









-
Lonicera japonica Thunb. (Caprifoliaceae), a perennial semi-evergreen vine is widely recognized for its dried buds and freshly opened flowers, which are integral to traditional Chinese medicine[1,2]. This species is a plant of significant importance in both medicine and food, which is not only renowned for its effective and extensive pharmacological effects but also plays an important value in cosmetics, food, and health drinks[3,4]. Various parts of L. japonica are employed in medicine applications, being incorporated into approximately 30%–35% of Chinese traditional medicine formulations and over 200 proprietary Chinese medicines products. L. japonica has a long history of heat-clearing and detoxifying effects and is commonly used in treating various infectious diseases, including heat stroke, heat-toxic blood dysentery, carbonosis, and throat arthralgia[5−7]. Moreover, extracts of L. japonica and its active ingredients exhibit antibacterial, anti-inflammatory, antitumor, antidiabetic, antioxidant, and liver-gallbladder protective effects and have been investigated for their potential to inhibit COVID-19[8−11]. Previous research has identified chlorogenic acid (CGA), luteolin, and cyclic ether ketone as important active components of L. japonica, with CGA recognized as the primary bioactive compound and a standard reference for assessing its chemical quality[6].
CGA, 3-O-caffeoylquinicid, is a phenylpropanoid compound synthesized through esterification between hydroxycinnamic acid and quinic acid[12]. This compound is predominantly generated via the shikimate pathway during plant respiration and is extensively found in various plant families, including Asteraceae and Solanaceae. The biosynthesis of CGA is regulated by the phenylpropanoid pathway, wherein the initial three enzymatic steps are catalyzed by phenylalanine ammonia-lyase (PAL), cinnamate 4-hydroxylase (C4H) and 4-coumarate-CoA ligase (4CL), leading to the production of p-coumaroyl-CoA, a central precursor for several important secondary metabolites[13]. Subsequently, p-coumaroyl-CoA can follow one of three possible biosynthetic pathways to produce CGA[14−16]. At the initial step of the pathway, hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase (HCT) mediates the transfer of a hydroxycinnamoyl group from p-coumaroyl-CoA to shikimic acid, resulting in the production of p-coumaroyl-shikimate[17]. This intermediate is enzymatically modified to caffeoyl-shikimic acid by the action of p-coumarate 3'-hydroxylase (C3'H), which subsequently reacts with shikimic acid via HCT to yield caffeoyl-CoA[18]. The final step involves the reaction of caffeoyl-CoA with quinic acid, catalyzed by hydroxycinnamoyl-CoA quinate transferase (HQT), to produce CGA[17]. The second pathway involves the direct reaction of p-coumaroyl-CoA with quinic acid to form p-coumaroyl-quinic acid, also facilitated by HCT. This intermediate is subsequently hydroxylated by C3'H[17,18]. In addition, hydroxycinnamoyl-CoA, quinic acid hydroxy cinnamoyl transferase (HCGQT) is also recognized as a crucial enzyme in the CGA synthesis pathway[19]. It catalyzes the direct esterification of quinic acid and caffeoyl-D-glucose to CGA[18,20]. HQT serves as a crucial enzyme that regulates the rate of the metabolic pathway of CGA. Research indicates that CGA accumulation in tomato[18]. Furthermore, inhibition of HQT expression in potatoes has been shown to lead to a reduction of over 90% in CGA levels[21]. In L. japonica, the biosynthesis and accumulation of CGA occur within chloroplasts and the cytoplasm, with subsequent storage in vacuoles. The dynamic fluctuation of CGA accumulation is closely associated with the spatiotemporal expression level of the HQT gene[22].
MYB transcription factors serve as central regulators in the transcriptional control of CGA-like phenolic compound biosynthesis[13,21,23]. The MYB transcription factor family is a highly diverse and functionally transcriptional regulator family in eukaryotes, with a demonstrated critical function in modulating phenylpropanoid metabolic pathways in plants. Recent studies mainly focus on the functions of MYB transcription factors in tomato, potato, tobacco, and other Solanaceae species. In tobacco, overexpression of NtMYB1, NtMYB2, NtMYB4, and NtMYB59 has been shown to reduce the levels of CGA, rutin, caffeic acid, and total flavonoids[24,25]. In potato, the MYB transcription factor gene StAN1 was transiently expressed in tobacco, leading to the upregulation of phenylpropanoid pathway genes, such as PAL1, PAL2, CHS, F3H, and DFR, thereby enhancing CGA and anthocyanin accumulation[21]. Furthermore, increasing the expression of LmMYB15 in tobacco plants significantly elevates CGA accumulation. LmMYB15 functions as a transcriptional activator by directly binding to the promoters of phenylpropanoid biosynthetic genes (4CL, MYB3, and MYB4), thus enhancing phenylpropanoid metabolism and promoting CGA synthesis[26]. Although MYB transcription factors are widely recognized to govern CGA biosynthesis and the phenylpropanoid pathway, the molecular mechanisms governing CGA biosynthesis remain less understood compared with those underlying lignin, anthocyanin, and flavonoid metabolism.
With the advancement of sequencing technology, transcriptome sequencing has become a widely utilized method for studying plant secondary metabolism and the mechanisms of transcriptional regulation. However, second-generation sequencing techniques are constrained by inherent limitations, including short read lengths, base-calling inaccuracies, and challenges in transcript structural annotation and functional characterization. In contrast, contemporary full-length sequencing technologies, especially single-molecule real-time (SMRT) sequencing, effectively address these limitations through long-read capabilities and high accuracy[27]. PacBio SMRT sequencing enables de novo reconstruction of full-length transcripts without assembly due to its ultra-long reads, thereby enhancing the quality of reference genomes[28,29]. Therefore, this study comprehensively analyzed the transcriptional regulation of CGA biosynthesis through full-length transcriptome sequencing combined with stage-specific RNA-seq data in L. japonica. By analyzing the transcriptome data, the expression pattern of HQT, a pivotal gene in CGA synthesis, will be elucidated, and candidate MYB transcription factors that regulate HQT transcription will be identified. This study will provide new insight into the molecular regulatory mechanisms that control CGA biosynthesis.
-
In this study, Huajin 6, a new variety of honeysuckle cultivated at the Botanical Garden of Shandong University of Chinese Medicine, was selected as the material. This variety was developed by the Research Institute of Chinese Medicinal Materials through several decades of targeted breeding. The plant exhibits an upright growth habit following auxiliary setting, with new branches appearing light green and old branches dark green. The leaves are papery and lanceolate in shape. Roots, stems, leaves, and flowers from different plants were randomly collected, covering five distinct stages of floral development. Every sample that was gathered was quickly frozen in liquid nitrogen and then stored at −80 °C.
