









-
Perennial ryegrass (Lolium perenne L.) is native to the Mediterranean region. It is a high-quality perennial cool-season forage and turf grass and is widely cultivated in temperate areas in the world[1]. The increasing frequency of extreme heat events driven by global climate change severely restricts the growth of perennial ryegrass and threatens the persistence of ryegrass pasture and turf. As a typical cool-season species, most perennial ryegrass varieties are sensitive to high temperatures[2]. Elucidating the molecular mechanisms underlying its heat responses and identifying key heat-tolerant genes are fundamental for the genetic improvement of perennial ryegrass and other related cool-season grasses.
Comparative transcriptomic analysis is a robust method to understand the heat stress-responsive genes in plants. Previously, several independent studies on transcriptomic alterations of heat-treated perennial ryegrass have been performed from different perspectives[3]. Wang et al. analyzed the transcriptomic alterations of perennial ryegrass after 6 h of heat treatment, and identified many known heat-responsive genes, including heat shock proteins (HSPs), heat shock factors (HSFs), and antioxidant related genes[4]. Nie et al. analyzed the effect of methyl jasmonate (MeJA) application on heat tolerance in perennial ryegrass and found that exogenous application of MeJA enriched photosynthesis-related pathways under heat stress with significantly increased expression of genes involved in chlorophyll (Chl) biosynthesis and antioxidant metabolism, and decreased expression of Chl catabolic genes[4]. Chen et al. analyzed the transcriptomes of perennial ryegrass for its acquired heat tolerance (gradually increased to 40/35 °C [day/night] at a rate of 3 °C every 24 h) and basal thermotolerance (subjected to 40/35 °C [day/night] for 24 h) and the expression levels of endoplasmic reticulum (ER) protein processing-related genes and plant hormone signal transduction genes were primarily induced in the acquired thermotolerance[5]. However, the degree of heat tolerance varies among the germplasm of perennial ryegrass. How heat-tolerant accessions respond to heat stress compared to heat-sensitive ones, and what distinct thermotolerance strategies exist among these varieties/accessions, remain unclear.
In a previous study, phenotypic and physiological traits associated with heat tolerance variations were determined, and the heat tolerance of 98 germplasm accessions was also evaluated[2]. The objective of this study was to understand transcriptomic differences between a heat-tolerant and heat-sensitive ryegrass varieties, and to identify specific hub genes potentially contributing to the tolerant trait in the heat-tolerant ryegrass variety. Two bred lines, 'NanHei-4#' and 'NanNong-6#' (abbreviated as 'X4' and 'X6' hereafter), newly developed through six years of hybridization and exhibiting superior turf characteristics were evaluated together with a widely used commercial turf-type cultivar 'Buena Vista' ('BV') as a reference control[6]. Based on the comparative transcriptome data, this study comprehensively explored the temperature adaptation mechanism of heat-tolerant and heat-sensitive varieties, identifying the candidate genes for stress resistance. This provides essential knowledge for molecular breeding of perennial ryegrass and other plants.
-
Three perennial ryegrass varieties were used in this study. 'BV', and two varieties bred by the group, 'X4' and 'X6'. 'X6' is a variety developed using 'BV' as a parental line. Field observations ranked their heat tolerance as 'X6' > 'X4' > 'BV'. Plants were grown in an automated greenhouse at Nanjing Agricultural University, China, under 25/20 °C (day/night) and a 14/10 h (light/dark) photoperiod. Plastic pots (8 cm diameter × 15 cm depth) were filled with 1.2 kg of peat moss (Pindstrup Mosebrug A/S, Denmark). Shoots were clipped to 8 cm every 7 d and fertilized with half-strength Hoagland solution.
Heat stress treatment
-
After 60 d of establishment, the potted grasses were transferred to growth chambers under a 14/10 h photoperiod, 65%–75% relative humidity, and a light intensity of 750 µmol·photons·m−2·s−1. Chamber settings were 25/20 °C (day/night) for control or 38/30 °C for heat-stress treatment for 30 d, each with four biological replicates.
Measurements of plant growth and physiological parameters
-
Turf quality was rated according to the National Turfgrass Evaluation Program (NTEP) criteria. The physiological index (Electrolyte leakage rate [EL], relative water content [RWC], chlorophyll [Chl] content, and Malondialdehyde [MDA] content) measurements were carried out following the same protocol as described before[2].
Determinations of Superoxide (SOD), catalase (CAT), peroxidase (POD), and Ascorbate peroxidase (APX) activities were carried out according to Wang et al.[7]. Production of superoxide anion (O2·−) was quantified using the Reactive Oxygen Species Assay Kit for Superoxide Anion with DHE (Cat No.: S0064S. Beyotime Biotech Inc., China), and hydrogen peroxide (H2O2) was quantified using chemical staining methods as described by Velikova et al.[8].
Transcriptome analysis
-
RNA sequencing (RNA-seq) was carried out to compare the transcriptomics of two ryegrass varieties ('X6' and 'BV') with contrasting heat tolerance traits. This extreme phenotypic contrast approach maximizes the detection of differentially expressed genes in core thermotolerance pathways. For RNA-seq, leaf samples of ryegrass were harvested after 24 h of heat stress in the growth chamber at 38/30 °C (day/night) or 25/20 °C for the control. Three biological repeats were taken for each group. The RNA-seq was outsourced to Guangzhou Genedenovo Biotechnology Co., Ltd. (Guangzhou, China) and performed on an Illumina NovaSeq 6000 platform. In short, differential expression between the two genotypes under heat treatment was assessed with DESeq2 (v.1.25.9). Differentially expressed genes (DEGs) were screened using DESeq2 software with the selection criteria set to |Log2FC| ≥ 1 and FDR < 0.01. The RNA-seq data were deposited to the NCBI SRA database (SRA Accession number: PRJNA1278810).
Gene Ontology (GO) enrichment analysis was performed to identify significantly overrepresented functional terms among differentially expressed genes (DEGs) relative to the genomic background[9]. The analysis followed a systematic approach: all DEGs were annotated using the GO database (
www.geneontology.org ) through term mapping; thereafter, the number of genes associated with each GO term was quantified. To determine statistical significance, the calculated p-values were adjusted for multiple testing using the false discovery rate (FDR) correction, with an FDR ≤ 0.01 considered statistically significant. GO terms satisfying this criterion were identified as significantly enriched in the DEG set.Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was carried out to elucidate the biological functions of DEGs for significantly enriched metabolic pathways and signal transduction pathways among DEGs relative to the whole genome background[10]. The resulting p-values were also subjected to FDR correction, with pathways meeting the significance threshold (FDR ≤ 0.01) considered biologically relevant.
For the weighted gene co-expression network analysis (WGCNA), genes with small or abnormal variations were filtered out after background correction and transcript normalization. The correlation strength among the 16,252 genes followed a scale-free distribution (Supplementary Fig. S1). Eight gene modules were identified and marked with different colors. The number of genes in the eight modules ranged from 280 to 5,282. When the optimal power value was set to 20, the R2 value was greater than 0.75, and the average connectivity was at its lowest (Supplementary Fig. S2). This power value was used to construct the scale-free network. Following network construction, the biological significance of identified gene modules was investigated through KEGG pathway enrichment analysis.
Protein-Protein Interaction (PPI) network analysis
-
Protein-protein interaction (PPI) network analysis of DEGs was performed using the STRING database[11], followed by network visualization and topological analysis using Cytoscape[12]. For species already annotated in STRING, known and predicted PPIs were directly extracted for the gene set. For unannotated species, Blastx alignment was first performed against reference proteomes in STRING to identify orthologous proteins, and then the interaction network was constructed based on these evolutionarily conserved relationships. All networks were built using high-confidence interactions to ensure biological relevance, and subsequently analyzed to identify key hub proteins and functional modules underlying the observed expression changes. Sequences of the identified hub genes are presented in Supplementary Data 1.
