined subfield, which helps reduce potential bias arising from highly standardized phrasing in one specific mycotoxin category.
Manual determination of target information for mycotoxin-degrading enzymes
-
We first manually identified 18 information targets related to mycotoxin-degrading enzymes in the literature. These include 'the name of the enzyme', 'the microbial source of the enzyme', 'the type of enzyme', 'the substrate for enzyme recognition', 'the all products of enzymatic degradation of toxins', 'the thermal stability of the enzyme', 'the optimal temperature of the enzyme', 'the pH tolerance range of the enzyme', 'the optimal pH of the enzyme', 'the conditions for enzyme degradation of all substrates', 'the amount of toxin used', 'the concentration of the enzyme', 'the reaction speed of degradation', 'the degradation reaction time', 'the degradation ratio', 'the biological sequence information of the enzyme', 'the enzyme-related databases id', and 'the DOI number'. The results for these 18 information targets were recorded promptly, and if any of the information was missing, it was set as empty in the table. To establish the reference answers for benchmarking, the target information from each of the 13 studies was manually extracted independently by two researchers using a predefined extraction form containing the 18 target items. Before formal extraction, the two researchers jointly reviewed a small subset of papers to harmonize the operational definitions of each target item and ensure a consistent understanding of the extraction criteria. After independent extraction, the two result sets were compared item by item, and discrepancies were resolved through discussion. When consensus could not be reached, a third researcher was consulted for adjudication. The final consensus dataset was used as the reference standard for evaluating LLM extraction accuracy.
Testing of LLMs via fair questioning
-
We strictly adhered to the controlled variable method to ensure that differences in querying LLMs were solely attributed to variations in target information. For each question entered into the tested LLMs, we uniformly used consistent syntax: "literature PDF" (or "literature text" for LLMs that do not support direct PDF input), please help me find "Question n" in the above text. For LLMs that natively support PDF input, the original PDF files of the 13 studies were directly uploaded; for LLMs that do not support PDF input, the pdf_to_text module of our ptol tool was employed to convert PDF documents into plain text files before submission. The content of the 18 questions was replaced with 'Question n' as the standard input for six LLMs (Supplementary Table S2). The efficiency of LLMs was estimated by timing from clicking the "Send" button until the complete output of all responses by the LLM. Similarly, we replaced the content of 'Question n' with the grammatical criteria of the 18 information target items separated by commas to complete the test of extracting all target information at once according to the above method. Finally, we maintained timely records of each LLM's answers and time spent on each question for each piece of literature.
Evaluation of LLMs performance
-
We used machine learning evaluation metrics such as accuracy, precision, recall, F1-score, and Matthews Correlation Coefficient (MCC) to compute a precise assessment of each LLM's performance in extracting the 18 information targets from the 13 studies. The formulae for the specific metrics are given below:
$ Accuracy=\dfrac{TP+TN}{TP+TN+FP+FN} $ (1) $ Recall=\dfrac{TP}{TP+FN} $ (2) $ Precision=\dfrac{TP}{TP+FP} $ (3) $ F1score=\dfrac{2TP}{2TP+FP+FN} $ (4) $ MCC=\dfrac{TP\times TN-FP\times FN}{\sqrt{(TP+FN)(TP+FP)(TN+FP)(TN+FN)}} $ (5) where, TP, FP, TN, and FN denote, respectively, the number of information targets correctly found to be present in the text, the number of information targets incorrectly found to be present in the text, the number of information targets correctly recognized to be absent from the text, and the number of information targets incorrectly found to be absent from the text. Furthermore, we also employed area under the curve (AUC) performance evaluation metrics, which comprehensively analyze the ability and accuracy of LLMs in recognizing target information in text.
Visualization and statistical analysis
-
We used GraphPad Prism 8.0.2 software to visualize the machine learning metrics of six LLMs for all information targets extracted from the 13 studies. The Friedman test was used to assess significant differences between the performance of the different models, and the Nemenyi post hoc test was further used for pairwise comparison. Comparisons with p < 0.05 were considered statistically significant.
Software implementation for pipeline
-
To accelerate the process of extracting target information from scientific literature using LLMs, we developed an auxiliary toolkit using Python 3.9.7. The dictionary in the nltk module (v3.7) was utilized to generate all variants of keywords; four modules, PyPDF2 (v2.12.1), pdfplumber (v0.9.0), fitz/PyMuPDF (v1.21.1), and pdfminer.six (v20221105) were implemented to provide users with different options for converting PDF files into text files. Finally, we programmed various subroutines within the toolkit to facilitate user operations.
Multimodal image analysis using large language models
-
To evaluate whether LLMs can effectively analyze genomic-derived information from visual inputs, we constructed three types of visual representations based on the sequence information of the 13 test enzymes:
(1) Phylogenetic tree: Sequences were aligned using the ClustalW program in MEGA 7.0.26 software, and a phylogenetic tree was constructed using the neighbor-joining (NJ) method with 1,000 bootstrap replicates.
(2) Domain architecture diagram: Sequences were submitted to the NCBI Conserved Domain Database (CDD) for domain prediction, and the results were visualized using the Chiplot website (
www.chiplot.online ).(3) Multiple sequence alignment: Sequences were aligned using the ClustalW program in MEGA 7.0.26 software, and the alignment results were visualized using Genedoc 2.6.002 software.