Library construction of PacBio SMRT sequencing
-
Total RNA was isolated from the Huajin 6 leaves samples, and RNA quality was assessed through agarose gel electrophoresis (purity), a Nanodrop 2,000 spectrophotometer (purity/concentration/absorption peak), and an Agilent 2,100 Bioanalyzer (RIN value). Poly A-containing mRNA enriched by oligo (dT) beads was synthesized into cDNA by the SMARTer PCR cDNA Synthesis Kit. Following PCR amplification with cycle optimization, fragments larger than 4 kb were identified via the BluePippin system and amplified. Libraries were constructed through end repair and SMRT bell adaptor ligation, followed by exonuclease digestion to remove unligated fragments for SMRT bell library preparation. Finally, the libraries were sequenced on the PacBio Sequel IIe platform to obtain full-length transcriptome data[30].
Functional annotation analysis of transcripts
-
Raw Iso-Seq sequencing data were processed through PacBio's SMRT Link software; the corrected and deduplicated transcripts were used for downstream analysis[31,32]. Unique transcripts were functionally annotated against seven databases: NCBI non-redundant protein (Nr) and nucleotide (Nt) sequences, Protein family (Pfam), Eukaryotic Orthologous Groups (KOG), Swiss-Prot (a manually curated protein database), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO)[33]. Protein-coding sequences (CDS) were predicted from cDNA using a long-read optimized ANGEL pipeline. The ANGEL algorithm was trained on high-confidence protein sequences from the target species or its close relatives, followed by CDS prediction using the same framework. Plant transcription factors were identified using the iTAK pipeline with default parameters. Long non-coding RNAs (lncRNA) were screened using four tools: CNCI, CPC, Pfam, and PLEK, to assess their coding potential[34,35]. Transcripts with predicted coding potential by any tool were discarded, retaining non-coding transcripts as the final lncRNA databases. Simple sequence repeats (SSRs) were identified with MISA software, which detects both perfect microsatellites and compound microsatellites.
Transcriptome data analysis
-
Clean reads from the 15 samples were compared with PacBio sequences. The normalized FPKM values were applied to assess gene expression levels. With reference to the full-length transcriptome, genes related to the phenylpropane metabolic pathway and HQT genes, a key gene for CGA biosynthesis, were screened through gene annotation. MYB transcription factors were retrieved from the full-length transcriptome, followed by phylogenetic analysis of their amino acid sequences in MEGA software[36]. Excel was used to process data to generate mean values and standard deviations (SD) to generate a bar graph. Analysis of Variance (ANOVA) and Fisher's Least Significant Difference (LSD) test were performed in SPSS software[37]. For Venn diagrams visualization, the online tool Jvenn (
www.bioinformatics.com.cn/static/others/jvenn/index.html ) was employed[38]. In R, gene expression levels were visualized via the pheatmap package, while principal component analysis (PCA) was performed on transcriptome data using the stats package. The corrplot package was utilized to examine the correlation among gene expressions[39].Determination of CGA
-
The CGA extraction method was slightly modified from a previously described protocol[40]. A precise preparation of 1 μmol/mL CGA standard solution was prepared in 95% (v/v) ethanol. After gradient dilution, absorbance at 330 nm was determined using the Synergy H4 microplate reader (BioTek). The standard curve was generated by plotting absorbance (y-axis) against CGA concentration (x-axis), from which the linear regression equation was derived. Fresh floral tissues (50 mg) were weighed, followed by extraction with 10 mL of 95% ethanol in a water bath at 85 °C for 4 h. A 50 μL aliquot of the extract was combined with 150 μL of 95% ethanol. With 95% ethanol as blank, the Synergy H4 microplate reader (BioTek) was used to measure absorbance. CGA concentrations in experimental samples were derived from the standard curve. Three biological replicates were analyzed per sample.
Verification of genes by real-time quantitative PCR (qRT-PCR)
-
Total RNA was extracted from L. japonica flowers at five developmental stages using established protocols for plant RNA isolation. Reverse transcription and RT-qPCR were performed according to previous studies[41]. LjActin served as an endogenous control, and relative gene expression was calculated via the 2−ΔΔCᴛ method[42]. All primers listed in Supplementary Table S1 were designed using the NCBI Prime-blast tool (
www.ncbi.nlm.nih.gov/tools/primer-blast )[43]. -
Firstly, after removing adapters and low-quality sequences from the raw data, a total of 10,061,167 full-length non-redundant reads were generated through PacBio SMRT sequencing. The sequencing produced reads averaging 2,250 bp in length, demonstrating an N50 statistic of 2,800 bp (Supplementary Fig. S1a). Additionally, a total of 447,437 CCS reads were obtained, exhibiting a mean length of 2,772 bp (Supplementary Fig. S1b). Following stringent quality control, 402,739 full-length non-chimeric reads (FLNC) were identified among the CCS reads (Supplementary Fig. S1c). Subsequently, following iterative redundancy removal and error correction, 205,091 high-quality isoforms were retained, exhibiting a mean length of 2,670 bp (Supplementary Fig. S1d). Finally, CD-HIT-processed transcripts were clustered at a 95% sequence identity threshold, resulting in 97,588 non-redundant transcripts averaging a length of 2,883 bp (Fig. 1a). The corresponding number of genes and transcripts after redundancy removal is shown in Fig. 1b. The above results indicate that the transcripts obtained through PacBio SMRT sequencing are of high-quality and reliable.
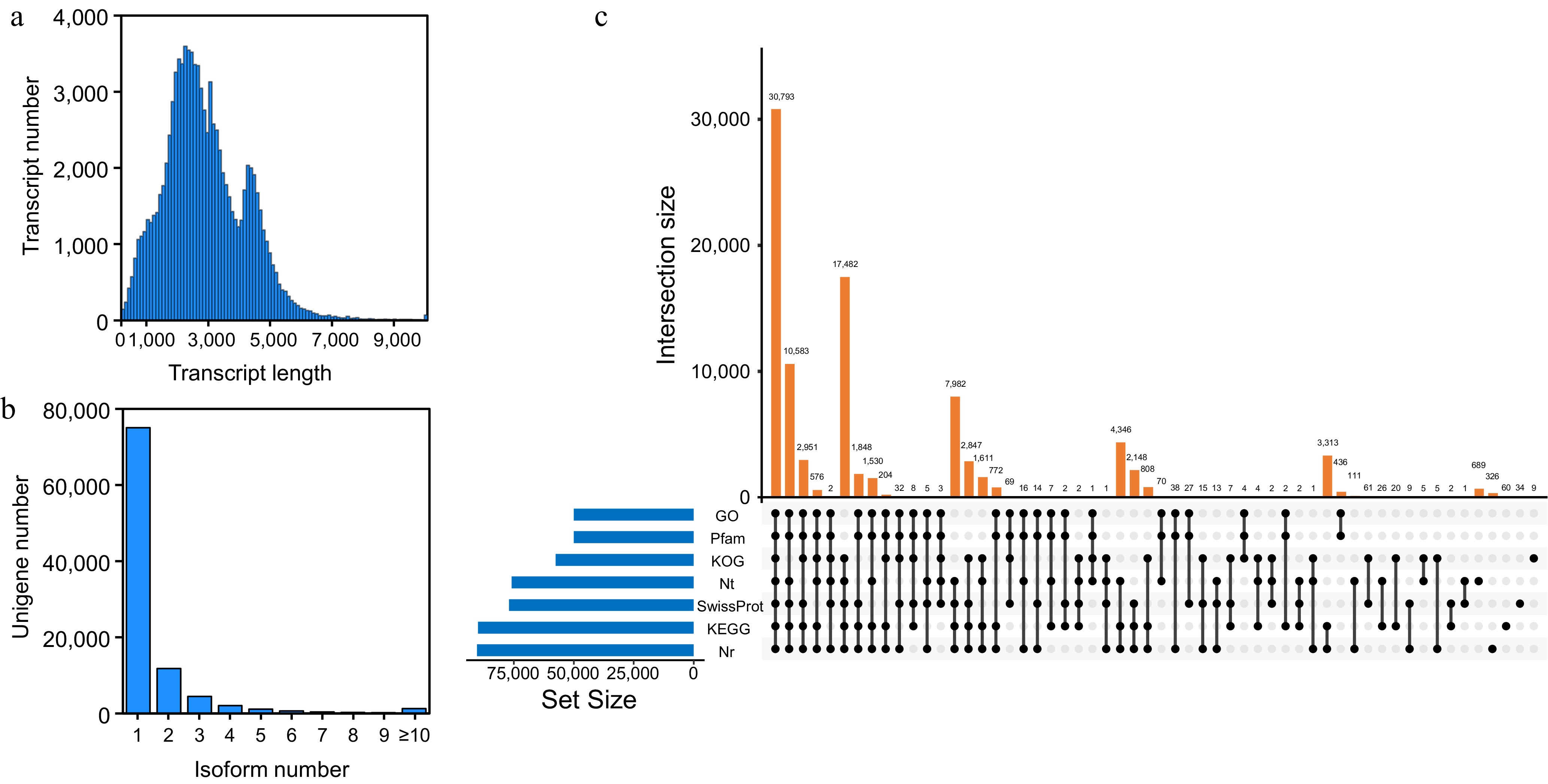
Figure 1.