RT-qPCR for analyzing relative gene expression levels
-
The total mRNA of leaves was extracted using the E.Z.N.A.® Plant RNA Kit with genomic DNA remover (OMEGA Bio-Tek Inc., USA) and used for the synthesis of cDNA using the First-strand cDNA Reverse Transcription Kit (Takara Bio Inc., China). The qRT-PCR reaction was performed with the SYBR® Green PCR Master mix (Applied Biosystems Inc., USA) using the LightCycler® 480 II Real-Time PCR System (Roche Ltd., USA). All PCR reactions were performed with four biological replicates. LpelF4a was used as the reference gene, and the gene's relative expression level was analyzed using the 2−ΔΔCᴛ method[13]. The sequences of primers used in this study are listed in Supplementary Table S1.
Statistical analysis
-
The physiological index and RT-qPCR data were analyzed using One-way Analysis of Variance (ANOVA) with SPSS 19.0 (SPSS Inc., Chicago, IL, USA) with statistical significance set at p ≤ 0.05. Graphs were prepared with SigmaPlot 10.0 (Systat Software Inc., Chicago, IL, USA). The heatmap of the gene relative expression data was performed using the R statistical software (R4.4.3 by R Development Core Team). Data are expressed as means ± standard error (SE).
-
After 30 d of continuous heat stress at 38/30 °C (day/night), three ryegrass cultivars displayed varying degrees of leaf wilting and chlorosis, where the variety 'X6' retained the best overall green and turgid foliage appearance while 'BV' had the least with pronounced leaf desiccation and yellowing (Fig. 1a).
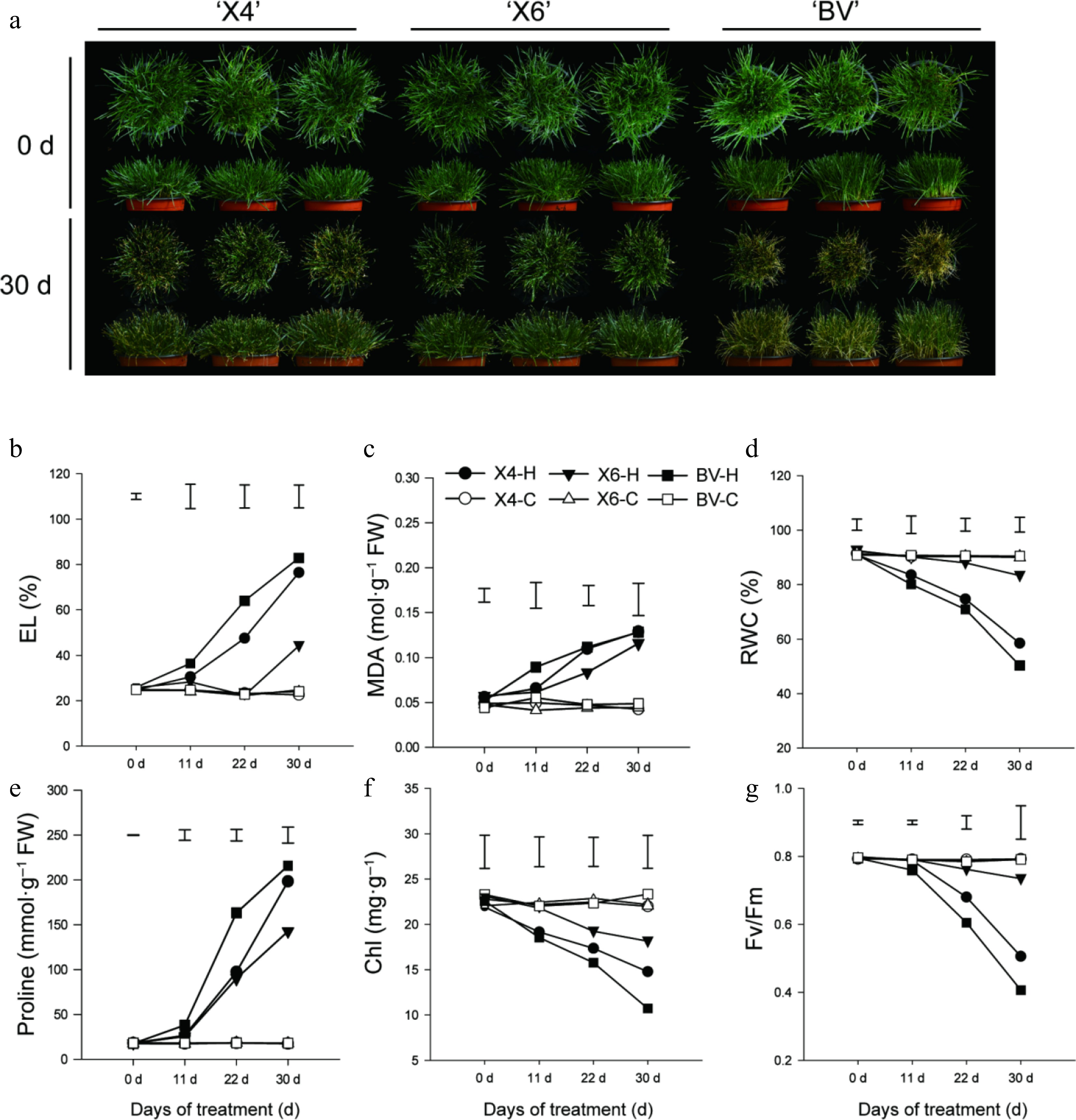
Figure 1.
Characterization of heat tolerance for three perennial varieties. (a) Phenotypes of three varieties before and after 30 d of heat tolerance. (b)–(g) Electrolyte leakage rate (EL), MDA content, relative water content (RWC), proline (Pro) content, chlorophyll (Chl) content, and photochemical efficiency (Fv/Fm) of three varieties subjected to heat stress at 38/30 °C (day/night) or under the control condition (25/20 °C). Data are means ± SE (n = 3), and the different letters above columns represent statistically significant differences at p = 0.05.
Six physiological indexes closely related to ryegrass heat tolerance[2] were measured on days 0, 11, 22, and 30 after the heat treatment. As shown in Fig. 2, under the control condition (optimal temperature), all six physiological indexes remained constant among the varieties. During the 30 d of continuous heat stress, significant differences among the varieties were detected, and the variabilities of these physiological indexes were more dramatic in 'BV' than 'X4' and 'X6' during the heat stress, suggesting that 'BV' was the most heat-sensitive variety while 'X6' was the most heat-tolerant one. Specifically, 'X6' showed the lowest EL and MDA content but the highest RWC, Chl content, and photochemical efficiency (Fv/Fm), suggesting that it maintained better membrane stability, leaf water status, and the lowest photosynthetic impairment than the others (Fig. 1b–g). It is interesting to note that 'X6' had the lowest proline content after 30 d of heat stress, which was the opposite of the RWC of ryegrass varieties (Fig. 1). Overall, 'X6' preserved better membrane integrity, water status, and photochemical efficiency than 'BV' and 'X4' under the prolonged high-temperature stress.