The three figures were independently analyzed by two researchers who summarized the overall characteristics of the 13 enzymes from a holistic perspective, including sequence conservation, evolutionary relationships, domain architecture features, and functional groupings, thereby establishing a reference answer set. The same images were then input to the LLM with the following prompt:
'Please comprehensively analyze the overall characteristics of these 13 enzymes based on the provided multiple sequence alignment, phylogenetic tree, and domain architecture diagrams, including sequence conserved regions, evolutionary clustering relationships, domain distribution patterns, and their possible functional groupings'.
The consistency between LLM outputs and the manually established reference answers was calculated to assess the LLM's capability for multimodal genomic information analysis.
Claude Sonnet 4 was selected for this analysis based on its superior performance in the preceding text extraction tasks. Among the six LLMs evaluated, Claude Sonnet 4 achieved the highest accuracy and F1-score in extracting structured information from mycotoxin-degrading enzyme literature, demonstrating strong proficiency in handling domain-specific biological information. Therefore, it was chosen as the representative LLM for the multimodal image analysis component of this study.
-
To accurately assess the performance of LLMs in extracting target information from literature texts, we selected six well-known advanced foundational LLMs (ChatGLM-3-Turbo, ChatGPT-4o, Claude2, Claude Sonnet 4, DeepSeek-V3, and Kimi Chat) for fair comparison, and then tested their ability to extract targeted information from 13 literature texts containing relatively complete key information about mycotoxin-degrading enzymes, using manually determined correct information as the criterion for quantitative and case-by-case analysis (Fig. 1a, Supplementary Tables S3–S15).
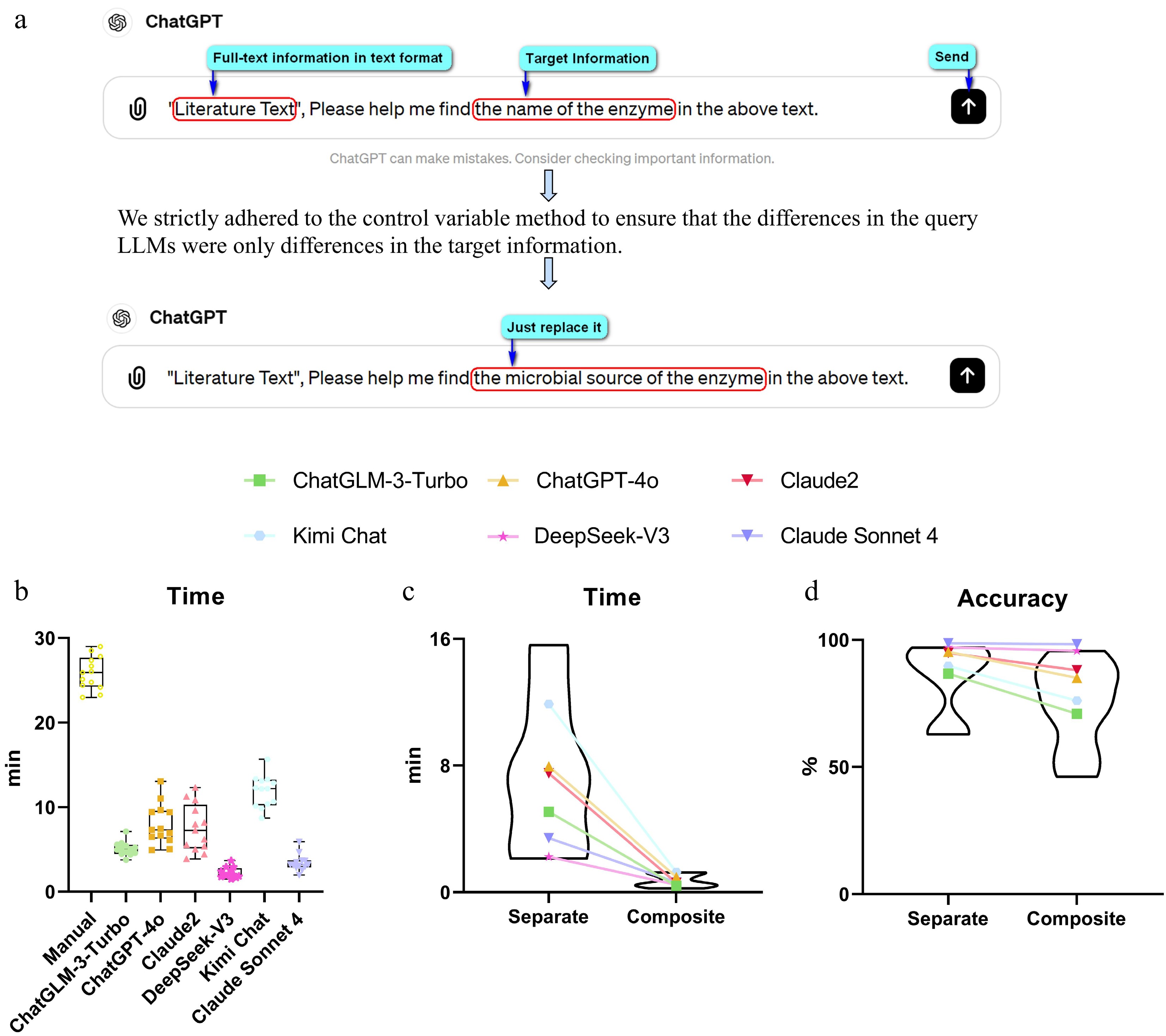
Figure 1.