PacBio single-molecule long-read sequencing of L. japonica. (a) Length distribution of unigenes. (b) Length distribution of transcripts after redundancy is removed. (c) Upset plot showing transcript annotations in Nr, SwissProt, KEGG, KOG, GO, Nt, and Pfam database.
Functional characterization of transcripts
-
To elucidate transcript functions, the aforementioned transcripts were annotated via cross-database comparisons (NR, SwissProt, KEGG, KOG, GO, NT, and Pfam). In seven databases, 30,793 transcripts were annotated. The number of transcripts annotated in each database was as follows: NR (90,382), SwissProt (76,945), KEGG (89,936), KOG (57,523), GO (49,993), NT (49,993), and Pfam (7,586) (Fig. 1c). Annotation results from the Nr database identified the five species exhibiting the highest degree of similarity to L. japonica transcripts were Vitis vinifera, Daucus carota, Sesamum indicum, Coffea canephora, and Cynara cardunculus (Fig. 2a). The transcripts were categorized by GO analysis into three functional categories: In the Biological Process (BP) category, the majority of transcripts were annotated with terms pertaining to metabolic processes, cellular processes, and single-organism processes. Within the Cellular Component (CC) category, the majority of transcripts were enriched for the term 'cell' and 'cell part'. Function characterization showed binding and catalytic activity molecular functions dominated the Molecular Function (MF) annotations (Fig. 2b). In addition, the transcripts in the KOG database were clustered into 26 categories, the majority of which fall into three main groups: general function prediction only, signal transduction mechanisms and secondary metabolites biosynthesis, transport and catabolism (Fig. 2c). The KEGG pathway analysis classified the transcripts into six distinct groups: metabolism, genetic Information processing, cellular processes, environmental information processing, organismal systems, and human diseases. Figure 2d highlights the top 20 pathways showing the strongest enrichment signals. A considerable number of transcripts were annotated to KEGG pathways, including signal transduction, carbohydrate metabolism, energy metabolism, translation, and so forth.
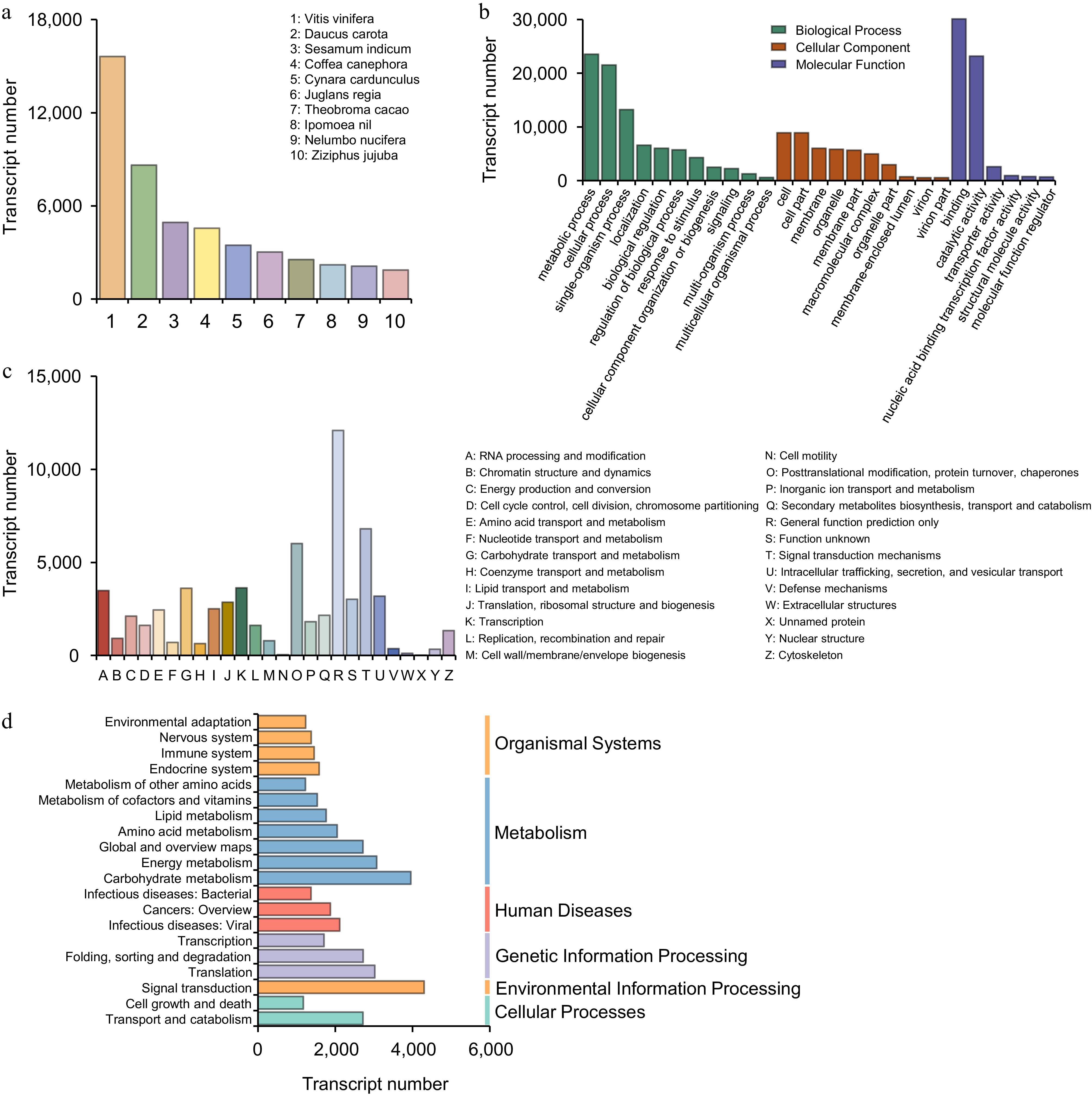
Figure 2.