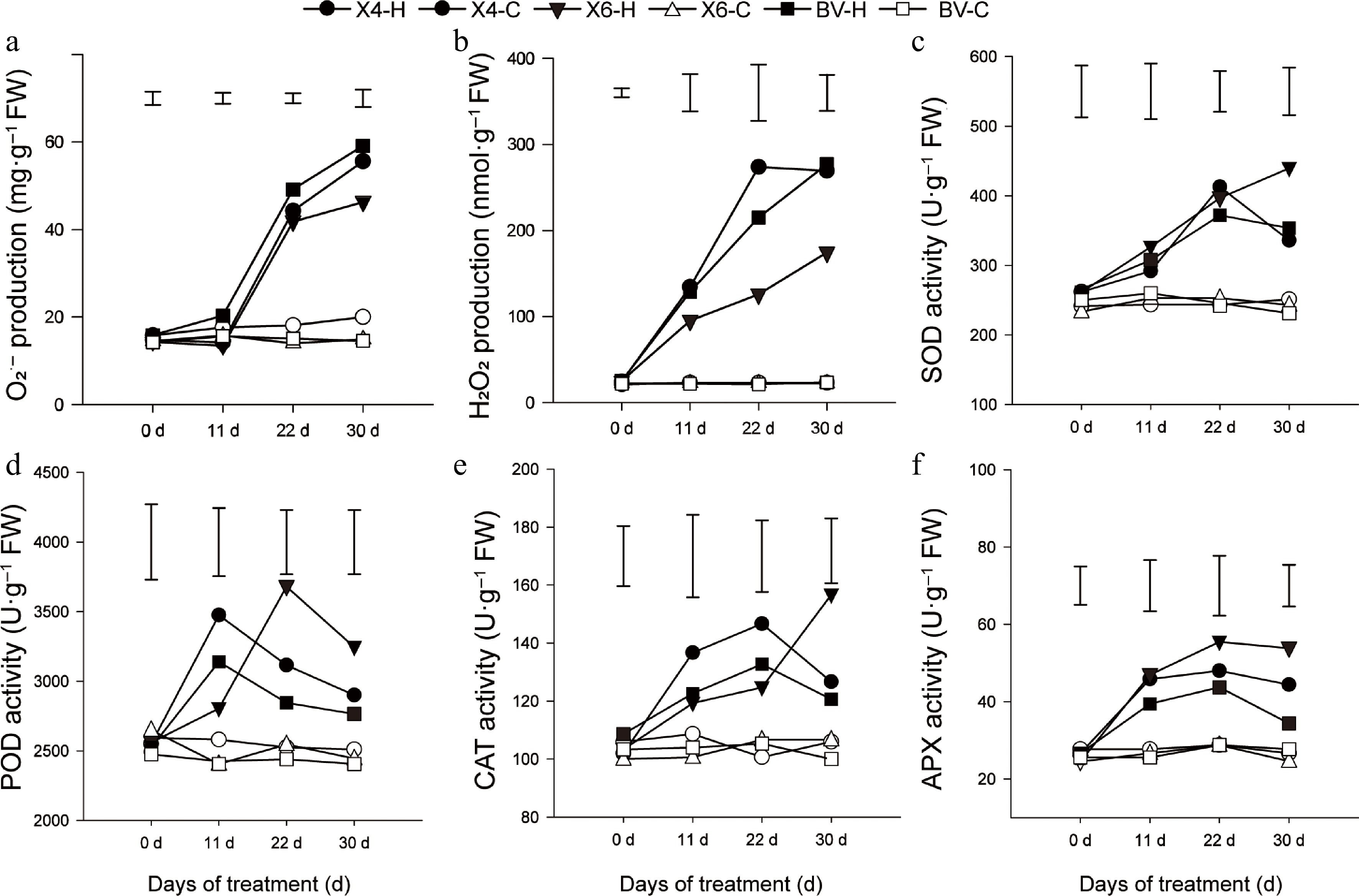
Figure 2.
Reactive oxygen species (ROS) status and activities of ROS-scavenging enzymes of three perennial varieties. (a), (b) Production of O2·– and H2O2 contents. (c)–(f) Activities of SOD, POD, CAT, and APX of perennial ryegrass subjected to heat stress at 38/30 °C (day/night) or under the control condition (25/20 °C). Data are means ± SE (n = 3), and the different letters above columns represent statistically significant differences at p = 0.05.
Since heat stress could trigger a marked, time-dependent surge in reactive oxygen species (ROS), the superoxide anion (O2·−) production and hydrogen peroxide (H2O2) contents were further measured, as well as the activities of four ROS-scavenging enzymes (Fig. 2a, b). The difference among varieties became evident after 11 d of heat treatment that reached significant differences after 22 to 30 d of the treatment: by day 30, 'BV' leaves showed the highest ROS burden, with O2·− and H2O2 levels climbing to roughly 55 % and 59 % above their respective controls, whereas 'X6' consistently maintained the lowest O2·− and H2O2 concentrations under heat stress among the tested varieties (Fig. 2a, b), highlighting its superior capacity to restrain ROS build-up during the prolonged heat stress.
Consistently, the heat stress elicited distinct antioxidant-enzyme dynamics among the ryegrass varieties (Fig. 2c–f): activities of superoxide dismutase (SOD), peroxidase (POD), catalase (CAT), and ascorbate peroxidase (APX) all rose above control levels but at different temporal patterns that 'X6' had the highest SOD and CAT activities only at day 30 and the highest POD and APX activities at 22 and 30 d after the heat treatment, while 'BV' had the lowest CAT, POD and APX activities after 30 d of the heat treatment (Fig. 2c–f).
Comparative transcriptomic analysis between two ryegrass varieties with contrasting heat tolerance
-
To understand the early heat-responsive differences between the ryegrass variety, the heat-tolerant variety 'X6' and the sensitive variety 'BV' for RNA-seq were selected after 24 h of heat stress. Samples of 'X6' and 'BV' treated under the optimal control condition or heat treatment were designated as 'X6-C' (C stands for control), 'X6-H' (H stands for heat), 'BV-C', and 'BV-H', respectively.
The derived RNA-Seq data were summarized in Supplementary Table S2. In brief, a total of 74.98 Gb of high-quality clean reads were obtained with the Q30 base content of each sample higher than 91.5%. Among the reads, 71.78%–77.81% were clean reads that could be aligned to the reference genome, among which 68.42%–74.49% were unique mapped sequences. There are 7,520 newly identified genes (listed in Supplementary Data 2) that were not shown in the published ryegrass genome annotation[14]. In addition, among the reads located on the reference genome, the proportion of reads distributed on exons was 88.51%, validating that the RNA-Seq data could be used for further analysis.
Differentially expressed genes (DEGs) between the two ryegrass varieties
-
Comparative transcriptomic analysis revealed that there were 12,279 differentially expressed genes (DEGs) in the comparison of 'X6-H' vs 'X6-C', among which 9,787 were up-regulated and 2,489 down-regulated, and 9,375 DEGs in the comparison of 'BV-H' vs 'BV-C', among which 6,661 were up-regulated and 2,714 down-regulated (Fig. 3a; Supplementary Fig. S3). Transcriptomic comparison between the two varieties showed that there were 6,877 DEGs in the 'X6-H' vs 'X6-C' group, 3,976 specific DEGs in the 'BV-H' vs 'BV-C' group, and 5,399 common DEGs between the two groups (Fig. 3b). The 5,399 common DEGs between the two groups could be regarded as common heat-responsive genes in ryegrass, while the number of variety-specific DEGs in the 'X6-H' vs 'X6-C' group was 73% more than the 'BV-H' vs 'BV-C' group (Fig. 3b).
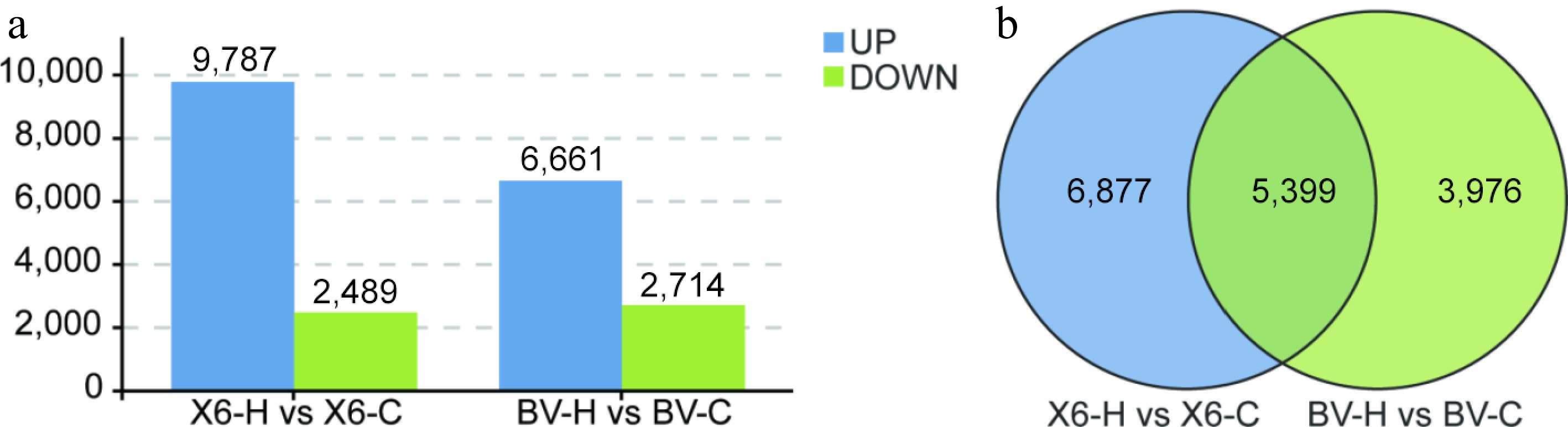
Figure 3.