Evaluation of LLM querying strategy and extraction performance. (a) Schematic of the standardized separate questioning approach, in which the target information term is systematically replaced across otherwise identical queries. (b) Box plots comparing processing time (min) between manual extraction and six LLMs under separate questioning modes. (c) Connected dot plot comparing processing time (min) for each LLM between separate and composite questioning modes, with violin plots showing the overall distribution. (d) Connected dot plot comparing overall extraction accuracy (%) for each LLM between separate and composite questioning modes, with violin plots showing the overall distribution. Statistical differences between models were assessed using the Friedman test followed by the Nemenyi post hoc test (p < 0.05).
We first compared the time spent on extracting key information by manual methods and LLMs (Fig. 1b, Supplementary Tables S16–S18). The results showed that LLMs were extremely efficient in information extraction, while the average time spent on manual information extraction was as high as 25.85 min. We then compared the effectiveness of extracting all information targets using LLMs in both composite questioning (18 target information items fused into one question) and separate questioning (extraction of each target item as one question) approaches (Fig. 1c, d). The results showed that separate questioning took more time than composite questioning, which was expected. However, the separate questioning approach could greatly improve the accuracy of target information extracted via LLMs, with the average accuracy rates under the separate questioning mode being: ChatGLM-3-Turbo (86.75%), ChatGPT-4o (95.30%), Claude2 (94.87%), Claude Sonnet 4 (98.72%), DeepSeek-V3 (97.01%), and Kimi Chat (89.75%) (Fig. 1d, Supplementary Tables S19 and S20).
Next, based on the results of separate questioning, we evaluated the differences in the ability of the six LLMs to extract the targeted information (Fig. 2, Supplementary Table S21). The results showed that Claude Sonnet 4, DeepSeek-V3, and ChatGPT-4o performed best in terms of accuracy. Particularly noteworthy was the performance comparison between the two versions of the Claude series: Claude Sonnet 4 achieved the highest average accuracy of 98.72%, significantly outperforming Claude2's 94.87%, demonstrating substantial progress through model iteration. Even more impressively, Claude Sonnet 4 excelled in handling information that was rarely mentioned in the literature. When given instructions to extract 'all' relevant information, it could sometimes identify and extract key information that was only briefly mentioned once or twice in articles, showcasing its outstanding advantage in detailed information capture capability.
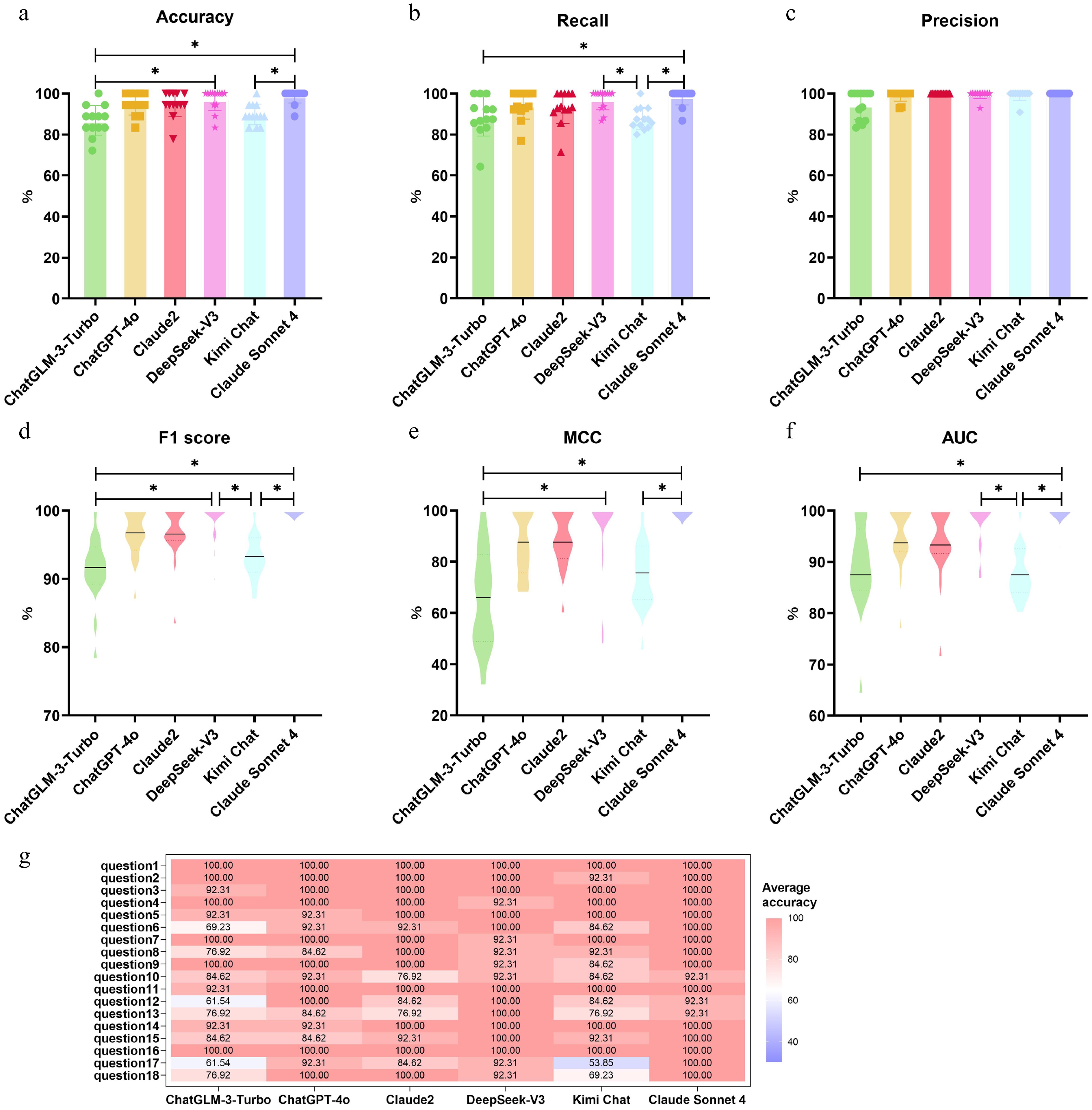
Figure 2.
