









-
High-throughput sequencing, omics technologies, large-scale and information-rich databases, and prominent bioinformatic tools enable researchers to extensively explore not only individual microbial entities but also entire communities in various environments. Taxonomic and functional structures, associations among microbial members, and environmental factors that shape community properties have been increasingly documented over the last decade, providing comprehensive biological and ecological information. This comprehensive biological and ecological information, including the genome, transcriptome, proteome, lipidome, and metabolome of the microbial communities, is collectively referred to as 'Microbiome'[1].
Among the environments, our understanding of the microbiomes associated with macroorganisms (plants, animals, and humans) has advanced significantly. Typically, in the macroorganism-associated microbiomes, host factors, including individual genetic[2−7], and epigenetic factors[8−12] within the host populations, host compartments[2,13,14], environmental cues[15−19], which are adjacent to the host, and their combined effects[7,17] have been examined. Successional changes and inheritance of host-associated microbiomes during the growth and development of the host entity have also been widely studied[14,20−23]. Evolutionary relationships between hosts and microbiomes have been investigated[24−28], paving the way to construct food production strategies combined with wild host genetic sources and associated microbial partners (known as microbiome rewilding)[29,30]. Recent studies have focused on disease-associated microbiomes (or host-associated microorganisms associated with reduced health status) called the 'Pathobiome'[31,32] to develop personalized therapeutics and health management platforms[33,34].
More recently, it has been evident that microorganisms, including fungi, as well as macroorganisms, can host distinct microbiomes within their unique microhabitats. For instance, filamentous fungi, including arbuscular mycorrhizal fungi (AMF) and ectomycorrhizal fungi (EcMF), are able to form a specialized microhabitat adjacent to their hyphae, called the hyphosphere[35]. Similar to the plant rhizosphere, fungi can recruit specific groups of microbes in the hyphosphere, which can support fungal growth and development[36,37], and even influence fungal fitness[38]. Recent studies revealed that the hyphosphere microbiomes are key players affecting soil food webs, soil resource cycling, and further impacting the physiology of the plants where the host fungi are associated[39−42]. Taken together, it can be inferred that the classical understanding of mycorrhizal systems, represented by binary associations between host plants and mycorrhizal fungi, should be expanded to include multi-domain associations among plants, mycorrhizal fungi, and microbiomes. Especially, the ecological similarity between the hyphosphere and plant microbiomes suggests the possibility of the agricultural applications of the hyphosphere microbiomes to improve the productivity of edible or medicinal fungi via sustainable cultivation approaches, as in the plant rhizosphere microbiomes.
In this context, numerous studies have aimed to reveal the microbial individuals[43] or microbiomes associated with edible and medicinal fungi, which form mushrooms or fruiting bodies[44−46]. Recent advances have shown that mushroom-forming fungi harbor distinct and diverse microbiomes in the hyphosphere and other compartments, including fruiting bodies[44,47,48]. Mounting evidence indicates that both the composition and dynamics of these microbiomes can directly affect fungal growth, health, and mushroom productivity[38], similar to the ecological impacts of plant microbiomes. In this review, recent advances and findings in our understanding of microbiomes associated with mushroom-forming fungi (hereafter mushroom microbiome) are comprehensively summarized, with particular focus on their diversity and functional roles. Current understandings are further synthesized to clarify how these microbiomes influence mushroom growth, health, and productivity through their dynamic relationships with the fungal hosts. By providing an integrated ecological perspective, this review aims to inform and guide the development of sustainable mushroom production systems under controlled (or semi-controlled) and natural conditions.
-
Fungal hyphae are evidence of vegetative growth of filamentous mushroom-forming fungi. Analog to plant roots, which play roles in absorbing nutrients and secreting metabolites, they are crucial for most fungal biological activities, including nutrient acquisition, environmental exploration, reproduction, and infection (for symbiotic or pathogenic fungi) (Fig. 1a)[49]. Given that fungal ecological success- reproduction and survival-highly relies on the hyphal growth[50], it is important to understand the microbiomes surrounding fungal hyphae, which directly affect fungal physiology. Hyphosphere is a specialized microhabitat directly influenced by fungal hyphae (Fig. 1b). Similar to plant root exudates in the rhizosphere, fungal exudates, including various carbohydrates (fructose, glucose, trehalose, etc.), organic acids (citric acid, succinic acid, oxalic acids, etc.), amino acids (aspartic acid, glutamic acid, leucine, etc.), phenolic compounds, and alcohols, shape soil microbiomes adjacent to hyphae[42]. These metabolites contribute to attracting and colonizing microbes distinct from those found in bulk soils, ultimately leading to the formation of specialized microbiomes known as the hyphosphere microbiome. Compared to the hyphosphere, the mycosphere is a broader space influenced by fungal mycelia (a mass of branching, thread-like hyphae). The hyphosphere was dealt with as the mycosphere, since previous studies on mushroom microbiomes did not examine the microbiomes in soils collected from extraradical hyphae.