Number of differentially expressed genes (DEGs) of two ryegrass varieties with contrasting heat tolerance. (a) Number of up- or down-regulated DEGs. (b) Venn diagram of DEGs in different groups between 'X6-H vs X6-C' and 'BV-H vs BV-C'.
Verification of RNA-Seq data by real-time quantitative RT-PCR
-
To further validate the reliability of the RNA-seq data, the relative expression levels of ten DEGs were quantified using real-time quantitative PCR (RT-qPCR). As shown in Fig. 4a, the expression patterns obtained by RT-qPCR mirrored those revealed by RNA-seq. For the DEGs identified in groups of 'X6-H' vs 'X6-C' and 'BV-H' vs 'BV-C', the correlation indexes between relative expression values (RT-qPCR) and TPM values (RNA-seq) were 0.85 and 0.80 (p < 0.01), respectively (Fig. 4b, c). This result confirmed the reliability of the RNA-seq data.
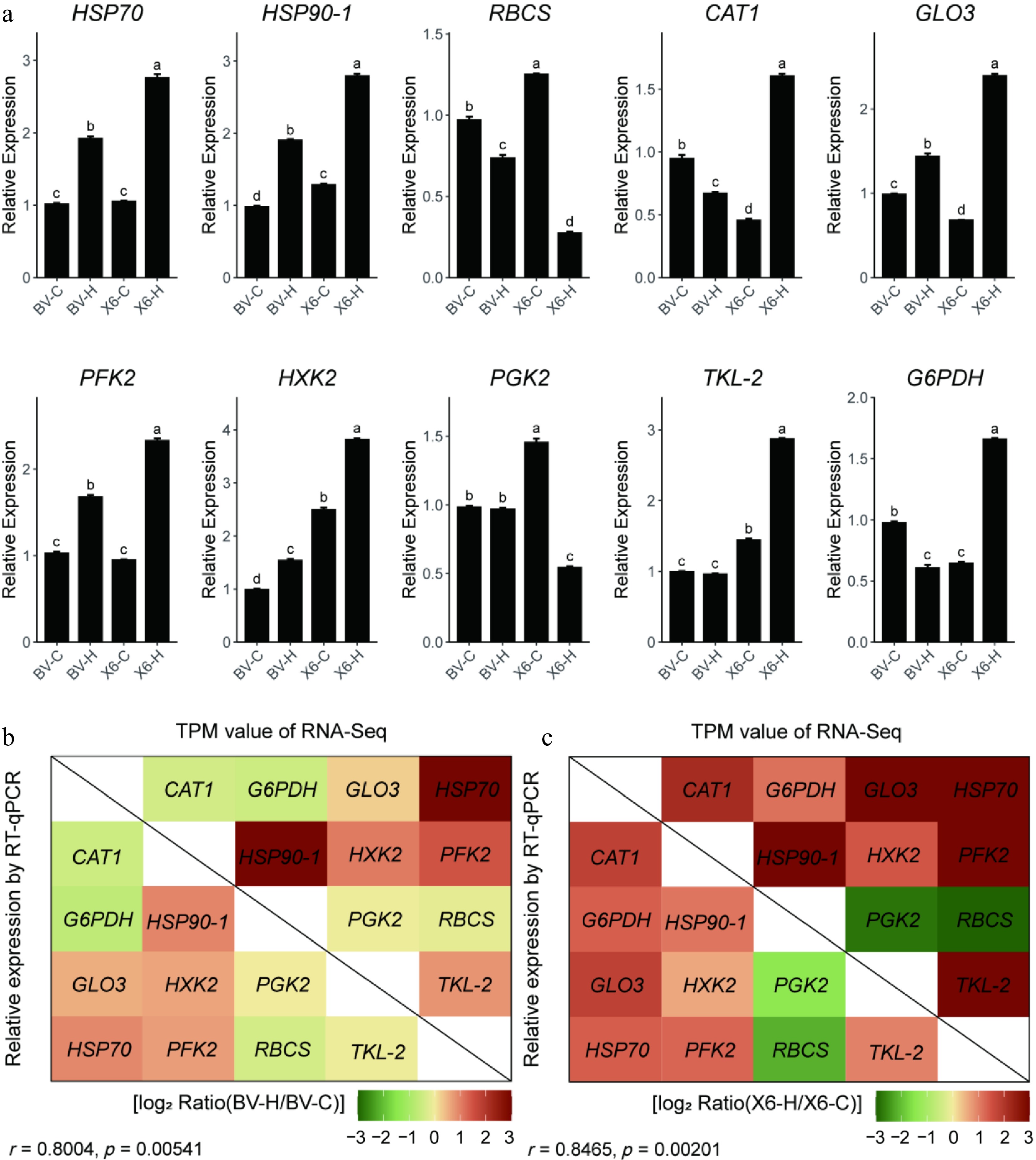
Figure 4.
Verification of RNA-Seq data by real-time quantitative RT-PCR. (a) Relative expression of ten genes identified as DEGs in the RNA-seq using real-time PCR. (b), (c) Correlation analysis between relative expression values of RT-qPCR and TPM values of RNA-seq. Data in (a) are means ± SE (n = 4) and the different letters above columns represent statistically significant differences at p = 0.05.
Classification of common heat-responsive DEGs between the two ryegrass varieties
-
The 5,399 common heat-responsive DEGs between the two ryegrass varieties were analysed using the GO and KEGG enrichment analysis. Accordingly, the GO analysis showed that the DEGs were mostly enriched in translation, RNA binding, ribosomal structural components, and general cellular metabolic processes (such as peptide synthesis, peptide metabolism, and amino acid synthesis) (Fig. 5a). The KEGG enrichment analysis showed that the significantly enriched pathways were mostly related to genetic information processing and cellular metabolism, including ribosomes, spliceosomes, ribosome biogenesis in eukaryotes, photosynthetic antenna proteins, RNA polymerase, nucleocytoplasmic transport, and RNA degradation (Fig. 5b). Additionally, many heat shock proteins (HSPs), nucleoporin proteins (NUPs), light-harvesting complex proteins (LHCs), and large ribosomal subunit proteins (RPLs) were among the common heat-responsive DEGs (Fig. 5c), suggesting protein folding and stability, nuclear export/entry regulation, photosynthesis, and protein biosynthesis were common early heat-responsive genes in both ryegrass varieties. Specifically, six commonly heat-inducible HSP genes (i.e., HSP21.9, HSPF70-8, HSP17.4, HSP70, HSP17.9, and HSP21) demonstrated higher up-regulated degrees in 'X6-H' vs 'X6-C' group than the 'BV-H' vs 'BV-C' group (Fig. 5c).