Performance comparison of six large language models (ChatGLM-3-Turbo, ChatGPT-4o, Claude2, Claude Sonnet 4, DeepSeek-V3, and Kimi Chat) across six evaluation metrics: (a) accuracy, (b) recall, (c) precision, (d) F1 score, (e) Matthews correlation coefficient (MCC), and (f) area under the ROC curve (AUC). Each data point represents the mean score of a model on 18 targeted information categories for 1 of the 13 literature sources; the central line within each violin plot indicates the median. Significant differences between models were assessed using the Friedman test followed by the Nemenyi post hoc test; * indicates p < 0.05. (g) Heatmap showing the mean extraction accuracy (%) of each model across the 18 individual targeted information categories, averaged over all 13 literature sources. Color intensity reflects accuracy level as indicated in the scale bar on the right.
Finally, we further evaluated the performance of the six LLMs on 18 target information extractions from an accuracy perspective, and the results showed that all six models performed erratically on questions corresponding to different target information (Fig. 2g, Supplementary Table S22). Overall, the six models performed well on simple questions such as extracting enzyme name, microbial source, and enzyme type, while their accuracy dropped significantly below 50% for questions with complex logical queries (involving logically calculated information such as toxin dosage, enzyme concentration, and degradation ratio). This suggested that the LLM had a better ability to extract target information existing in the direct text, but a poor ability to extract the target information through more complex logical thinking, such as inference.
Systematic review pipeline: integrating AI and Python
-
Based on the above tests, we found that advanced LLMs such as Claude Sonnet 4, DeepSeek-V3, and ChatGPT-4o can serve as auxiliary tools for manually extracting target key information. To improve the efficiency of scientific research, we developed a semi-automated pipeline integrating Python scripting with large language models to enhance the efficiency of this process (Fig. 3).
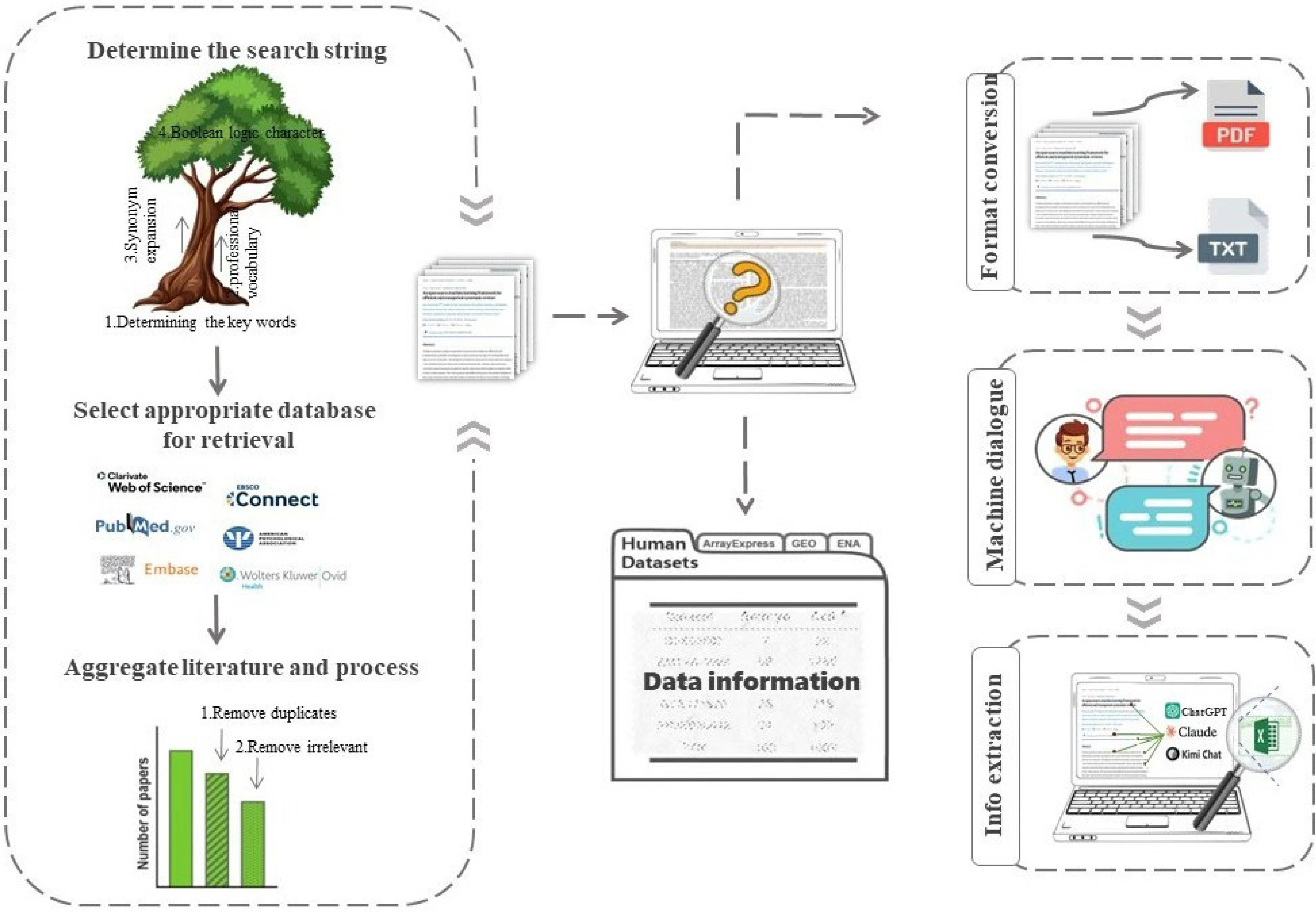
Figure 3.