Functional analysis of full-length transcripts. (a) Function classification annotated in the Nr database. (b) Function classification annotated in the GO database. (c) Function classification annotated in the KOG database. (d) Function classification annotated in the KEGG database.
Gene structure analysis
-
A total of 97,018 CDS were predicted out of 97,588 transcripts, which ranged in length from 145 to 7,815 bp and were predominantly distributed between 200 and 600 bp (Fig. 3a). A total of 5,222 transcription factors were identified. The 30 most prevalent transcription factor families are shown in Fig. 3b. The top three families were C3H, SNF2, and MYB-related. A total of 115,299 SSRs were identified within the transcripts of L. japonica. Among the six SSR motif types, the mono-, di-, and tri-nucleotide repeats were the most prevalent. Mono-nucleotide repeats constituted 57,435, representing 49.81% of all SSRs, followed by di-nucleotide repeats with 39,774, accounting for 34.50%. A total of 15,690 tri-nucleotide repeats were identified, representing 13.61% of all SSRs. The remaining repeated sequence types were exceedingly scarce, accounting for a mere 2.08% (Supplementary Fig. S2a). Most mono-nucleotide SSRs consisted of 9-12 repeats, while the majority of di-nucleotide SSRs comprised 5-8 repeats. Tri-nucleotide SSRs predominantly exhibited fewer than 12 repeats (Fig. 3c). CNCI, Pfam, PLEK, and CPC analyses predicted 34,407, 43,055, 17,095, and 11,616 transcripts, respectively, to be non-protein-coding. Additionally, 4,819 transcripts were identified as lncRNA through the above four methods (Fig. 3d). The average length of lncRNA is 1,971.06 bp, with the majority of lncRNA falling within the range of 300−3,500 bp (Supplementary Fig. S2b).
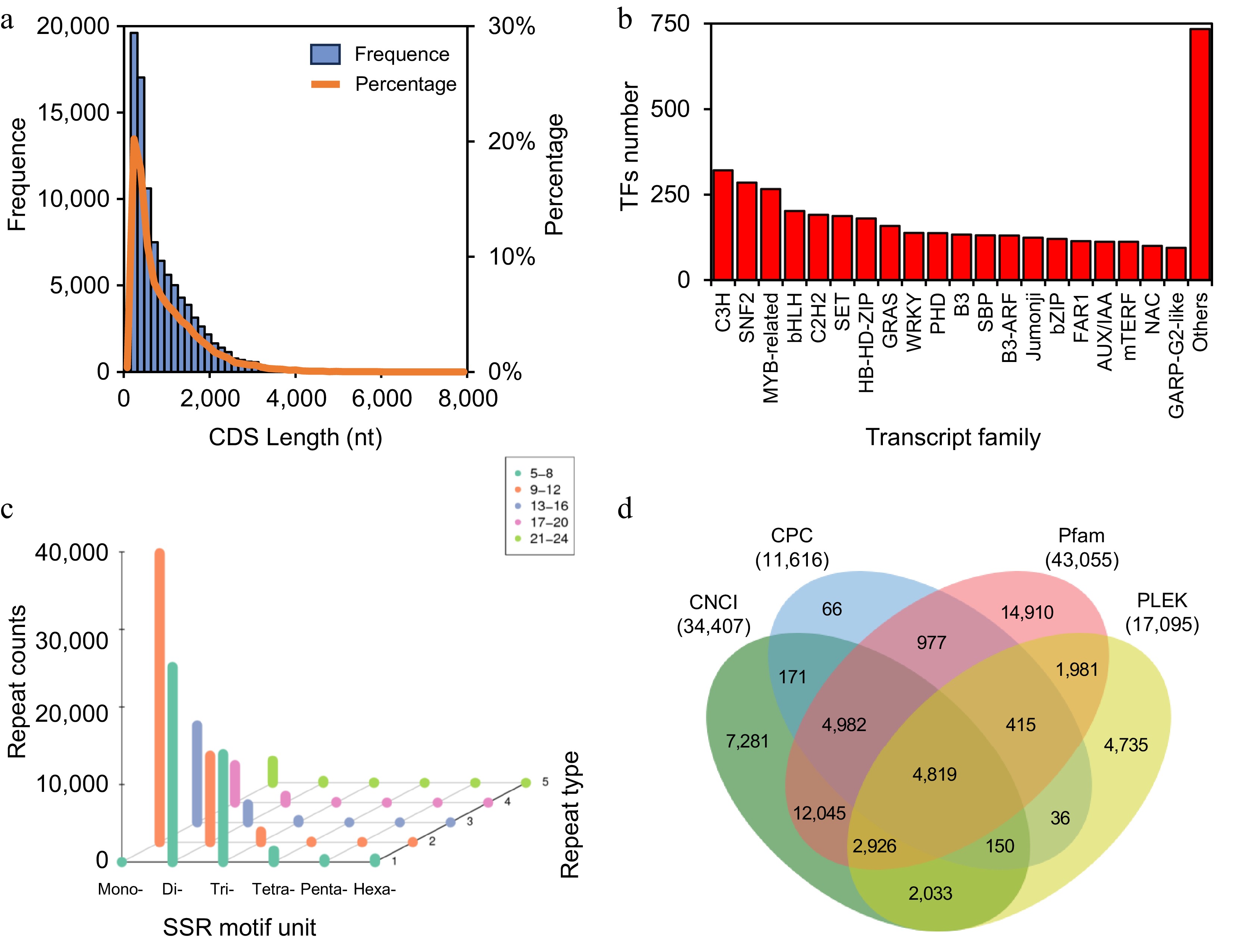
Figure 3.
Gene structure analysis. (a) Length distribution of CDS. (b) Top 20 transcription factor families. (c) Repeat numbers of SSR motifs. The X-axis represents SSR type (number of repeated bases), the Y-axis shows the number of base repeats (corresponding to color and legend), and the Z-axis indicates the number of SSRs. (d) Venn diagram of lncRNA prediction.