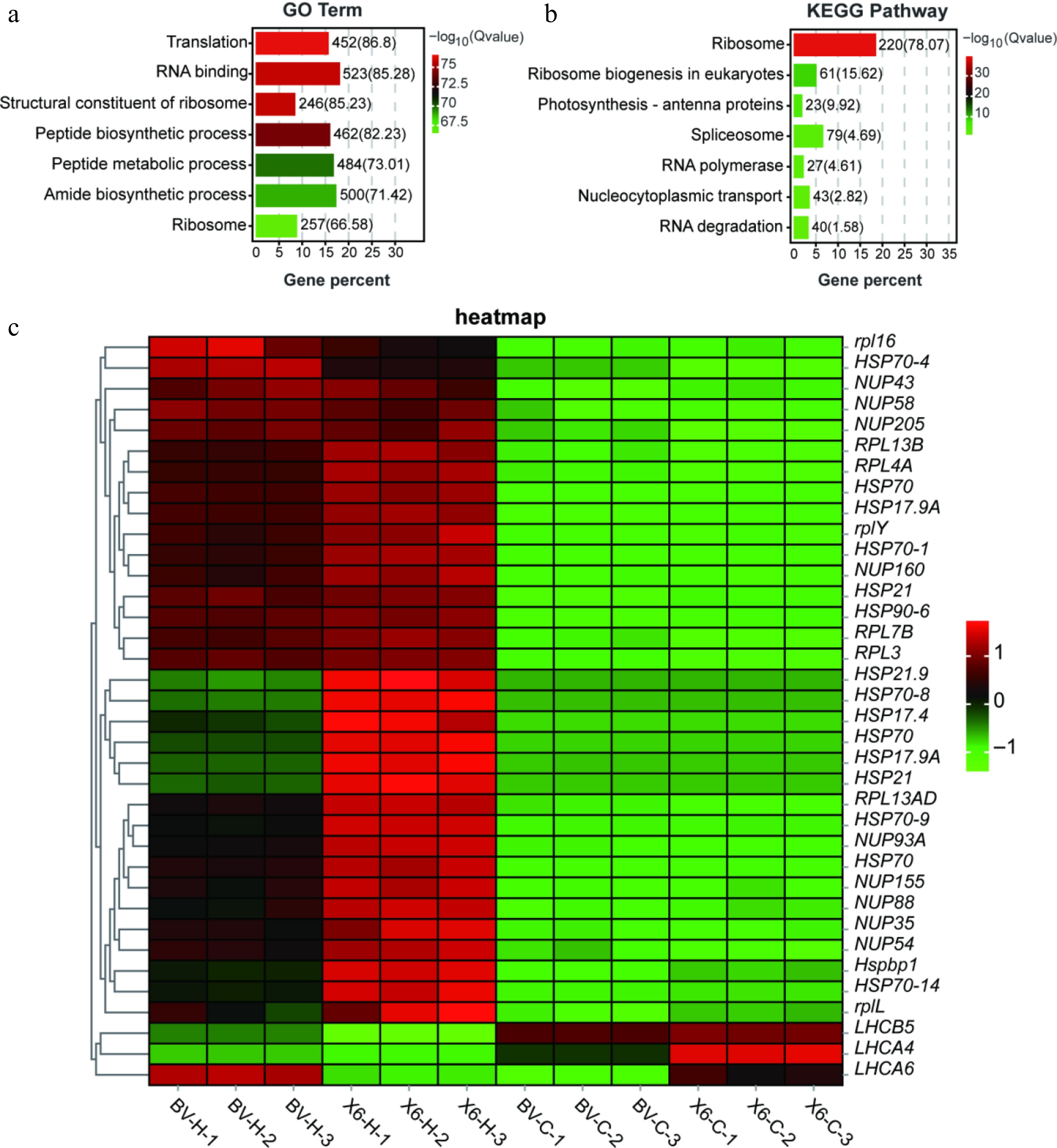
Figure 5.
Gene Ontology (GO) and KEGG analyses of common heat-responsive DEGs between two ryegrass varieties with contrasting heat tolerance. (a), (b) GO and KEGG analyses of the 5,399 common heat-responsive DEGs between two ryegrass varieties. (c) Heatmap of representative common DEGs among the samples.
Weighted gene co-expression network analysis (WGCNA) on specific DEGs in the heat-tolerant ryegrass variety
-
Using weighted gene co-expression network analysis (WGCNA), all DEGs were classified with different expression patterns into eight gene co-expression modules named after different colors (Fig. 6a). Correlation analysis showed that there were two modules significantly correlated with 'X6-H': the Red module (correlation coefficient was 0.99, p < 0.001) and the Dark turquoise module (correlation coefficient was 0.87, p < 0.001), supporting that DEGs in these two modules could be regarded as specific ones in the group of 'X6-H' (Fig. 6b).
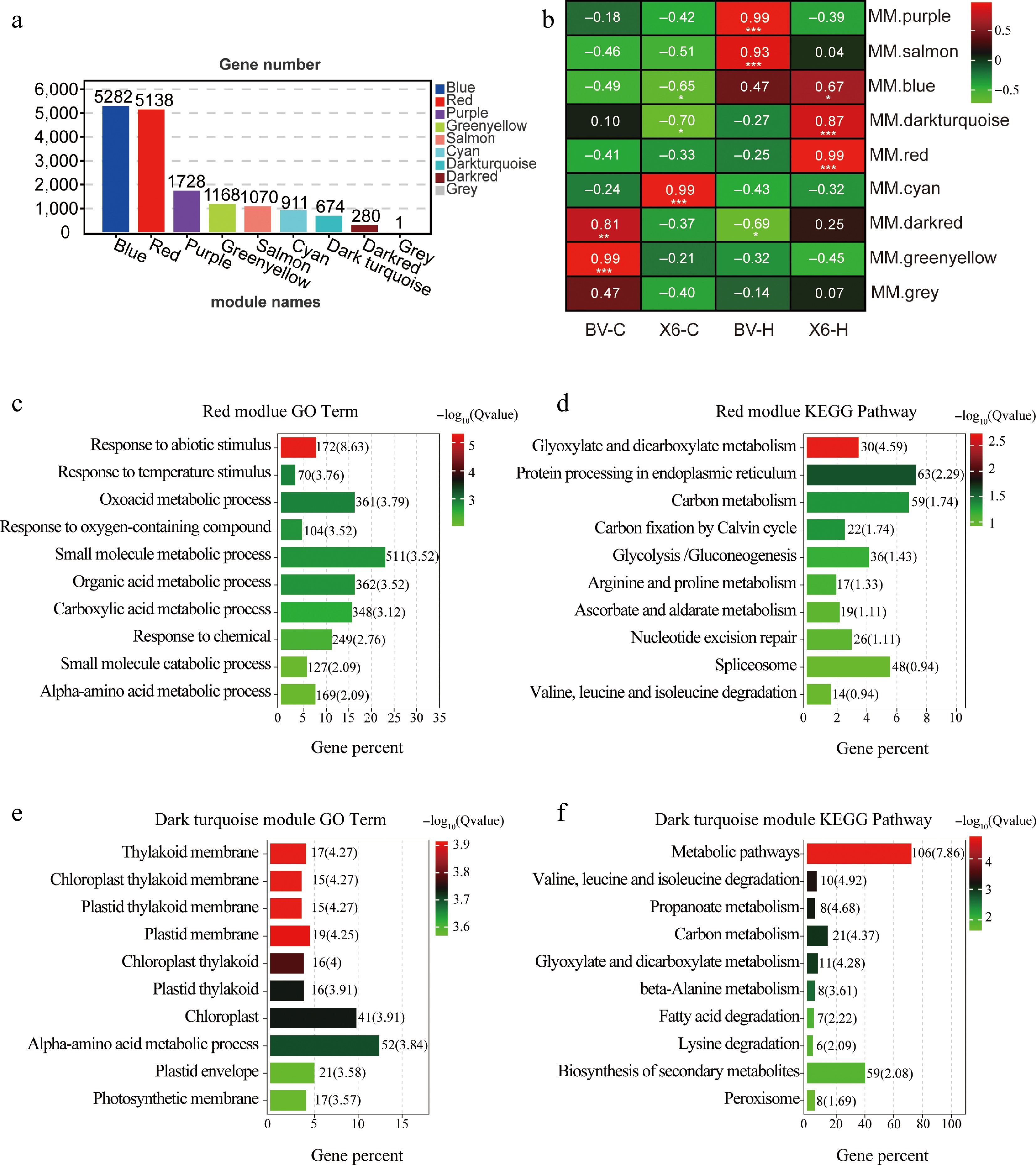
Figure 6.
Weighted gene co-expression network analysis (WGCNA) of modules comprising specific DEGs in heat-treated 'X6' (X6-H) and GO and KEGG analysis of these specific DEGs. (a) Number of DEGs in eight modules generated from WGCNA. (b) Modules and traits correlation analysis pinpointed that the Red module and the Dark turquoise module had significant correlations with 'X6-H'. (c), (d) Top ten GO and KEGG enriched terms for DEGs in the Red module. (e), (f) Top ten GO and KEGG enriched terms for DEGs in the Dark turquoise module.