Workflow for extracting information from scientific literature using large language models. The left panel illustrates the literature retrieval stage, comprising search string construction (including keyword identification, professional vocabulary, synonym expansion, and Boolean logic), database selection, and literature aggregation with duplicate and irrelevant paper removal. The center panel represents the screening and data organization stage, in which retrieved literature is reviewed and structured into a dataset. The right panel illustrates the information extraction stage, in which documents are converted into a format compatible with the target large language model (PDF or plain text), submitted via machine dialogue, and processed for structured information extraction using ChatGPT, Claude, or Kimi Chat. Arrows indicate the sequential flow between stages.
In the retrieval phase, our system begins with natural language search terms provided by the user and utilizes Python to automatically expand, standardize, and concatenate these terms, thereby constructing queries tailored to various databases. This setup provides convenient query links, facilitating relatively efficient searches. For the preliminary set of retrieved literature, the system employs computational methods such as text similarity assessments to automatically identify and eliminate duplicate documents. If certain formats are not recognized by the language models, Python scripting can convert them into compatible text formats.
In the information extraction phase, we employ a user-guided semi-automated strategy based on large language models (LLMs). For example, to identify specific enzymes mentioned within a document, users can inquire: 'Please list all the enzymes mentioned in this document'. For more complex tasks such as extracting reported enzyme degradation rates, users need to guide the LLM through appropriate prompting phrases such as: 'Please list the enzyme degradation rates reported in this text, along with the associated experimental conditions (such as enzyme concentration, quantity of toxins, reaction duration, etc.)'. If initial results are insufficient, users can further refine their query strategy. Based on the model's feedback, users need to iteratively perfect and specify their natural language inquiries to accurately capture the required data dimensions. This semi-automated approach provides certain efficiency advantages over fully manual procedures, while still requiring active user participation and professional judgment.
We compare our proposed pipeline with existing theoretical pipelines (Table 1). The results show that our theoretical pipeline has the most steps.. For comparison among other pipelines, the pipelines proposed by van de Schoot et al.[14], Timmins & McCabe[28], and Bramer et al.[11] have more steps. Therefore, our proposed pipeline can serve as a recent alternative theoretical pipeline.
Table 1. Comparison of existing pipelines.
Pipeline Ours Ref. [28] Ref. [29] Ref. [11] Ref. [30] Ref. [14] Ref. [27] Ref. [31] Ref. [32] Ref. [33] Ref. [34] Ref. [35] Ref. [36] Determining the
key words√ √ √ √ √ Term validation √ √ √ √ √ Semantic enrichment √ √ √ √ √ √ Applying boolean
operators√ √ √ √ Utilizing truncation
and wildcards√ √ √ Selecting appropriate
databases√ √ √ √ √ Compiling databases √ √ Removing duplicate
literature√ √ Excluding irrelevant
literature√ √ √ Information extraction √ √ √ √ √ Model evaluation √ √ √ √ √ √ √ Information retrieval
techniques√ Note: √ indicates that the proposed pipeline has this property. Software implementation for pipeline
-
To help streamline the literature review process and reduce some manual input, we developed an auxiliary software (ptol,
https://github.com/cauBioinformation/ptol ). This tool is designed to assist in generating search queries for academic databases such as PubMed and Web of Science. It operates based on user-defined keywords, generating relevant keyword permutations and providing access to corresponding download links. This may help improve the efficiency of accessing relevant academic literature. The ptol source code is provided with documentation under an open-source license to promote community improvements and transparency. The software can be integrated into local environments across Windows, Linux, and Mac OS platforms via the Python Package Index (PyPI) by executing 'pip install ptol' in a command-line interface.The ptol is a Python Package that is divided into four main modules, namely query_syntax, remove_duplicates, download_pdfs, and pdf_to_text. The query_syntax module is developed based on the literature keywords specified by the researcher, initially identifying their potential variations and then automatically generating search URLs that correspond to the literature results in the PubMed database. This feature allows users to access comprehensive details related to the desired literature, including titles and publication data. Furthermore, the module constructs keyword-based search syntax for both PubMed and Web of Science, catering to different user preferences and enhancing search flexibility.
Subsequently, users need to consolidate target documents obtained from various bibliographic databases and sources, for example, using MS Excel software to merge title and DOI number links. The remove_duplicates module is then used to attempt to eliminate redundancies within this collated dataset. Users need to download documents in PDF format following the guidelines described in the pipeline and observing copyright norms, typically leveraging university library subscriptions or open-access repositories. The download_pdfs module provides an option for users while observing literature copyright, built on the principle of accessing free literature databases.
Finally, the accumulated PDF documents were directly uploaded to LLM interfaces that natively support PDF input (such as Claude Sonnet 4, ChatGPT-4o, and DeepSeek-V3). For LLMs that do not support direct PDF input, the pdf_to_text module was employed to batch convert PDF documents into plain text files prior to submission. Researchers can then interact with these models using the query strategy proposed in our manuscript and allow for personalized variations, ultimately extracting the target information from the documents quickly.