CGA content and transcriptome analysis of flowers in L. japonica
-
To uncover the regulatory mechanisms governing CGA biosynthesis during flower development in L. japonica, the content of CGA was determined across five developmental stages: S1 (Juvenile bud stage): The upper end of the bud begins to expand and curve, appearing yellow-green with dense fuzz. S2 (Third green stage): The upper end of the bud becomes significantly swollen and remains yellow-green. S3 (Second white stage): The upper end of the bud turns white, while the lower part retains a yellow-green color. S4 (Complete white stage): The entire bud is white except for the yellowish-green base. S5 (Golden flowering stage): The bud turns golden and rapidly begins to fade (Fig. 4a). The findings indicated a significant increase in CGA concentration in the corolla during the S2 and S3 stages, with levels reaching 22.86 and 24.58 mg/g, respectively. The concentration of CGA in the corolla at the S1 and S5 stages was relatively low, with values of 15.17 and 12.54 mg/g, respectively. The lowest concentration of CGA in the corolla was observed at the S4 stage, measuring 9.76 mg/g (Fig. 4b). This indicated that the optimal harvesting period for corolla tubes is the S2-S3 stage. To further investigate the underlying molecular mechanisms, transcriptome sequencing was conducted at five distinct stages of floral development. A total of 94,507 transcripts were identified, and PAC analysis was further used to evaluate inter-sample diversity and similarity among biological replicates. The S1, S2, S3, and S4 transcriptome data exhibited a high degree of similarity and overlap while displaying a notable divergence from the S5 transcriptome data. This indicated that the unopened transcripts of corolla tubes exhibited high similarity, whereas those after opening displayed marked divergence (Fig. 4c). Concurrently, the analysis of a Venn diagram showed that 38,751 genes were expressed across all five stages (Fig. 4d).
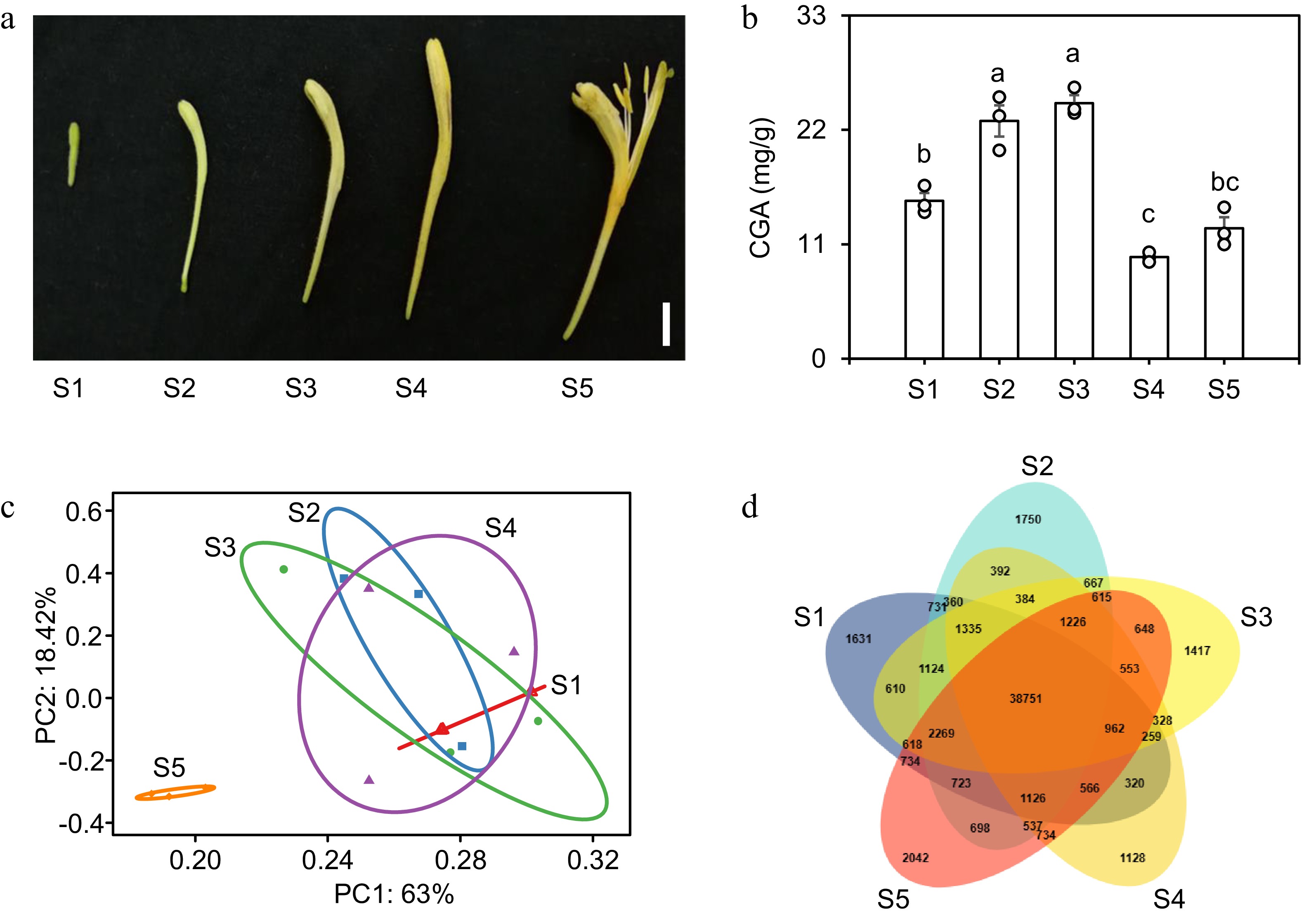
Figure 4.
Transcriptome analysis of flowers at five developmental stages in L. japonica. (a) Phenotypes of flowers at five developmental stages: juvenile bud stage (S1), third green stage (S2), second white stage (S3), complete white stage (S4), and gold flowering stage (S5). Scale bar = 1 cm. (b) CGA content in flowers at five developmental stages. Data are mean ± SE (n = 3). Different letters indicate significant differences (one-way ANOVA, Fisher's LSD, p < 0.05). (c) PCA of transcriptional profiles in L. japonica flowers at five developmental stages. (d) Venn diagram of transcript expression in flowers at five developmental stages.
Functional annotation of genes involved in CGA biosynthesis
-
To investigate the molecular basis of CGA biosynthesis in L. japonica, this study identified homologous genes of key enzymes from the CGA biosynthesis pathway through gene annotation of the full-length transcriptome and transcriptome data (Fig. 5a). The results showed that 6 PAL, 2 C4H, 12 4CL, 2 HCT, 3 C3H, and 8 HQT genes were involved in the pathway. The heatmap analysis of the 47 genes using FPKM values from the transcriptome data showed that most genes had varying expression levels throughout the five flower developmental stages. Notably, the expression abundance of HQT was observed to be higher in the early stages of flower development (Fig. 5b). Further corrplot analysis showed a correlation between HQT and the PAL, C4H, and 4CL genes expression levels (Fig. 5c). This suggests that these genes may be crucial for the production of CGA in L. japonica.