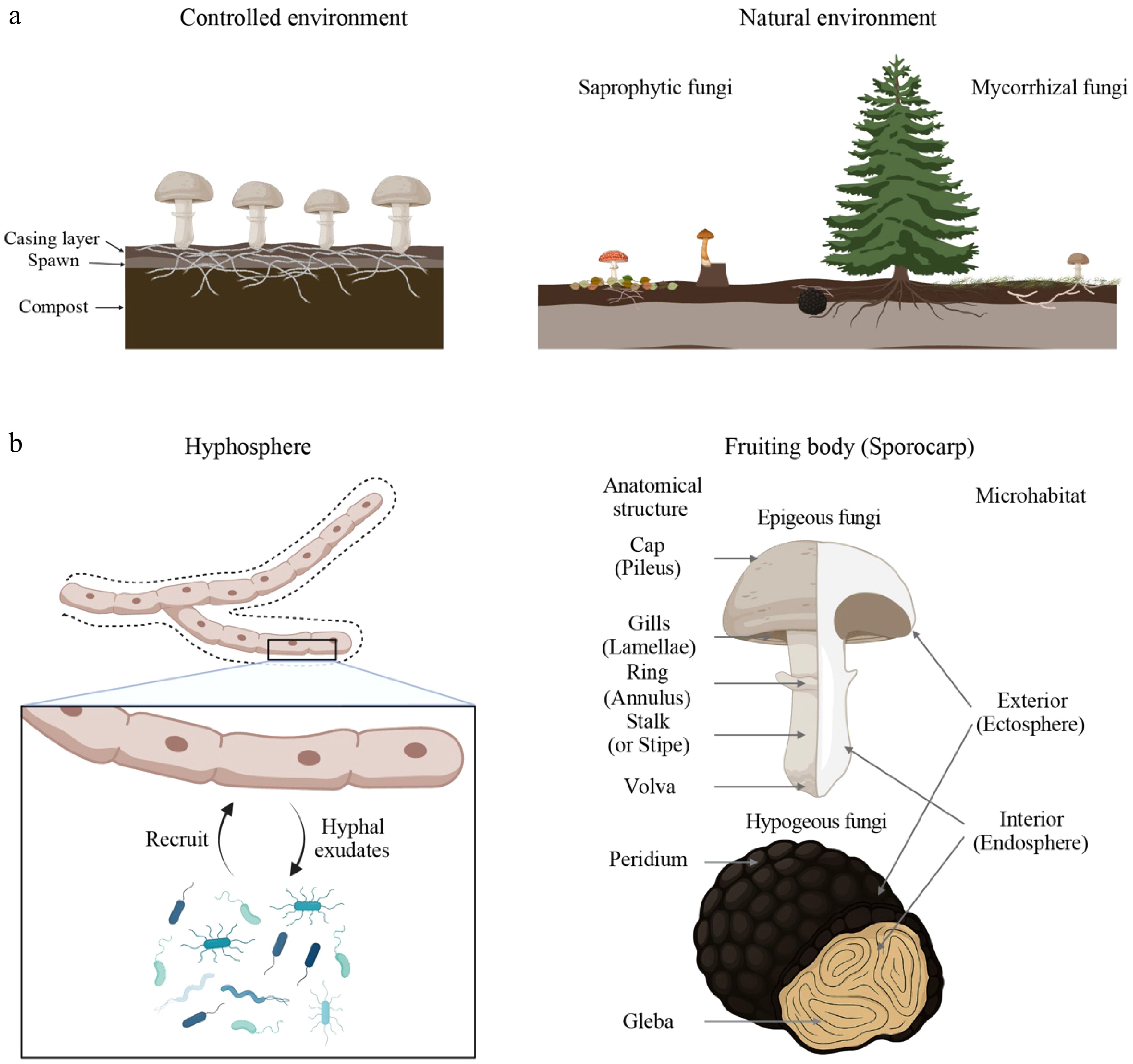
Figure 1.
Macro- and micro-habitat structures of mushroom-forming fungi. (a) Macrohabitats in which mushroom-forming fungi reside. Cultivated mushroom fungi, such as Agaricus bisporus, are generally grown under the standardized conditions consisting of compost soils, spawn, and casing layers (left panel). In contrast, most wild mushroom-forming fungi naturally inhabit forest environments, where saprotrophic species are found in the litter and humus layers, and mycorrhizal species associate with the root zones of host plants (right panel). (b) Microhabitats where mushroom fungi-associated microbiomes are generally found. The hyphosphere is the soil region affected by fungal hyphae (left panel; space indicated with the dashed line). Mushroom fungi secrete hyphal exudates acting as nutrient sources or signal molecules to attract microbes that can provide nutrients or protect fungi from other harmful microbes, similar to the plant microbiomes in the rhizosphere soils. Another microhabitat is the fruiting body (sporocarp). The distinguished anatomical structure of the fruiting bodies is depicted in the right panel. Mushroom-associated microbiomes could be found on the outer layer of the fruiting bodies (ectosphere) as epiphytes or inside them (endosphere) as endophytes. The figure was created in BioRender (
https://biorender.com ).Bacteria Previous studies have mainly examined the microbiomes in the mycosphere (Supplementary Table S1). Previous studies conducted under natural conditions, such as forests, reported that the mycosphere of mushroom-forming EcMF and saprophytic fungi (SAP) is typically dominated by Proteobacteria (present Pseudomonadota), Acidobacteriota, and Actinobacteriota (present Actinomycetota), although the abundances varied with fungal species, sampling sites, and sampling points[44,51−54]. Previous studies commonly reported that mycosphere soils host distinct microbiomes with fungal species-associated taxonomic variations, compared to bulk soils or soils where mushroom-forming fungi were less colonized. For example, Actinomycetota and Bacteroidota were generally more abundant in the mycosphere soils[51]. In the shiro of Tricholoma matsutake, Firmicutes (present Bacillota) were significantly higher, compared to the non-shiro soils[53]. Meanwhile, Pseudomonadota, Acidobacteriota, Planctomycetota, and Verrucomicrobiota were significantly higher in the mycosphere soil of Russula griseocarnosa[55]. At the genus level, however, Bradyrhizobium, Pseudomonas, Burkholderia (including Burkholderia-Caballeronia-Paraburkholderia), Mycobacterium, Bacillus, and Paenibacillus were mostly identified as the bacterial genera enriched in the mycosphere or similar soil environments, irrespective of geographical regions and fungal species[44,51−54]. Considering that taxonomically close bacteria normally share functional characteristics, the taxonomic similarity in the enriched bacterial genera across various mushroom-forming fungi suggests the commonality in the metabolic demands of fungi under natural conditions.
Similar to the mycosphere in natural conditions, the microbiomes in compost and casing where mushrooms are artificially grown have been studied. Compost, a product of the natural microbial breakdown processes of substrate materials (rice straw, manure, etc.), is a nutrient medium where fungal mycelia are grown[56]. Casing is a cover material, including peat and calcium carbonate, for fruiting body formation[57]. Several studies identified bacterial communities inhabiting compost or casing soils, as the artificial bed materials act like soil environments. In compost, typically, Pseudomonadota (Pseudomonas), Actinomycetota, and Bacillota (Bacillus) were abundant during and after the mycelium colonization in the compost of Agaricus bisporus, A. subrufescens, and Pleurotus ostreatus[58−61]. Meanwhile, Pseudomonadota (Pseudomonas), Bacteroidota (Flavobacterium), Bacillota (Bacillus), and Actinomycetota dominated the bacterial communities in the casing layer, irrespective of casing materials, cultivation conditions, and even fungal species[62−65]. Surprisingly, the compost and casing microbiomes showed higher compositional homogeneity regardless of the fungal species, compared to the mycosphere in the natural conditions. Previous studies suggest that the observed homogeneity may be driven by substrate-driven filtering during the compost fermentation (microbes able to degrade and utilize recalcitrant organic matter are selected), facility standardization (leading to the homogeneous external environments), and interactions between mushroom-forming fungi and microbes[58,66−68].