Preliminary exploration of multimodal genomic information analysis using Claude Sonnet 4
-
This study conducted a preliminary exploration of Claude Sonnet 4's capability to process genome-derived visual information. Three types of visual representations were constructed based on the sequence information of the 13 test enzymes: a phylogenetic tree (Fig. 4), a domain architecture diagram (Fig. 5), and a multiple sequence alignment (Fig. 6). Claude Sonnet 4 was then prompted to comprehensively analyze the overall characteristics of the 13 enzymes across all three figures simultaneously. In this analysis, Claude Sonnet 4 demonstrated the ability to identify major branching patterns in the phylogenetic tree, including clustering of fungal laccase sequences (Pleurotus and Trametes-derived sequences), sister relationships of Alcaligenes faecalis sequences, and evolutionary associations of Devosia genus sequences. Regarding domain architecture interpretation, the model showed basic recognition capabilities for different functional modules, successfully describing multi-copper oxidase domain combinations, PQQ_2_superfamily, and other key structural features. In multiple sequence alignment analysis, Claude Sonnet 4 was able to identify conserved region distribution patterns and establish correlations with phylogenetic groupings. Notably, the model exhibited a tendency for cross-modal information integration, attempting to correlate three distinct types of biological data to form coherent interpretative frameworks. This analytical capability transcends traditional single data-type processing approaches, demonstrating AI's potential in comprehensive multi-evidence analysis within bioinformatics.

Figure 4.
Phylogenetic tree of the 13 mycotoxin-degrading enzyme candidates. The tree was constructed using the neighbor-joining (NJ) method based on multiple sequence alignment generated by ClustalW in MEGA 7.0.26. Bootstrap values from 1,000 replicates are shown at each branch node. Scale bar represents 0.10 substitutions per site.
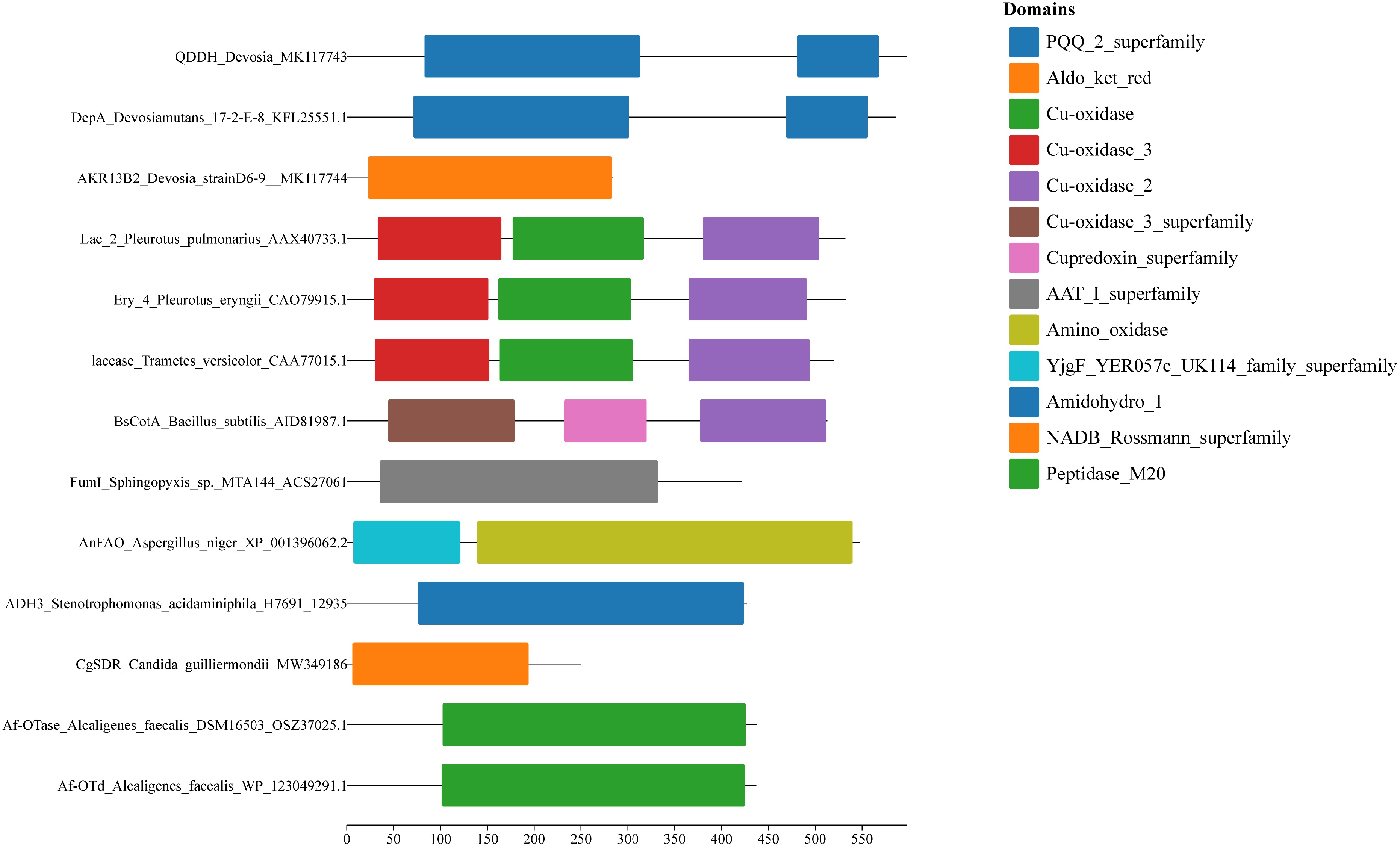
Figure 5.