To functionally classify these 'X6-H'-specific DEGs in the two modules (Red and Dark turquoise), GO and KEGG analyses were performed with these DEGs (Fig. 6c–f). As for DEGs in the Red module, the top ten GO enrichment processes included responses to abiotic stimuli, temperature stimuli, oxidative acid metabolism, organic acid metabolism, carboxylic acid metabolism, α-amino acid metabolism, and small molecule metabolic processes (Fig. 6c). The top ten KEGG enriched pathways were glyoxylate and dicarboxylate metabolism, protein processing in endoplasmic reticulum, spliceosome, nucleotide excision repair, metabolism of aldehyde acid and dicarboxylate, carbon metabolism, and the ascorbate and glutathione cycle, etc. (Fig. 6d; Supplementary Figs S4–S6).
As for DEGs in the Dark turquoise module, GO analysis revealed significant enrichment in chloroplast and photosynthesis-related terms, such as thylakoid membranes, plastid membranes, and chloroplasts (Fig. 6e). The top ten KEGG enriched pathways included propionate metabolism, fatty acid degradation, and secondary metabolic pathways (Fig. 6f).
Core gene co-expression network analysis on specific DEGs in the heat-tolerant ryegrass variety
-
To functionally classify these 'X6-H'-specific DEGs in the two modules (Red and Dark turquoise), the hub genes were then identified with high interconnectivity and correlation through a co-expression network analysis. The connectivity of the top 100 nodes and the connections between nodes with weights greater than 0.5 in the Red and Dark turquoise modules were analyzed. In the Red module, the gene co-expression network of the top 100 connectivity pairs included four hub genes: LOC127291855, LOC127302184, LOC127294868, and MSTRG.17145 (Fig. 7a; Sequences of these genes were presented in Supplementary Data 1). In the Dark turquoise module, AVT1A encoding a vacuolar amino acid transporter and EDA2 encoding a probable serine protease were the hub genes in the co-expression network of the top 100 connectivity pairs (Fig. 7c).
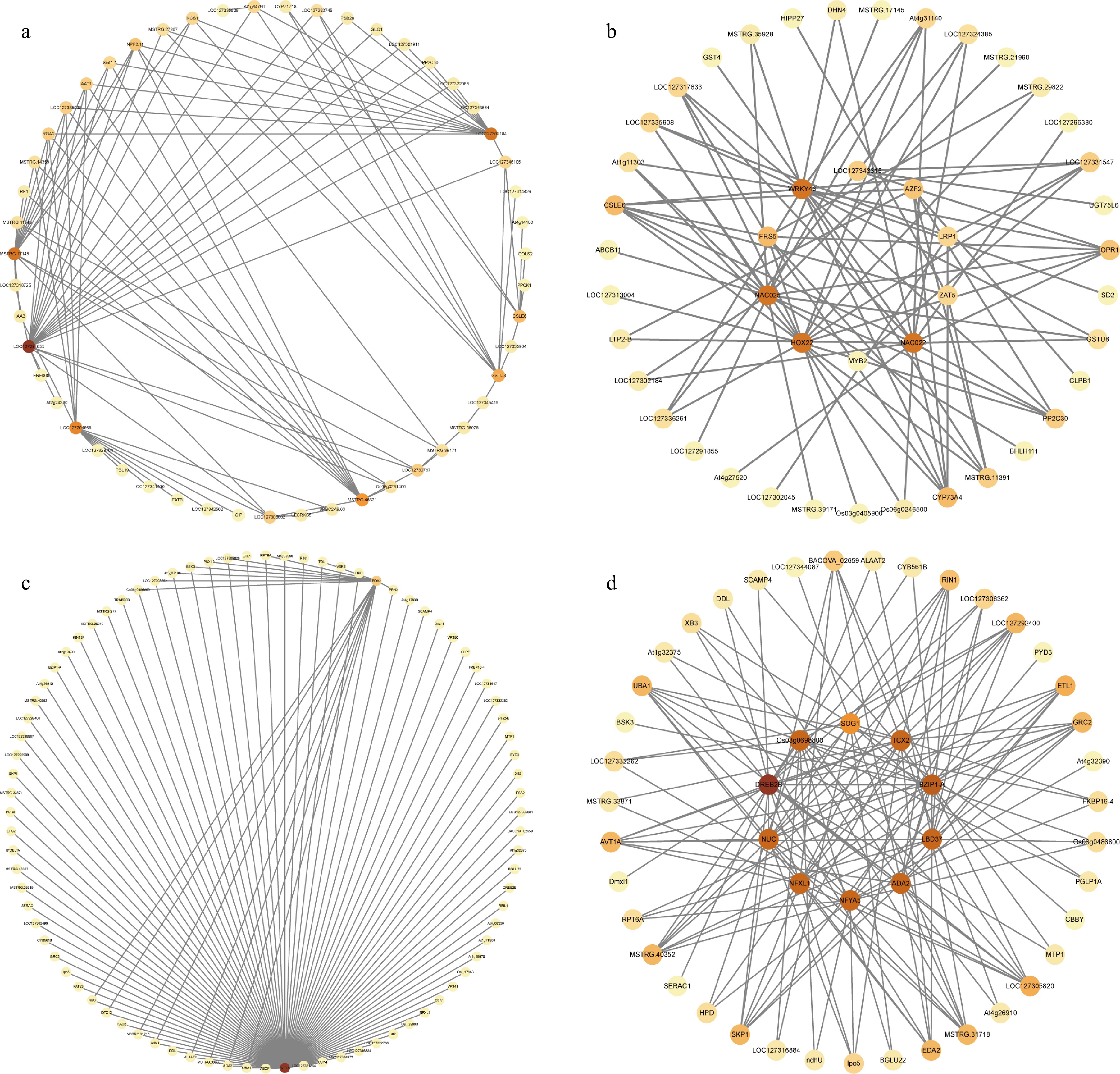
Figure 7.
Gene co-expression network analysis of modules comprising specific DEGs in heat-treated 'X6' (X6-H). (a), (b) Co-expression network of the top 100 genes with connectivity and the top ten transcription factors with connectivity in the Red module. (c), (d) Co-expression network of the top 100 genes with connectivity and the top ten transcription factors with connectivity in the Dark turquoise module.
Furthermore, the hub transcription factors were also identified: in the Red module, the top ten transcription factors in the co-expression network were NAC022, NAC025, HOX22, WRKY46, FRS5, AZF2, ZAT5, MYB2, LRP1, and LOC127343315 (Fig. 7b); while in the Dark turquoise module, the top ten transcription factors in the co-expression network included ADA2, NUC, BZIP1-A, DREB2B, NFXL1, LBD37, TCX2, NFYA5, Os03g0698800, and SOG1 (Fig. 7d).
-
Perennial ryegrass is one of the most important cool-season grasses used for both forage and turf purposes in the world. Originating from the Mediterranean area, most perennial ryegrass germplasm resources are sensitive to heat stress[2]. In this study, different ryegrass varieties were utilized with contrasting heat tolerance to explore their heat-tolerance mechanism at the transcriptomic level by categorizing the common heat-responsive DEGs and specific heat-responsive DEGs in a heat-tolerant ryegrass variety. Furthermore, not only key GO terms and KEGG pathways but also specific hub genes and transcription factors associated with the heat-tolerant trait of the variety 'X6' were identified.
Selection of two extreme types with contrasting heat-tolerance traits for the comparative transcriptomic study
-
In general, consistent with field observations, the heat tolerance of the three tested varieties was: 'X6' > 'X4' > 'BV'. Under normal physiological conditions, the metabolic systems of plants are in dynamic equilibrium and work synergistically. When plants encounter environmental stress, this stable system undergoes significant disruption. High temperature conditions not only trigger basic physiological changes such as alterations in cell membrane permeability and water metabolism imbalance, but also lead to damage to the photosynthetic system and oxidative stress responses, causing a series of cascading effects. Therefore, multiple physiological indicators were used to accurately and comprehensively assess the heat tolerance of perennial ryegrass varieties[2]. EL (measuring the permeability of plasma membrane) and MDA content (a byproduct of oxidized membrane) were used to evaluate damage degrees of the cell membrane; the dynamic changes in RWC can effectively reflect how plants respond to heat stress in terms of leaf water retention; Chl content and Fv/Fm ratio were used to evaluate plants' photosynthetic capacity. ROS serves as an early stress signaling molecule, but excessive ROS in the cell leads to the oxidation of unsaturated fatty acid chains, proteins, and nucleic acids; antioxidant enzymes scavenge ROS, maintaining the continuous ROS scavenging ability within plant cells[15,16].