Fungi Most studies of mushroom microbiomes focus on bacterial communities, while fungal communities associated with mushrooms or individual fungi have received less attention. This may be due to the predominance of the mushroom-forming fungi under natural and controlled conditions. Previous studies on T. bakamatsutake and T. matsutake have shown that the fungus outcompetes other fungi, resulting in a decrease in fungal richness and diversity[53,54] (Supplementary Table S1). This phenomenon has also been reported in another ECM fungus, Russula griseocarnosa[69]. The fungal communities of the compost and casing layers also showed the dominance of a mushroom-forming fungus[58,60,61,70] (Supplementary Table S1). Despite the predominance of mushroom-forming fungi, a few fungi are frequently co-detected. For example, in the mycosphere of Tricholoma spp., Mucoromycota (Umbelopsis and Mortierella), and Ascomycota (Penicillium) were frequently found in conjunction with each other[52−54]. This suggests that these minor fungi, which survive from competitive exclusion, may provide additional or complementary metabolic activities to the microbial members, including the predominant fungus, potentially contributing to the stability of microbial associations. Moreover, the initial fungal community composition in natural soils, compost, and casing layers has the potential to predetermine mushroom productivity and yield, as suggested in the study of the bacterial community[61]. Previous studies reported that the initial compost and casing layers harbor diverse fungal members, including Lecanicillium, Mycothermus, Thermomyces, Collectotrichum, Pichia, Trichoderma, Fusarium, Cladosporium, and Mortierella[65,71]. Although these fungi are outcompeted by the mushroom fungi, their debris can act as necromass, and their metabolites can positively or negatively influence subsequent colonization of the mushroom fungi. Considering these ecological and agricultural aspects, the fungal communities in the initial compost, casing layer, and even natural soils need to be examined as extensively as the bacterial ones.
Fruiting body (or sporocarp)
-
The fruiting body or sporocarp is the most important fungal structure for reproduction, leading to the persistence of their populations. Depending on fungal species, fruiting bodies can be dissected into several anatomical structures. The fruiting bodies of many Basidiomycota fungi consist of a cap (also known as pileus), a hymenophore (such as gills/lamellae, pores, or teeth), and a stalk (or stipe), with some species, including Amanita muscaria, also possessing a ring (or annulus) and/or volva. In contrast, hypogeous fungi (e.g., truffles), which have a closed fruiting body, possess different structures called peridium (outer wall) and gleba (interior spore-bearing tissue) (Fig. 1b). Similar to the seeds and reproductive tissues of plants, which are colonized by diverse microbes[72−74], fungal fruiting bodies are not sterile but form additional microhabitats or niches for microbes. In this section, we will address previous findings of the fruiting body (or sporocarp) microbiomes of mushroom-forming fungi.
Bacteria Compared to soil microhabitats, including bulk and mycosphere soils, fruiting body microbiomes are more simplified[75,76]. Pent et al.[75] suggested that the differences in chemical properties, including pH and nutrient levels, may be involved in the diminished diversity in the fruiting bodies. Typically, bacterial microbiomes associated with fruiting bodies are frequently represented by Pseudomonadota, Actinomycetota, Bacteroidota, Bacillota, and Acidobacteriota[45]. These fruiting body microbiomes are known to be derived from external soil environments, both in natural and controlled conditions[44,46,64,75,76]. Among them, Pseudomonadota has been identified as the most abundant bacterial phylum, though variations exist depending on the fungal species, environmental conditions, and fungal lifestyles[44,47,77,78]. In particular, the bacterial genera Bradyrhizobium, Burkholderia, Pseudomonas, Acinetobacter, Sphingomonas, and Massilia are frequently found in the fruiting bodies (Supplementary Table S2). This taxonomic similarity suggests similar factors enriching specific groups of bacteria.
Fungi The fruiting body can also host other fungal members, although limited information is available. In the sporocarps of wood-decaying fungi, fungi belonging to Ascomycota (Helotiales and Hypocreales), Basidiomycota (Atheliales and Cantharellales), and Mucoromycota (Mucorales and Umbelopsidales) are detected with species-dependent variations[79]. A previous study of T. magnatum and T. macrosporum demonstrated that yeast-like fungi, including Geotrichum and Diutina, are commonly distributed on the surface and in the gleba of the truffle fruiting bodies of both fungi[80] (Supplementary Table S2). As in bacteria, the compartments of fruiting bodies also influence the fungal community composition[46,80]. Compared to the mushroom-forming fungi in natural environments, such information is barely available in those commercially grown in controlled facilities. While most fungal community analyses of mushroom cultivation systems focus on compost or casing layers, recent metagenomics and functional gene profiling studies are beginning to reveal subtle but potentially important differences in the low-abundance fungal taxa present in different tissues of fruiting bodies[79,81]. The exceedingly low abundance of non-host fungi in fruiting bodies, except for notable pathogens or contaminants, limits the detection and ecological analysis of the fruiting body-associated fungal communities—underscoring the need for high-resolution, multi-omics or targeted approaches in future work.
Factors influencing mushroom microbiomes
-
As reported in other macroorganism-associated microbiomes, the composition and diversity of the mushroom microbiomes are influenced by external environments and host factors, such as endogenous genetic differences (Fig. 2). Biogeography and the external environment are the shaping factors of the mushroom microbiome composition and diversity. Ge et al.[82] showed that the bacterial composition in both the mycosphere and fruiting bodies of Cantharellus cibarius is shaped by geographic differences. In particular, the geographic differences in soil chemical factors, such as available nitrogen and total phosphorus levels, are significantly involved in the observed compositional variations of the mycosphere bacterial communities[82]. Despite the geographic differences in the fruiting body chemistry in the original study, we found that the total nitrogen (FTN) and phosphorus levels (FTP) in the fruiting bodies were significantly varied depending on the sampling sites (one-way ANOVA: FTN, p = 0.0305; FTP, p = 0.00108), when we analyzed it using a publicly available dataset of the work by Ge et al.[82]. Combined with the previous findings, this suggests that geographic differences may induce the variations in fungal properties, influencing the composition and assembly of fruiting body microbiomes. Considering that chemical environments and surrounding microbiomes influence each other interactively, the combined effects of both factors should be considered.