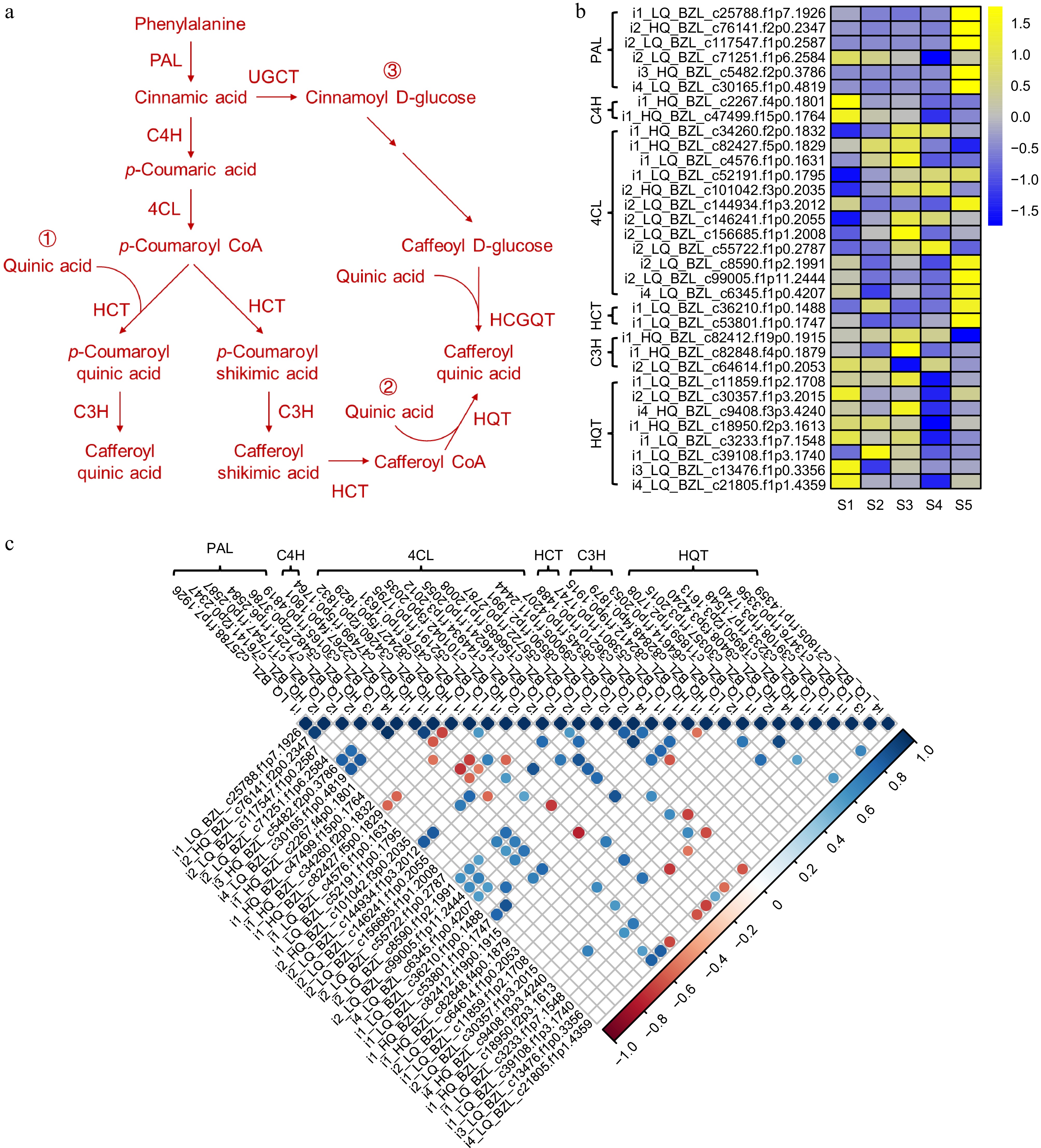
Figure 5.
Visualization of gene expression levels associated with CGA biosynthetic pathways in L. japonica. (a) Schematic diagram of CGA biosynthetic pathways. (b) Heatmap of gene expression related to CGA biosynthetic. (c) Correlation analysis of gene expression in CGA biosynthetic pathways.
Phylogenetic and gene expression patterns analysis of MYB transcription factors
-
Studies have demonstrated that MYB transcription factors were critical regulators of phenolic metabolism, including CGA synthesis and accumulation[44,45]. Therefore, 252 MYB transcription factors were identified from full-length transcriptome sequences in this study. The phylogenetic tree was constructed using the protein sequences of the relevant organisms. The phylogenetic tree indicates that MYB transcription factor proteins can be divided into six clades, with groups A, B, C, D, E, and F comprising 25, 52, 44, 34, 37, and 60 members, respectively (Fig. 6a). The heatmap analysis revealed distinct variations in the expression patterns of MYB transcription factor genes during the development of corolla in L. japonica (Fig. 6b).
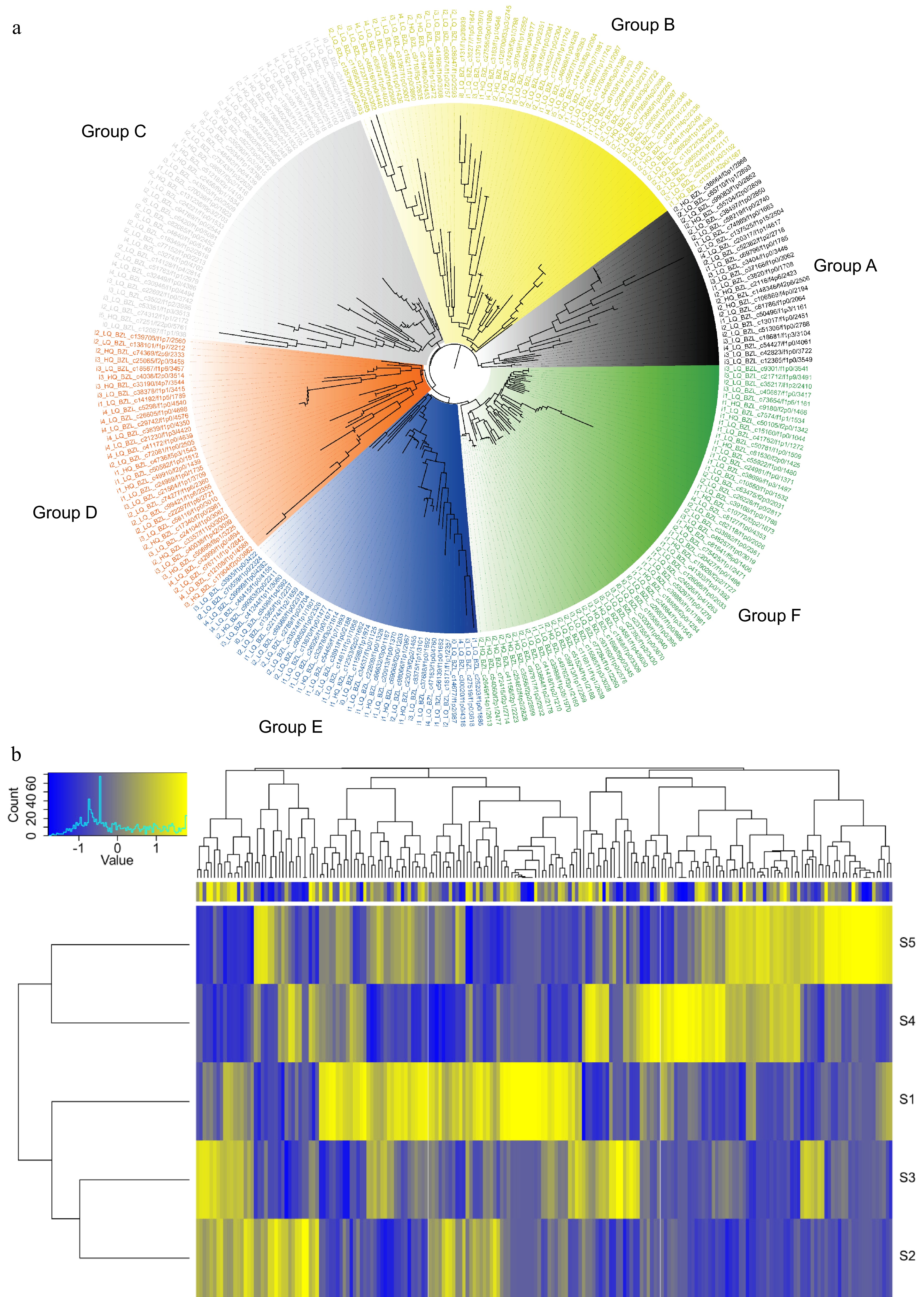
Figure 6.