Proline, as a key osmotic regulator, is a key factor for maintaining cellular osmotic balance and an efficient antioxidant. In this study, under heat stress, the proline content showed an increasing trend that is consistent with the known role of increased proline content in enhancing stress resistance. Yet, the proline content of 'X6' was lower than that of 'BV' and 'X4'. Other studies also revealed that the correlation between proline levels and thermotolerance is not strictly positive: certain highly heat-tolerant plants do not show significantly higher proline concentrations. For example, in thermotolerance studies on bermudagrass (Cynodon dactylon, a heat-tolerant warm-season grass) and tall fescue (Festuca arundinacea, a less heat-tolerant cool-season grass), while proline generally contributes positively to heat resilience, the heat-tolerant bermudagrass displays proline contents comparable to or even lower than tall fescue[17]. This suggests that highly thermotolerant plants may employ alternative mechanisms, such as enhanced aquaporin activity, cuticular wax deposition, or stress memory priming, to maintain hydration and stability, supporting the physiological plausibility of a 'low-proline high-RWC' phenotype in specific contexts. The elevated RWC in 'X6' indicates superior cellular water retention and minimal dehydration, attributable to integrated strategies such as enhanced stomatal regulation, optimized root hydraulic conductivity, and rapid osmoregulatory reprogramming. In contrast, the heat-sensitive control cultivar 'BV' exhibited higher leaf proline accumulation but lower RWC, reflecting inefficient water conservation despite compensatory proline synthesis.
'X6' and 'BV' were chosen as heat-tolerant and heat-sensitive varieties for the transcriptomic analysis after 24 h of heat stress to select against potential DEGs influenced by the circadian rhythm. The exclusion of intermediate variety X4 would avoid substantial data heterogeneity. Focusing on comparison between extremotypes enabled effective deconvolution of core regulatory networks underlying complex traits. The total number of obtained DEGs was 12,279. Considering the total number of genes was 38,868 in the annotated perennial ryegrass genome[14], the number of these identified DEGs was quite high with the selection criteria set to |Log2FC| ≥ 1 and FDR < 0.01. Similar RNA-seq studies on heat-treated ryegrass identified even more DEGs: for example, a total of 20,183 DEGs were identified in perennial ryegrass between heat-treated ones and the control after 6 h of heat-treatment[3], and 3,994 DEGs were identified in heat-treated vs control ryegrass after the 12 h of heat treatment[4] with similar DEG selection criteria. These large numbers of DEGs were classified into common heat-responsive DEGs between the varieties and specific heat-responsive DEGs in the heat-tolerant variety 'X6' for better understanding.
Classification of common heat-responsive DEGs between the two varieties
-
As for the common heat-responsive DEGs between the varieties, their GO and KEGG enrichment analyses emphasized the roles of general cellular processes (such as translation, RNA binding, ribosomal structural components, peptide synthesis, peptide metabolism, and amino acid synthesis) and molecular processing and metabolism (i.e., ribosomes, spliceosomes, ribosome biogenesis in eukaryotes, photosynthetic antenna proteins, RNA polymerase, nucleocytoplasmic transport, and RNA degradation). These results were highly similar to the ones revealed by previous RNA-seq studies[3,4,14]. Functional studies supported the roles of genes in these basic cellular processes in plant heat tolerance. For example, the RNA-binding protein GLYCINE-RICH RNA-BINDING PROTEIN 7 (GRP7) in Arabidopsis can initiate the translation process through phase separation to modulate plant temperature resilience[18]. High temperatures cause ribosome stalling at the 5' end of mRNA in Arabidopsis, leading to the degradation of mRNA that encodes heat shock proteins, which affects the plant's heat tolerance[19].
Additionally, differential expression of photosynthetic antenna proteins was also observed[20]. Key genes, such as LHCB5 and LHCA4, encode light-harvesting complex proteins that have irreplaceable functions in light energy capture and were closely related to PSII heat stress memory[21]. Rubisco is the most abundant enzyme in plants and one of the most nitrogen-intensive proteins. High-temperature stress typically coincides with global suppression of protein synthesis and accelerated protein degradation. Consistently, small subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase genes (RCBS) were more down-regulated in X6-H than in Bv-H (Fig. 4). Under stress conditions, plants prioritize the synthesis of defensive molecules such as heat shock proteins (HSPs) to protect cellular structures, while reducing the production of photosynthetic proteins—a typical adaptive response[22]. Downregulating RBCS expression allows reallocation of nitrogen resources and energy toward synthesizing protective compounds like HSPs and antioxidant enzymes. Since Rubisco's activity is inherently constrained under high temperatures, transient reduction of its abundance has minimal short-term impacts on survival. This 'defense-over-growth' strategy is particularly pronounced in heat-tolerant cultivars. For instance, studies on thermotolerant ryegrass revealed strong induction of the heat shock transcription factor HsfA2, which up-regulates small HSP18.2 and ascorbate peroxidase APX1, thereby mitigating protein misfolding and excess reactive oxygen species accumulation, ultimately enhancing thermo-tolerance[23]. These findings suggest that heat-adapted plants preferentially reduce investments in carbon assimilation machinery while bolstering protective mechanisms to acclimate to elevated temperatures.
Classification of specific heat-responsive DEGs in the heat-tolerant variety revealed key pathways and hub transcription factor genes
-
An emphasis in this study was on the specific heat-responsive DEGs in the heat-tolerant variety 'X6'. Further clustering analysis divided the DEGs in the RNA-seq data into eight modules, establishing a co-expression regulatory network based on WGCNA. The Red and the Dark turquoise modules were significantly correlated with the heat-treated 'X6' group. For DEGs in the Red modules, KEGG analysis showed glyoxylate and dicarboxylate metabolism, protein processing in ER, spliceosome, carbon metabolism, and glycolysis/gluconeogenesis as the top five enriched pathways. Key genes of the 'glyoxylate and dicarboxylate metabolism' pathway regulate the secretion of organic acids (such as acetate, citrate, and malate) in response to heat stress. It is known that the cellular increase of acetic acid can ameliorate heat stress in rice[24], and citric acid and malate both confer multiple abiotic stress tolerances in plants[25,26].
Furthermore, enriched DEGs in the Red module also revealed the importance of carbon metabolism, including carbon fixation by the Calvin cycle, glycolysis, and the ascorbate-glutathione cycle in the regulation of heat tolerance (Supplementary Figs S4 & S5). For example, the expression of Hexokinase2 (HXK2), Phosphofructokinase2 (PFK2), and PFK5 in the glycolysis pathway in the heat-tolerant variety 'X6' was significantly up-regulated after heat treatment, suggesting that the 'X6' variety has a more sensitive metabolic regulation system (Supplementary Fig. S4). Similarly, in soybean, the expression level of G6PDH is closely related to the heat tolerance of soybean anthers[27]. Under high-temperature stress, the Calvin cycle of photosynthesis often exhibits a 'source-sink imbalance'—on one hand, excess light energy is absorbed by photosystems, while on the other hand, CO2 assimilation becomes restricted (see reduced expression of RBCS, PGK, and GAPB in Supplementary Fig. S5). This mismatch prevents the normal consumption of surplus excitation energy and reduces power through carbon fixation, leading to the overproduction of reactive oxygen species. To mitigate this, plants may downregulate both the abundance and activity of Rubisco, thereby reducing carbon assimilation capacity (Supplementary Fig. S5).
Furthermore, the ascorbate-glutathione cycle protects redox-sensitive proteins from uncontrolled oxidation by ROS, particularly hydrogen peroxide (H2O2)[28]. Consistently, most DEGs in the ascorbate-glutathione cycle showed higher expression levels in the heat-tolerant variety 'X6' than the heat-sensitive one 'BV' (Supplementary Fig. S6).