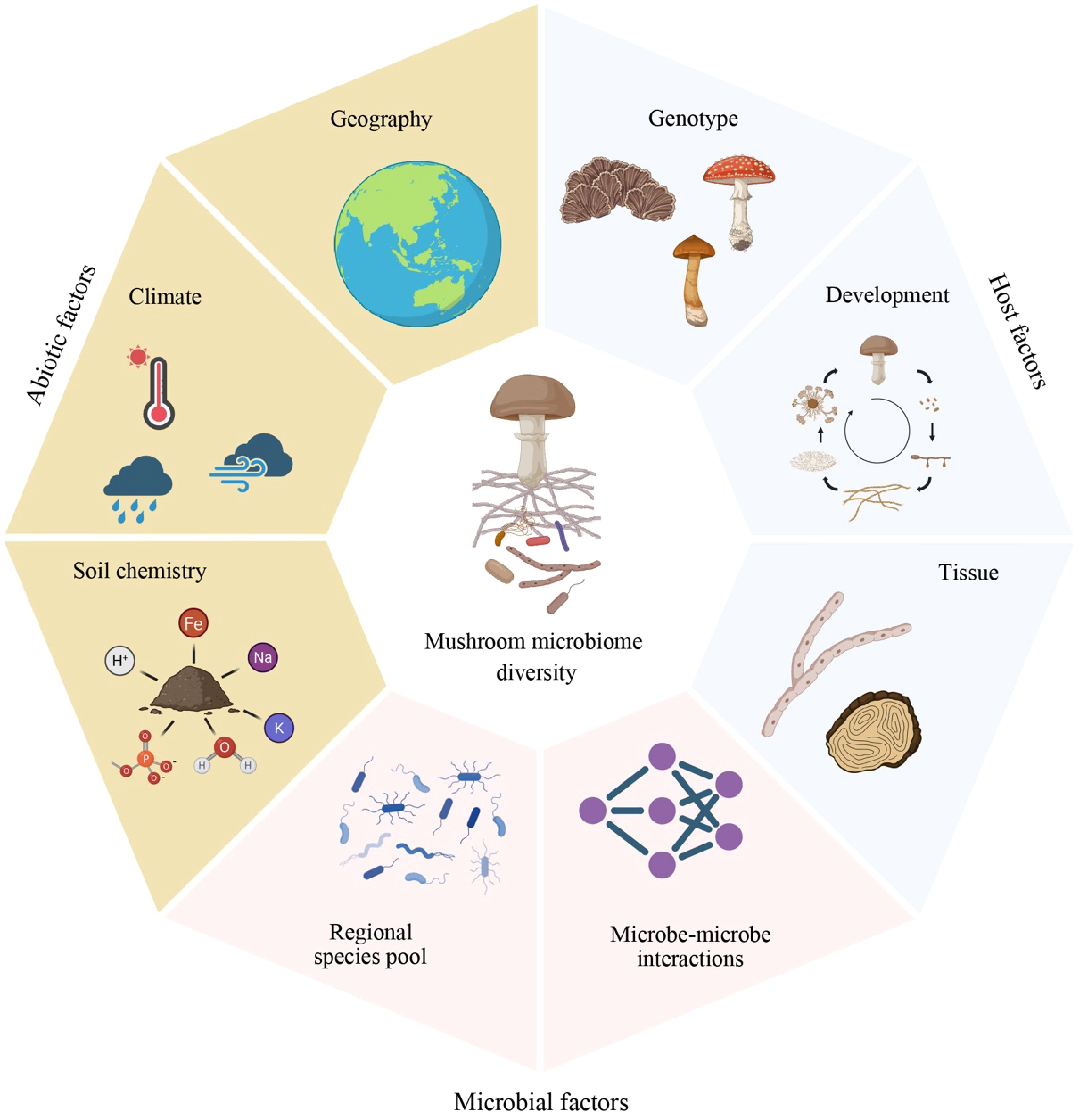
Figure 2.
Abiotic and biotic factors influencing mushroom microbiome diversity. Microbiomes associated with mushroom-forming fungi are shaped by abiotic (brown-filled cells), host-related (blue-filled cells), and microbe-associated factors (red-filled cells). Although each factor is depicted separately, these factors are tightly linked to each other. The figure was created in BioRender.
External environments, such as soil, atmosphere, hydrosphere, and other micro- and macro-organisms, act as regional microbial pools, where fungicolous bacteria and fungi originate. Previous studies repeatedly reported that fruiting body microbiomes are derived from nearby soil[46,47,75,77,79,80], and casing layer microbiomes[56]. Compared to macroorganism-associated microbiomes, mushroom microbiomes derived from external environments, except for soils, have received less attention. Previous studies of pathogenic fungi infecting mushrooms showed that fungal taxa, such as Trichoderma and Aspergillus, to which known mushroom pathogens belong, are found in the indoor atmosphere of Lentinula edodes cultivation factories[83,84]. Considering the facts that external environmental microbiomes form surface microbiomes, including skin[85] and phylloplane[86], a holistic exploration of the whole mushroom microbiome, including microbiomes in or on mushroom-forming fungi and in the external environment where the fungi encounter, is necessary.
Host factors, such as host genetic variations, host tissues (or compartments), and growth and development, also significantly affect associated microbiomes. By minimizing environmental variations, Maurice et al.[79] revealed that fungicolous fungal community composition and diversity were primarily shaped by the host species. The life strategies of mushroom-forming fungi (EcMF and SAP) also influence community composition and diversity[79]. Specifically, using fungal-fungal cooccurrence patterns, long-lived EcMF are generally associated with specialists, which show host specificity. Meanwhile, short-lived SAP showing higher fungal diversity than EcMF form significant co-occurrence connections with generalists, which are associated with multiple host fungi[79]. Maurice et al.[79] proposed that physical durability (fleshy and soft with high water content in short-lived SAP vs resistant with low water content in long-lived EcMF) and chemically distinct features, including secondary metabolites, of the fungal fruiting bodies play essential roles in determining fungicolous fungal diversity and community structures. Similar to fruiting body microbiomes, the fungal species-driven discrepancy of the exudates leads to the differentiation of mycosphere microbiome structures and diversity. While such studies in mushroom-forming fungi remain limited, this pattern has been clearly demonstrated in AMF. For instance, Zhou et al.[87] used a split-root system to inoculate different AM fungal species (Funneliformis mosseae and Gigaspora margarita, or Rhizophagus intraradices and G. margarita) onto a single plant root system under controlled conditions, demonstrating that different AM fungal species recruit distinct hyphosphere microbiomes with different predicted functional potentials through species-specific hyphal exudates. Species-level differences in the hyphal exudates of Rhizophagus clarus and R. irregularis under P starvation conditions, including sugars, amino acids, and amines, have also been proven using metabolite profiling[88]. These previous findings suggest that hyphal exudates are likely to be different in other EcMF or SAP fungi, driving species-level differences in bacterial and fungal community compositions. To prove this speculation, further in-depth experiments combined with meta-omics and metabolomics under controlled or semi-controlled conditions are necessary. Collectively, host fungal species-associated selection pressure, such as physically and chemically driven variations in niche environments, leads to the differentiation of host-associated microbiomes in diverse mushroom-forming fungi.