Domain architecture diagrams of the 13 mycotoxin-degrading enzyme candidates. Domain predictions were obtained from the NCBI Conserved Domain Database (CDD) and visualized using Chiplot. Each colored block represents a conserved domain, with domain types indicated in the legend on the right. The x-axis represents amino acid position, and domain lengths are shown to scale.

Figure 6.
Multiple sequence alignment of the 13 mycotoxin-degrading enzyme candidates. Sequences were aligned using ClustalW in MEGA 7.0.26 and visualized using Genedoc. Conserved residues are highlighted with shading, with darker shading indicating higher conservation across sequences. Gaps represent insertions or deletions relative to the consensus alignment.
-
This study systematically evaluated six mainstream LLMs on a fine-grained biological information extraction task, using mycotoxin-degrading enzyme literature as a domain-specific benchmark. Across 234 independent extraction tests spanning 13 publications and 18 target information categories, the results demonstrate that top-performing models can achieve accuracy rates of 95%−99% with processing times well under 10 min per article, compared to a manual average of 25.85 min. This represents a substantial efficiency gain and confirms that LLMs are capable of serving as effective auxiliary tools for domain-specific literature mining. While previous studies have documented LLM utility in biomedical literature screening and systematic review automation[19,20], fine-grained extraction of biochemical parameters, such as enzyme kinetic data, substrate specificity, and sequence identifiers, from specialized microbiology literature had not previously been systematically benchmarked. The present work fills this gap and establishes empirical performance references that can guide LLM adoption in similar annotation contexts.
A key contribution of this work is the development and evaluation of a semi-automated literature mining pipeline that combines LLM-based extraction with Python-based automation for literature retrieval, deduplication, and format conversion. A persistent challenge in enzyme functional annotation is that valuable biochemical characterization data remain dispersed across vast amounts of primary literature, making systematic utilization difficult and rendering manual extraction both time-consuming and susceptible to subjective variability[15,16]. The ptol toolkit developed in this study directly addresses this bottleneck by providing a structured workflow that guides users from keyword-based database search through duplicate removal, PDF acquisition, and standardized LLM input generation. Comparison with existing systematic review pipelines (Table 1) shows that our proposed pipeline covers the most properties among all evaluated frameworks and is the only one to include information retrieval techniques as a dedicated component. Although mycotoxin-degrading enzymes serve as the proof-of-concept domain here, the pipeline is not inherently domain-specific and could be extended to other enzyme families or microbial systems where comprehensive structured databases are currently lacking.
The benchmarking results reveal clear and practically meaningful performance differences among the six models. Claude Sonnet 4 achieved the highest accuracy at 98.72% under separate questioning, followed by DeepSeek-V3 at 97.01% and ChatGPT-4o at 95.30%, while Kimi Chat (89.75%) and ChatGLM-3-Turbo (86.75%) formed a lower-performing tier. The within-family comparison between Claude Sonnet 4 and Claude2 is particularly informative: the newer model outperformed its predecessor by 3.85% in separate questioning mode, and by 10.26% in composite questioning mode. The larger performance gap in composite questioning suggests that architectural advances in newer model generations are especially beneficial for multi-task and contextually complex scenarios, consistent with the broader trend of capability gains observed across recent LLM iterations[17]. A consistent task complexity stratification was observed across all models. Simple, directly stated information categories, including enzyme name, microbial source, and enzyme type, were extracted with 95%–100% accuracy across all models. Moderately complex categories, such as degradation products, reaction conditions, and temperature and pH parameters, showed an accuracy of 80%–95%, with emerging divergence between models. Tasks requiring numerical calculation or cross-passage inference, including enzyme concentration, degradation ratio, and reaction speed, dropped below 50% accuracy even for the best-performing model. This finding is consistent with documented limitations of current LLMs in numerical reasoning and multi-step logical inference, and indicates that human expert verification remains essential for quantitative biochemical parameters regardless of which model is selected.
The choice between separate questioning and composite questioning warrants careful consideration in practical applications. Composite questioning is, on average, 5–13 times faster than separate questioning, but the associated accuracy cost varies substantially across models. For Claude Sonnet 4, the two modes differed by only 0.43 percentage points (98.72% vs 98.29%), making composite questioning a reasonable option when time efficiency is the primary concern. For ChatGLM-3-Turbo, however, the accuracy difference reached 15.81 percentage points (86.75% vs 70.94%), substantially undermining the practical value of composite questioning for that model. These results indicate that the optimal questioning strategy is model-dependent and cannot be determined without prior performance evaluation. For applications where extraction accuracy is critical, separate questioning with a high-performing model is the recommended approach.
The preliminary multimodal analysis using Claude Sonnet 4 demonstrated that the model could identify major features of the 13 enzymes from phylogenetic trees, domain architecture diagrams, and multiple sequence alignments in a manner broadly consistent with expert manual analysis, including recognition of fungal laccase clusters derived from Pleurotus and Trametes, sister relationships among Alcaligenes faecalis sequences, and multi-copper oxidase domain combinations. These results suggest that multimodal LLMs hold potential as auxiliary tools for biological visual information interpretation beyond pure text extraction. However, several important caveats apply. The present exploration was based on a single analysis instance, and the consistency observed between model output and expert analysis does not exclude the possibility that the model relied on pre-trained biological knowledge rather than genuine visual reasoning from the provided images. More rigorous controlled experiments, such as analysis of modified or deliberately perturbed figures, are required before definitive conclusions about LLM multimodal capabilities can be drawn.