The core-gene co-expression network analysis also pinpointed the hub genes and transcription factors correlated with the heat tolerance trait of the variety 'X6'. Most of these hub genes and their orthologous genes have not been functionally characterized yet. In contrast, orthologs of several identified transcription factors were known as regulators in plant heat or other abiotic stress tolerance already. For example, the rice OsHOX22 functions in ABA-mediated drought and salt tolerances[29]; WRKY46 modulates plant osmotic/salt stress in Arabidopsis[30] and cold tolerance in cucumber (Cucumis sativus)[31].
NAC022/025 were identified as two hub transcription factors within the Red co-expression module. However, functions of NAC022/025 have not yet been experimentally characterized in ryegrass or other model plant species. Insights can be drawn from other close NAC family transcription factors. For instance, overexpression of ANAC019 enhances thermotolerance in Arabidopsis by up-regulating downstream genes such as the heat shock transcription factor HSFA1. Similarly, the membrane-associated NAC factor NTL4 exhibits up-regulated expression under heat stress, while ntl4 mutants display higher cell survival rates and reduced hydrogen peroxide accumulation under high temperatures, suggesting that wild-type NTL4 may act as a negative regulator suppressing certain heat-resistant responses[32]. Additionally, in rice (Oryza sativa), SNAC3 enhances plant tolerance to high temperature, drought, and oxidative stress upon overexpression[33]. In wheat (Triticum aestivum), TaNAC2L is also heat-inducible, and its overexpression improves acquired thermotolerance in Arabidopsis by regulating heat stress-related gene expression[34]. Collectively, as members of the NAC family, NAC022 and NAC025 are speculated to participate in regulating the expression of heat stress-responsive defense genes. However, due to the current lack of direct experimental evidence, NAC022 and NAC025 remain functionally uncharacterized, requiring further functional validation to elucidate their precise roles.
WRKY46 was a hub transcription factor of the Red co-expression module. The Arabidopsis WRKY46 is a positive regulator of leaf senescence by modulating ROS scavenging and ABA pathways, aligning with its potential to coordinate oxidative stress mitigation and hormonal crosstalk during heat stress adaptation[35]. ICE1 interacts with WRKY46 to suppress its activation of WRKY6 to delay SA-mediated senescence[35].
FRS5 was a hub transcription factor of the Red co-expression module. In Populus, FRS5 directly binds and regulates the expression of a superoxide dismutase gene, PagSOD2a, that serves as a potential photoreceptor-antioxidant defense nexus that integrates light signaling with redox homeostasis[36].
ZAT5 was another hub transcription factor within the Red co-expression module. Many members of the ZAT family, such as ZAT10 and ZAT12, are known to play critical regulatory roles in plant stress responses, typically by modulating ROS-scavenging and stress-protective genes[37,38]. The Arabidopsis ZAT5 enhances cold tolerance by up-regulating antioxidant gene expression under low-temperature stress[39]. Meanwhile, tomato (Solanum lycopersicum) SlZAT5 delays fruit ripening by interacting with the phosphatase SlPP2C2, undergoing dephosphorylation at the Ser-65 residue, and directly repressing ripening-associated genes[40]. Integrating these findings with analysis of ZAT5's heat-responsive role in ryegrass, it is proposed that ZAT5 and its regulatory module exhibit cross-species universality in the maintenance of cellular homeostasis under diverse stresses by orchestrating antioxidative and gene expression networks.
NFXL1 was identified as the hub transcription factor of the Dark turquoise co-expression module, indicating its central role in orchestrating the regulation of photosynthesis-related and heat-responsive genes under high-temperature conditions. This is corroborated by the discovery of the NFXL1-DREB2A cascade in Arabidopsis, which regulates thermotolerance in floral organs. The Arabidopsis NFXL1 functions as an upstream activator that is up-regulated under heat stress, directly binds to the DREB2A promoter, and initiates the DREB2A-HSFA3-HSP17.6 heat-response cascade[41]. Further functional characterization of these hub genes would be necessary using experimental approaches to verify whether some of them are valid molecular targets for gene editing to improve ryegrass heat tolerance in the future.
-
In summary, by selecting two perennial ryegrass varieties with contrasting heat tolerance ('X6' and 'BV'), transcriptomics were compared after heat stress. The result revealed that, unlike 'BV', 'X6' mounts several metabolic pathways in response to heat, including activated 'glyoxylate and dicarboxylate metabolism' pathway for the secretion of organic acid, 'glycolysis, and pentose phosphate pathway' to attain a more active energy/carbon allocation strategy under the heat stress. Such mechanistic insights and identified hub genes not only validate 'X6' as an elite heat-tolerant variety but also provide molecular targets for breeding perennial ryegrass toward enhanced resilience to future climate extremes.
This project was supported by grants from the National Natural Science Foundation of China (Grant Nos 32271755, 32441041).
-
The authors confirm contributions to the paper as follows: experiments: Zha W, Zeng L, Zhang Z; analysis, interpretation of results, and draft manuscript preparation: Zha W, Xu B. All authors reviewed the results and approved the final version of the manuscript.
-
The RNA-seq dataset that supports the findings of this study is available in the NCBI SRA database (SRA accession number: PRJNA1278810). The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Primer sequences used for qRT-PCR analysis.
- Supplementary Table S2 Assembly quality statistic.
- Supplementary Data 1 Sequences of identified hub genes.
- Supplementary Data 2 Newly identified genes from the RNA-seq.
- Supplementary Fig. S1 Weighted gene co-expression network analysis of WGCNA.
- Supplementary Fig. S2 Value curves between soft-thresholding power and scale-free topology fit index (left panel) and between soft-thresholding power and mean connectivity (right panel).
- Supplementary Fig. S3 Volcano plot for DEGs of two ryegrass varieties with contrasting heat tolerance. (a) X6-H vs X6-C; (b) BV-H vs BV-C.
- Supplementary Fig. S4 Depiction of DEGs in the glycolysis pathway and pentose phosphate pathway. Using LOG2FC values for plotting. Red solid arrows indicate up-regulation in the X6-H vs X6-C comparison, whereas green solid arrows indicate down-regulation.
- Supplementary Fig. S5 Depiction of DEGs in the Calvin cycle. Using LOG2FC values for plotting. Red solid arrows indicate up-regulation in the X6-H vs X6-C comparison, whereas green solid arrows indicate down-regulation.
- Supplementary Fig. S6 Depiction of DEGs in the ascorbate-glutathione cycle. Using LOG2FC values for plotting. Red solid arrows indicate up-regulation in the X6-H vs X6-C comparison.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zha W, Zeng L, Zhang Z, Zhang J, Xu B. 2025. Comparative transcriptomic analysis revealed specific modules and hub heat-responsive genes in perennial ryegrass. Grass Research 5: e024 doi: 10.48130/grares-0025-0021 

Comparative transcriptomic analysis revealed specific modules and hub heat-responsive genes in perennial ryegrass
- Received: 12 May 2025
- Revised: 29 June 2025
- Accepted: 28 July 2025
- Published online: 10 October 2025
Abstract: Perennial ryegrass (Lolium perenne) is one of the most important cool-season grasses used for forage and turf purposes. Heat stress is the primary abiotic factor threatening its pasture/turf persistence. In this study, heat-responsive genes common between or specific to two ryegrass varieties with contrasting heat tolerance traits were analyzed at the transcriptomic level. The result identified 5,399 common heat-responsive differentially expressed genes (DEGs) between the two ryegrass varieties enriched in KEGG pathways such as ribosomes, spliceosomes, ribosome biogenesis, photosynthetic antenna proteins, etc. Weighted gene co-expression network analysis (WGCNA) revealed specific DEGs in the heat-tolerant ryegrass variety that were mainly enriched in glyoxylate and dicarboxylate metabolism, glycolysis, protein processing in the endoplasmic reticulum, spliceosome, and ascorbate-glutathione cycle, etc. Furthermore, heat-tolerant variety-specific hub DEGs and transcription factors were also identified using Core Gene Co-expression Network analysis. Knowledge gained from this study provides a basis for the genetic improvement of heat-tolerant ryegrass and other cool-season perennial grass species.
-
Key words:
- Lolium perenne /
- High temperature /
- Thermotolerance /
- RNA-seq /
- Genotypic difference /
- Hub genes