As in plants, compartments or host tissues are key factors resulting in microbial variations within each host. Liu et al.[81] reported that fruiting body compartments of Tuber indicum contribute to the bacterial and fungal microbiome differentiation. In detail, from bulk soil to gleba, microbial diversity and the complexity of microbial associations are significantly decreased. A total of four bacterial phyla, including Rokubacteria, Nitrospirae, Chloroflexi, and Planctomycetes, are excluded from the gleba. Based on these findings, the authors suggested that niche-based selection occurs during the microbial colonization inside and outside of the fungal fruiting body. This compartment effect has also been verified in other Tuber spp.[80]. The peridium may act as a primary physical barrier restricting microbes from indiscriminately invading the fruiting body, as the rhizoplane of plants' roots does[89]. Bacterial isolates obtained from the surface-sterilized fruiting body of T. matsutake, belonging to Paenibacillus, Serratia, and Stenotrophomonas, possess fungal cell wall-degrading enzymes, including chitinase[90]. The presence of the chitinase has also been reported in the endobacteria that colonize the interior of fungal hyphae[91]. These findings suggest that bacteria that possess the ability to break the physical obstacle may be selected first, and other bacteria that do not have the ability may use the pre-built gate. After the invasion, changes in niche conditions may contribute to microbial selection. A recent advance suggests that changes in nutrient status between the soil (nutrient-poor condition) and the fruiting body (nutrient-rich condition) may contribute to microbial selection in the fruiting bodies[79]. To colonize in the interior of the fruiting body, furthermore, microbes should evade fungal defense systems[92,93], such as antimicrobial peptides, phenolic compounds, toxins, etc., which are accumulated in the fruiting body. Given that some endobacteria possess type II (T2SS) and type III secretion systems (T3SS) to release effectors, fungicolous bacteria and fungi in the fruiting body may also have a similar ability. How endofungal microbes in the fruiting bodies can evade these fungal defenses is another interesting question to be addressed in future work.
Functional landscape of mushroom microbiomes
-
Understanding microbial functions at both individual and community levels is crucial for understanding why the observed microbiome structures are assembled and further developing microbiome-driven or engineered management systems. To uncover the functional properties of microbiomes in diverse environments, culture-dependent approaches and meta-omic tools are extensively introduced and applied. Despite the limitations of culturing natural microbes, culture-dependent approaches have revealed that fungicolous bacteria and fungi influence host fungal growth and development, including fruiting body formation, via biochemical interactions. For example, A. bisporus can produce self-inhibitory volatile compounds, such as 1-octen-3-ol or 2-ethyl-1-hexanol[94]. Pseudomonad populations consisting of Pseudomonas putida, P. veronii, and P. poae metabolize these compounds, promoting primordium formation as well as vegetative growth[95] (Fig. 3). Previous studies also revealed that the mycelial growth of the mushroom fungus can be promoted by mycelium growth-promoting bacteria via removing ethylene with the 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase activity[94,96]. N-fixing Bradyrhizobium japonicum improved mycelial growth and protein contents in P. osteratus[97]. Apart from removing mushroom-inhibitory compounds and providing nutrients, some mushroom fungi-associated microbes are reported to improve the growth of mushroom fungi by inhibiting mushroom-pathogenic fungi, such as Trichoderma spp. Previous studies reported that Bacillus spp. can hinder the colonization of Trichoderma spp. by producing antifungal lytic enzymes, such as protease, chitinase, and cellulase, and metabolites, including siderophore and HCN[98,99]. Similar to artificially cultivated mushroom fungi, microbial functions similar to those in mushroom-forming fungi have been proposed in natural habitats. A culture-dependent study showed that bacterial isolates obtained from the fruiting body of T. matsutake, belonging to Paenibacillus, Serratia, and Stenotrophomonas, possess fungal cell wall-degrading enzymes, including chitinase[90]. Some isolated bacteria belonging to Ewingella, Pseudomonas, and Serratia further showed antifungal activities against other fungi, such as Mucor, Penicillium, and Sarocladium, under in vitro conditions, although exact mechanisms were not addressed[90]. Truffle-associated culturable bacteria taxonomically affiliated to Mesorhizobium, Klebsiella, Ochrobactrum, Acinetobacter, and Arthrobacter have also been reported to show N-fixing or P-solubilizing activity[100], proposing the potential to improve nutrient accessibility of mushroom-forming fungi under natural conditions. Specifically, these bacteria showing beneficial effects on the growth and development of mycorrhizal fungi are called 'mycorrhiza helper bacteria (MHB)'. Among the mushroom-forming mycorrhizal fungi, the Laccaria bicolor-Pseudomonas fluorescens association model has shown molecular mechanisms underlying the fungal-bacterial interactions. The MHB P. fluorescens alters the expression of the fungal genes represented by the upregulation of transport and protein synthesis-related genes and downregulation of stress response and post-translational modification-related genes[101]. Deveau et al.[102] revealed that L. bicolor accumulates trehalose in its mycelia, attracting P. fluorescens and promoting the growth of the bacterial population, while the bacterium produces thiamine, which improves the fungal growth (Fig. 3). Cusano et al.[103] further showed that the T3SS of P. fluorescens is necessary for promoting the mycorrhizal formation of L. bicolor. These findings suggest that the molecular mechanisms underlying mycorrhizal-bacterial interactions should be further explored through integrated approaches combining genomics, metabolomics, and ecological studies. Apart from these MHB activities, recent evidence shows the potential contribution of fungi to the biology of the mycorrhiza. Some microfungi, which belong to Mortierella, Mucor, Penicillium, and Umbelopsis, isolated from the fairy ring of T. matsutake promoted the mycelial growth of T. matsutake under in vitro conditions[104]. Another study also showed that P. citreonigrum could promote the β-glucosidase activity of T. matsutake, suggesting the improved cellulose-degrading ability of the host fungus by adjacent microfungi[105]. However, the compounds produced by P. citreonigrum that are involved remain unclear. Although mushroom-forming fungi generally predominate their habitats after colonization, microfungi research in T. matsutake habitats suggests the need to explore the functional properties of fungi associated with mushroom-forming fungi.