Characterization of MYB transcription factors in L. japonica. (a) Phylogenetic tree of 252 MYB transcription factors. (b) Heatmap showing the expression profiles of MYB transcription factors in flowers at five developmental stages.
Correlation analysis between MYB transcription factor and HQT gene expression profiles
-
To further analyze the biosynthesis mechanism of CGA, 74 MYB transcription factors were subsequently selected from 38,751 co-expressed genes and correlated with eight HQT genes and five stages of CGA content. The results demonstrated a positive connection between CGA accumulation and two HQT genes as well as five MYB transcription factors, whereas a negative association was observed with eleven MYB transcription factors (Supplementary Fig. S3). The heatmap displayed the expression patterns of 18 genes across five flower developmental stages. Among these, two HQT and five MYB genes exhibited a downward trend, whereas 11 MYB transcription factors showed an upward trend (Fig. 7a).
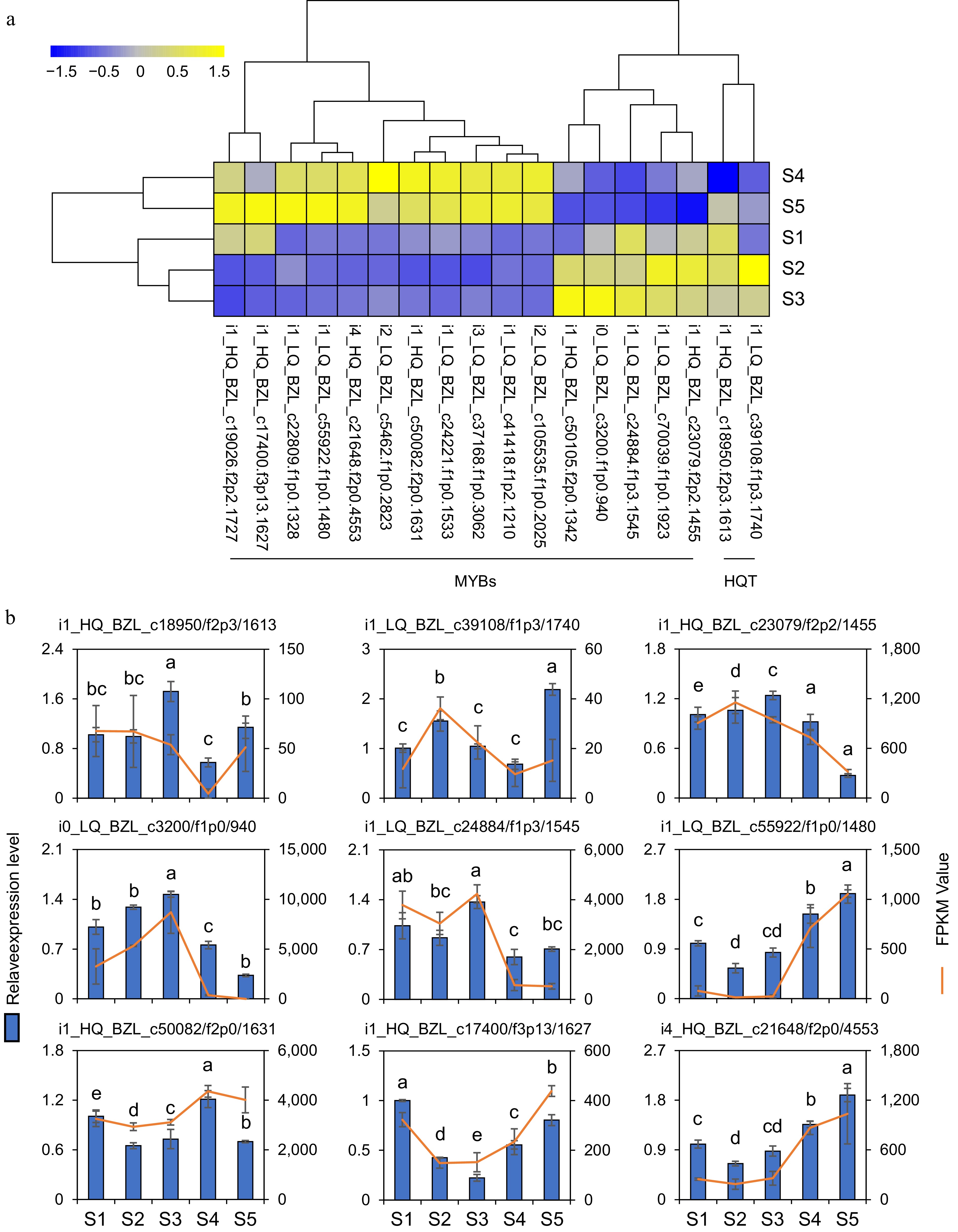
Figure 7.
Gene expression analysis. (a) The heatmap shows the expression profiles of 16 MYB transcription factors and two HQT genes based on FPKM values. (b) Expression changes of two HQT genes and 7 MYB DEGs in flowers at five developmental stages. The line charts represent gene expression levels derived from RNA-seq data using FPKM normalization. The bar charts represent corresponding gene expression levels quantified by qRT-PCR. The data represent the mean ± SE of three replicates (n = 3). Different letters in the graph indicated significant differences between groups (one-way ANOVA, Fisher's LSD test, p < 0.05).
Finally, in order to confirm the reliability and correctness of the transcriptome data, two HQT and seven MYB genes were selected for qRT-PCR analysis. The findings indicated that the relative expression level correlated with the FPKM values determined in the RNA-Seq analysis (Fig. 7b). Pearson correlation coefficients between FPKM and qRT-PCR values for the nine genes were calculated, revealing a pronounced correlation between the log2 (fold-change) values of FPKM and those of qRT-PCR (Supplementary Fig. S4). This provides further evidence of the accuracy and reliability of the transcriptome data.