From a practical perspective, these findings provide clear guidance for LLM applications in bioinformatics. For basic information extraction tasks, most modern LLMs can provide acceptable performance; for applications requiring high precision, high-performance models like Claude Sonnet 4 should be selected with separate questioning strategies; for tasks involving numerical calculations and complex reasoning, current LLMs still require human verification and supplementation. Our semi-automated workflow improves efficiency while retaining necessary human professional judgment, and this human-AI collaboration model may represent best practices for current-stage LLM applications.
-
This study benchmarked six mainstream LLMs on fine-grained biochemical information extraction from mycotoxin-degrading enzyme literature, demonstrating that LLM-assisted literature mining can effectively complement genome functional annotation by systematically mobilizing published experimental data beyond the reach of conventional sequence-based methods. Clear performance tiers and a consistent task complexity stratification effect were identified, and the semi-automated ptol pipeline was developed accordingly to improve extraction efficiency while retaining necessary human oversight. Preliminary multimodal analysis using Claude Sonnet 4 showed promise for interpreting genome-derived visual information but requires further validation. This work provides empirical benchmarks and practical guidance for LLM-assisted literature mining as a means of enriching genomic functional annotation in specialized biological domains.
-
The authors confirm their contributions to this study as follows: writing − original draft, visualization: Gao L, Chen H; writing − review and editing: Gao L, Chen H, Xu G; supervision: Gao L, Ding X, Nussio L, Yang F; formal analysis: Gao L, Chen H, Ni K, Yang F; resources: Gao L, Yang F; validation: Chen H, Ma Q; methodology, investigation: Chen H; project administration: Huang S, Xu G; funding acquisition, conceptualization: Xu G. All authors reviewed the results and approved the final version of the manuscript.
-
Code is available as an open-source Python package 'ptol' on the PyPI (https://pypi.org/project/ptol/) and Github (https://github.com/cauBioinformation/ptol). Example data required for software execution is available from github (https://github.com/cauBioinformation/ptol/tree/main/test_data).
-
This research was funded by the National Key Research and Development Programs of China (2023YFD1301004). We thank High-performance Computing Platform of China Agricultural University for its computing services.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/gcomm-0026-0010.
-
# Authors contributed equally: Lu Gao, Huilong Chen
- Supplementary Table S1 Information on the 13 tested literature on the topic of mycotoxin-degrading enzymes.
- Supplementary Table S2 Questions with the corresponding mycotoxin-degrading enzyme targeting information entered into the large language model dialog box
- Supplementary Table S3 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature1.
- Supplementary Table S4 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature2.
- Supplementary Table S5 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature3.
- Supplementary Table S6 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature4.
- Supplementary Table S7 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature5.
- Supplementary Table S8 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature6.
- Supplementary Table S9 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature7.
- Supplementary Table S10 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature8.
- Supplementary Table S11 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature9.
- Supplementary Table S12 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature10.
- Supplementary Table S13 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature11.
- Supplementary Table S14 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature12.
- Supplementary Table S15 Details of the results of testing six large language models for accuracy and time spent on all mycotoxin degrading enzyme target information from literature13.
- Supplementary Table S16 Comparative details of the mean time of the six large language models in the two questioning modes.
- Supplementary Table S17 Comparison of time for extracting information manually and for extracting information by a large language model.
- Supplementary Table S18 Comparison of the average time spent on the six large language models in the two questioning modes.
- Supplementary Table S19 Comparative details of the mean overall accuracy of the six large language models in the two questioning modes.
- Supplementary Table S20 Comparison of the average overall accuracy of the six large language models in the two questioning modes.
- Supplementary Table S21 Machine learning evaluation metrics for the performance of six large language models for extracting 18 target information from all 13 mycotoxin-degrading enzyme literatures.
- Supplementary Table S22 Comparative details of the average accuracy of the six large language models on 18 questions.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gao L, Chen H, Ding X, Huang S, Ma Q, et al. 2026. Large language models accelerate the acquisition of mycotoxin-degrading enzyme information. Genomics Communications 3: e011 doi: 10.48130/gcomm-0026-0010 

Large language models accelerate the acquisition of mycotoxin-degrading enzyme information
- Received: 07 February 2026
- Revised: 06 May 2026
- Accepted: 08 May 2026
- Published online: 29 May 2026
Abstract: Microbial genome sequencing data are growing exponentially, yet comprehensive functional annotation is challenged by the dispersed nature of enzyme characterization information across scientific literature. Using mycotoxin-degrading enzymes as a case study, we evaluated the performance of six mainstream large language models, ChatGLM-3-Turbo, ChatGPT-4o, two versions of the Claude series (Claude2 and Claude Sonnet 4), DeepSeek-V3, and Kimi Chat, in extracting structured information from 13 representative publications across 18 categories of enzyme-related data. The top-performing models (Claude Sonnet 4, DeepSeek-V3, and ChatGPT-4o) achieved accuracy rates of 95%–99% with extraction times under 10 min, representing a potential improvement over manual approaches. To preliminarily explore LLM capabilities beyond text analysis, we assessed Claude Sonnet 4's ability to interpret genome-derived visual information, including phylogenetic trees, multiple sequence alignments, and domain architectures, comparing its outputs with expert analysis. The model showed basic pattern recognition capabilities for these multimodal inputs, though comprehensive validation is still needed. Based on these findings, we developed ptol, a workflow tool for literature retrieval, format conversion, and LLM input generation. This work establishes a benchmark for LLM-assisted information extraction in the microbiology domain and may provide methodological reference for similar specialized applications.