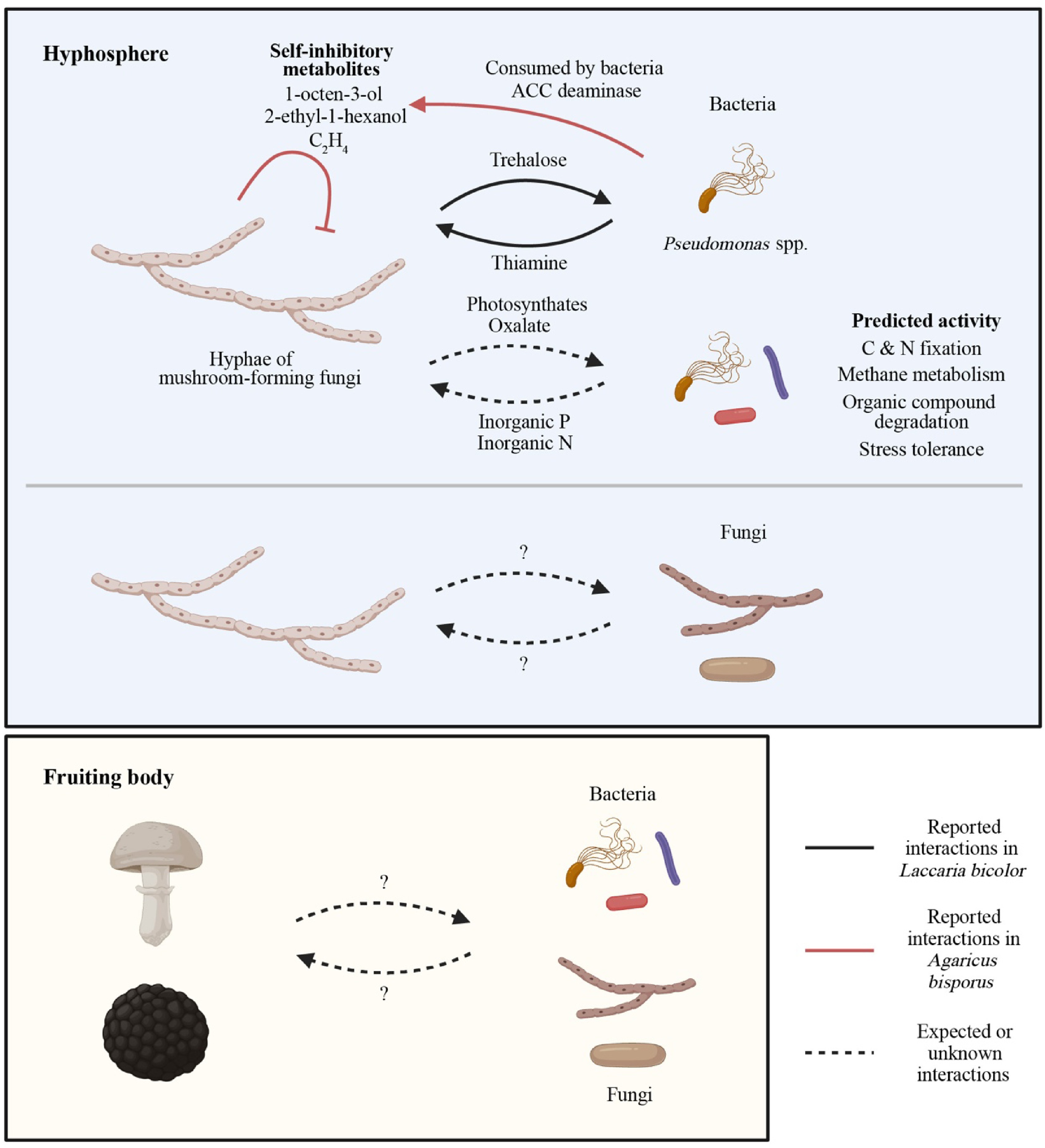
Figure 3.
Functions of mushroom-forming fungi-associated microbiomes. The functions of mushroom microbiomes have been investigated using culture-dependent and independent methods. Culture-dependent studies revealed that some bacteria, including Pseudomonas fluorescens, remove self-inhibitory metabolites produced by mushroom-forming fungi, such as Agaricus bisporus, improving the fungal growth and fruiting body formation. In addition, the Laccaria bicolor-P. fluorescens model demonstrated that fungal trehalose attracts the bacterium, which can produce thiamine required for fungal growth. Shotgun metagenomics, marker gene-based functional prediction, and metabolomics have further expanded our knowledge of the roles of mushroom microbiomes. A metabolic approach showed that mushroom-forming ectomycorrhizal fungi can provide plant-derived carbohydrates (photosynthates) to nearby soil microbes. The general functionality of the hyposphere bacterial microbiomes, including C and N fixation, methane metabolism, degradation of organic compounds (chitin, cellulose, peptidoglycan, aromatic compounds, etc.), and stress tolerance, has been reported. In particular, from the findings of arbuscular mycorrhizal fungi-associated hyphosphere microbiomes (the provision of inorganic P by hyphosphere microbiomes), it is expected that microbes associated with mushroom-forming fungi may show similar functional roles. On the other hand, the knowledge of biological mechanisms governing the interactions between a mushroom fungus and other fungi in the hyphosphere or adjacent bacteria and fungi inhabiting the fungal fruiting bodies remains lacking, although some studies showed that they promote host fungal growth under in vitro culture conditions. The figure was created in BioRender.