-
In recent years, PacBio SMRT, a third-generation sequencing technology, has seen significant advancements. The data generated from third-generation sequencing technologies are notably better than those obtained from second-generation sequencing technologies[29]. Moreover, complete transcripts can be obtained without requiring sequence assembly, thereby providing comprehensive genetic information for species lacking complete genomic data and for newly developed cultivars[46]. This study represents an inaugural investigation into the full-length transcriptome of the honeysuckle variety, Huajin 6. The research generated a total of 97,588 high-quality transcripts with a mean length of 2,883 bp (Fig. 1a). Annotation analyses were conducted with reference to following databases: NR, GO, KOG, and KEGG (Fig. 2). Subsequently, the following analyses were conducted: CDS prediction, transcription factor prediction, SSR analysis, and lncRNA prediction (Fig. 3). The flower buds of L. japonica are rich in flavonoids and phenolic compounds, rendering them a significant traditional Chinese medicinal ingredient. The full-length transcriptome data generated in this work serve as valuable genomic datasets and genetic resources for exploring the biosynthetic mechanisms of these natural compounds.
The flower bud, recognized as the medicinally active part of the component of L. japonica, contains CGA, whose concentration directly correlates with the plant's pharmaceutical value[47,48]. Consequently, investigating the alterations in the expression of CGA and its pivotal regulatory gene HQT during flower development helps elucidate the regulatory mechanisms of CGA synthesis and accumulation in L. japonica. Research has indicated that the biosynthetic pathway mediated by HQT in L. japonica is the main pathway for CGA synthesis[49]. The dynamic changes in HQT transcript levels during flower development show significant correlations with variations in CGA content, indicating that the HQT genes exert a regulatory function in CGA synthesis and accumulation in L. japonica[22]. Consequently, the content of CGA at five flower development stages was measured, and transcriptomic analyses were performed using RNA-seq technology. The highest concentration of CGA in the corolla was observed at stages S2 and S3 (Fig. 4b). Additionally, eight HQT genes were identified in the transcriptomic data. Among these, the expression levels of two genes (i1_HQ_BZL_c18950/f2p3/1613 and i1_LQ_BZL_c39108/f1p3/1740) were closely related to CGA content (Supplementary Fig. S4). This provides further confirmation that the HQT gene directly regulates the CGA content during the developmental stages of the flower in L. japonica.
It is generally accepted that transcription factors exert a significant regulatory influence on the biosynthesis and accumulation of flavonoids and phenolic compounds[50]. The MYB transcription factor family is a common regulatory factor of structural genes in the phenylpropanoid metabolic pathway[51,52]. Previous studies have indicated that the MYB transcription factor family participates in regulating the biosynthesis of CGA[51,53]. The expression of MYB16, MYB61, and MYB54 were positively correlated with the expression of PAL, C4H, and HCT genes in the CGA synthetic pathway. These transcription factors are likely to enhance the biosynthesis and accumulation of CGA[54]. In this study, 252 MYB transcription factors were retrieved from the full-length transcriptome, and further analysis showed that 16 of them were closely associated with CGA content. Following association analysis with the two HQT genes previously screened, five MYB transcription factors were discovered to promote the synthesis and accumulation of CGA by triggering the expression of these HQT genes. This research provides a theoretical understanding of CGA biosynthesis at the molecular level.
-
A total of 97,588 high-quality transcripts of L. japonica were obtained through full-length transcriptome sequencing with comprehensive annotation. The content of CGA in the corolla of L. japonica initially increased but subsequently declined across different development stages. Integrated analysis of the full-length transcriptome and stage-specific corolla transcriptomes identified five candidate MYB transcription factor genes that may positively regulate CGA content in the corolla through direct activation of HQT gene expression.
We acknowledge the support of the key research and develop-ment program of Shandong province (Grant No. 2022TZXD0036) to Jia Li, (Grant No. 2024LZGC012) to Zhilong Bao, and the Construction Engineering Special Fund of Taishan Scholars of Shandong Province (Grant No. tsqn202211135) to Jia Li.
-
The authors confirm contributions to the paper as follows: conceptualization, supervision: Ma F, Li J, Bao Z; methodology, data curation, investigation: Zhang P, Duan T; resources: Li Q, Huang Y, Liu Z, Ma F, Li J, Bao Z; funding acquisition: Li J, Bao Z; writing-review & editing: Zhang P, Duan T, Ma F, Bao Z. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Peng Zhang, Tongyao Duan
- Supplementary Table S1 Primer information used in the present study.
- Supplementary Fig. S1 PacBio single-molecule long-read sequencing of L. japonica. (a) Length distribution of subreads. (b) Length distribution of CCS. (c) Length distribution of FLNC. (d) Length distribution of consensus reads.
- Supplementary Fig. S2 Gene structure analysis of L. japonica. (a) Proportion distribution of SSR types. (b) Length distribution of lncRNA.
- Supplementary Fig. S3 The corrplot represents the correlation between CGA content and the expression levels of 75 MYB transcription factor and 8 HQT genes.
- Supplementary Fig. S4 Scatter plots represent the comparison of fold change relationships of FPKM and qRT-PCR data. The regression equation proves the correlation between the methods.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang P, Duan T, Li Q, Huang L, Liu Z, et al. 2025. Full-length transcriptome sequencing provides insights into chlorogenic acid biosynthesis in Lonicera japonica. Ornamental Plant Research 5: e032 doi: 10.48130/opr-0025-0029 

Full-length transcriptome sequencing provides insights into chlorogenic acid biosynthesis in Lonicera japonica
- Received: 13 December 2024
- Revised: 30 April 2025
- Accepted: 19 May 2025
- Published online: 20 August 2025
Abstract: Lonicera japonica Thunb. is a significant medicinal resource within traditional Chinese medicine, exhibiting a variety of pharmacological properties. Previous studies reported that the major bioactive compound in L. japonica was chlorogenic acid (CGA), while the mechanism of CGA biosynthesis remained unknown due to the limited genome information. In the present study, PacBio single molecule real-time (SMRT) sequencing technology was employed to conduct the inaugural full-length transcriptome sequencing of the honeysuckle cultivar 'Huajin 6'. A total of 97,588 high-quality transcripts, with an average length of 2,883 base pairs, were generated and annotated using the NR, GO, KOG, and KEGG databases. The coding sequences (CDS), transcription factors, simple sequence repeats (SSR), and long non-coding RNA (lncRNA) transcripts were predicted. The key genes encoding hydroxycinnamoyl-CoA quinate transferase (HQT) in CGA biosynthesis were isolated through the combination analysis of transcriptomic data and CGA content in flowers at different development stages. MYB genes that potentially regulated the transcription of HQT were further identified. Taken together, these results provide valuable genetic resources to assist the molecular study of L. japonica genes, which will facilitate the quality improvement of L. japonica cultivars.