To date, to broaden our understanding of microbial communities' functions in a particular environment, meta-omics-based approaches have been extensively applied. Although such research in the mushroom microbiome field remains insufficient compared to other microbiome fields, some functional features of mushroom microbiomes have been uncovered. Liu et al.[60] revealed the successional dynamics of the compositions and functions of the compost microbiome of P. ostreatus using shotgun metagenomics. In particular, during the mushroom cropping, bacterial functions related to glycine metabolism, carbon fixation, methane metabolism, and degradation of recalcitrant compounds, such as cellulolytic, hemicellulolytic, chitinolytic, and peptidoglycanlytic enzymes, were enriched in the short composting substrate conditions[60]. Another metaproteomic approach of Tuber melanosporum-inhabiting soils showed that microbial functions involved in abiotic stress responses, organic compound degradation, and carbohydrate and sulfur metabolisms are enriched compared to non-inhabiting soils[106]. In addition to the meta-omics-based findings, marker gene-based functional prediction has also provided insights into the functional properties of mushroom fungi-associated microbiomes. Oh et al.[53] showed that bacterial functions involved in amino acid uptake, carbohydrate metabolism (two-component regulatory system), and T3SS are more abundant in T. matsutake-dominant soils compared to T. matsutake-minor soils. Such an approach was also applied in the T. bakamatsutake microbiome study. In the soil where T. bakamatsutake was colonized, bacterial genes linked to glucan and chitin degradation, fatty acid and beta oxidation, and stress tolerance, as well as increased chemoheterotrophy and aromatic compound degradation were abundant, while more N-fixation-related functions were less abundant[54]. Although these studies mainly rely on gene abundance predictions, which do not always directly translate to active functions, they collectively suggest that bacterial microbiomes associated with mushroom-forming EcMF and SAP possess stress-tolerant traits and may perform specialized saprophytic activities. These functions could support nutrient scavenging from organic substrates, potentially aiding host fungi in nutrient acquisition. The finding that AMF-associated hyphosphere bacteria can solubilize inorganic P, which the fungi consume, with phytases or phosphatases[38,107] suggests the similar roles of EcMF-associated hyphosphere bacteria. A recent study reported that oxalotrophic bacteria in the ectomycorrhizosphere (soil space affected by ectomycorrhizal fungi) contribute to P mobilization under P-deficient conditions, and their activity was promoted with the exogenous oxalate[108]. It was proposed that EcMF may secrete oxalate as a part of hyphal exudates to promote the P-solubilizing activity of oxalotrophic bacteria; meanwhile, the bacteria provide inorganic P to the fungi[108] (Fig. 3). Compared to bacteria, community-level functional traits of the fungi associated with mushroom-forming fungi remain poorly explored. Given culture-dependent study findings, microfungi associated with mushroom-forming fungi might play yet underappreciated ecological roles, highlighting an important avenue for future research. To gain a comprehensive understanding, more meta-omics-based, large-scale studies are warranted to reveal common functional characteristics of mushroom microbiomes across diverse host fungi and geographical regions. This knowledge can aid in developing microbiome-informed cultivation strategies for mushroom fungi that are difficult to cultivate artificially.
-
Although a deeper understanding of bacteria and fungi associated with mushroom-forming fungi is still necessary, a more holistic framework that incorporates microbial components across domain of life in the environments of mushroom-forming fungi, functional metabolic analyses, and predictive modeling approaches is essential to broaden our knowledge of mushroom-forming fungi and surrounding microbes at the community and ecosystem levels. In this section, we propose a conceptual framework that can unravel ecological mechanisms underlying mushroom–microbiome interactions and ultimately guide the design of stable, productive microbial communities for sustainable mushroom cultivation.
Phage community, a significant component in environmental microbiomes
-
Phages are key members of environmental microbiomes. They modulate bacterial populations through direct lysis[109], contributing to carbon and nutrient recycling via the viral shunt[110]. Phages also influence ecosystem functions by transferring auxiliary metabolic genes (AMGs), which enhance host metabolic capacities, to their hosts through lysogeny[111]. Thus, phages affect processes such as nutrient cycling, energy flow, and stress responses in soil ecosystems. The importance of phages was initially recognized in aquatic environments[112], such as oceans, where the availability of soluble nutrients is limited. More recently, increased attention has been directed toward the role of phages within terrestrial ecosystems[113,114]. Considering that terrestrial fungi, including mycorrhizal fungi, are associated with soil bacteria, phages are likely to play critical roles in those terrestrial fungi-mediated microbial associations. The latest theoretical advancement emphasized the importance of phages in mycorrhizal symbiosis systems with three hypotheses: (1) leaky goods hypothesis (phage lysis releases nutrients that enhance mycorrhizal-plant symbioses through direct nutrient provisioning or indirect pathogen suppression); (2) auxiliary recruitment hypothesis (lysogenic conversion of MHB introduces AMGs that promote mycorrhizal symbioses); and (3) phage attenuation hypothesis (phage-mediated lysis of MHB weakens mycorrhizal symbioses, but may increase bacterial diversity with potential indirect benefits)[115] (Fig. 4). Together, these hypotheses suggest that phages can influence nutrient accessibility for mycorrhizal fungi and nearby plants, while also regulating the population dynamics and metabolic capacities of MHB. As described in the above sections, mushroom-forming fungi are also intimately associated with diverse bacteria, which influence fungal growth and fruiting body formation, under natural and controlled conditions. It can be inferred that similar phage-mediated mechanisms are likely to occur in the mushroom fungi-microbiome association systems. Nutrient leakage from phage lysis could directly or indirectly influence fungal nutrient acquisition, while lysogenic phages may shape microbial functional networks through AMG transfer. Despite these plausible interactions, phage dynamics and their ecological functions remain largely unknown in mushroom-forming fungal systems. Phage research in the mushroom-forming fungi is mainly limited to the phage therapy against bacterial pathogens, such as Pseudomonas tolaasii, a causal agent of brown blotch disease[116,117]. A deeper understanding of phage–bacteria–fungus interactions will provide new insights into nutrient cycling, fungal physiology, and potentially offer innovative strategies for improving the stability and productivity of mushroom-associated ecosystems.
Microbial community-scale metabolic modeling
-
With the advancement of meta-omics, particularly shotgun metagenomics, corresponding bioinformatic methodologies, such as metagenome assemblers, metagenome-assembled genome (MAG) binners, taxonomic classifiers, and gene prediction and annotation tools, are being improved. One of the emerging methodologies is the metabolic modeling based on genomic information, called a genome-scale metabolic model (GEM) (Fig. 4). GEMs are built by coupling an annotated genome to a database of known gene-transcript-protein-metabolic reaction associations[118,119]. GEMs are potent tools for predicting and analyzing metabolic flows (or fluxes) and identifying critical metabolic pathways affecting microbial behavior under abiotic and biotic environmental changes[119]. Such models help researchers expand the knowledge of the physiology of individuals regardless of organism types[120−123], although there are hurdles to overcome for generating accurate and precise GEMs[119]. In particular, the advent of binning tools that enable obtaining high-quality MAGs provides promising opportunities to explore the metabolic potentials of each genome in a particular community[124]. Recently, researchers have aimed to understand microbial communities in diverse environments, including soils and human guts[125−128], by combining microbial GEMs at the community level (known as microbial community-scale metabolic models; MCMMs). Given that MCMMs are constructed by integrating each genome based on its metabolic capabilities and potential metabolic exchanges among GEMs, MCMMs are powerful tools for predicting metabolite-level microbial interactions and designing optimal synthetic microbial communities for further applications. In a human gut microbiome study, individual-specific short-chain fatty acid (SCFA) production profiles, which vary with dietary, prebiotic, and probiotic inputs and are essential as a health index, were successfully predicted using the MCMM, finally proposing a rational microbiome framework for potentially applying precision nutrition and personalized healthcare[127]. A similar approach was also performed in the plant rhizosphere research. Mataigne et al.[129] constructed a metabolic network using the MCMM, revealing metabolic dependencies and cooperation among bacteria contributing to compensating environmental constraints and maintaining co-existence in the complex Arabidopsis root microbiome. Thus, such previous studies have enhanced our understanding of microbes, their metabolic roles, and interactions in a particular environment. In this context, MCMMs of mushroom microbiomes will provide ecological and biological insights into metabolic interactions among mushroom-associated microbes under natural and controlled conditions. Compared to higher organisms (humans, animals, and plants), in particular, many mushroom-forming fungi have practical experimental advantages for genomic studies, including relatively smaller and less complex genomes, which facilitate the integration of host genomes into MCMMs. This integration enables the identification of microbiome-derived metabolites that support host fungal growth and development, the mapping of nutrient exchange between the fungi and fungicolous microbes, and the prediction of how microbial metabolic activities and interactions influence host fungal physiology. Collectively, MCMMs provide a predictive scaffold to translate genomes into metabolite exchange networks, enabling hypothesis-driven tests of how specific microbes and metabolites affect fungal growth, fruiting, and stress tolerance under natural and controlled conditions.
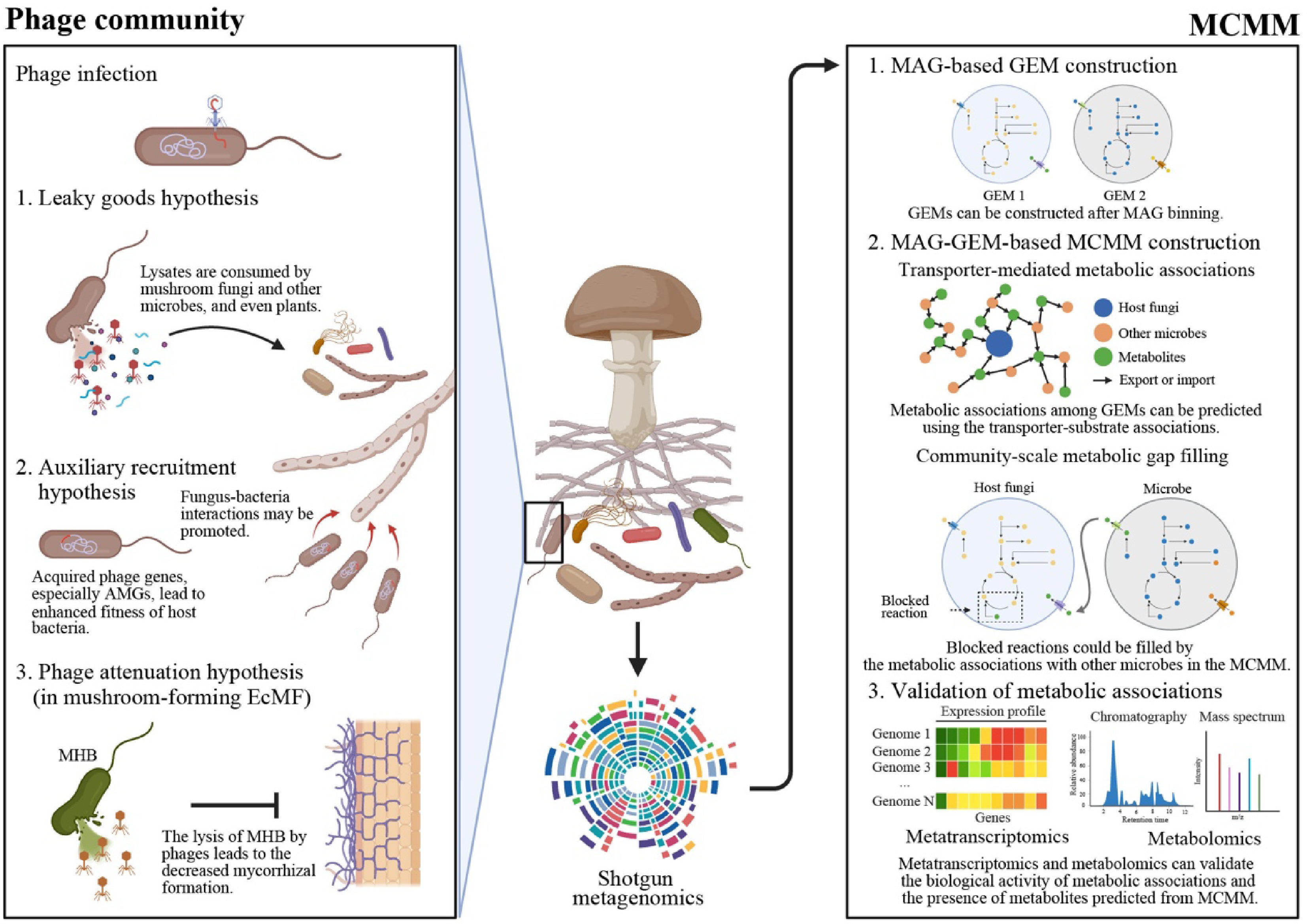
Figure 4.
The proposed holistic framework for a comprehensive understanding of mushroom-microbiome associations. To expand our knowledge of mushroom microbiomes, we proposed two agendas: phage communities (left panel) and microbial community-scale metabolic model (MCMM) (right panel). The phage communities are expected to contribute to the ecological functions of mushroom microbiomes by directly regulating bacterial populations and introducing auxiliary metabolic genes (AMGs) to bacterial populations. Phage-induced bacterial cell lysis can improve soil nutrient conditions by viral shunt (Leaky goods hypothesis). Furthermore, during lysogenic interactions, phage's AMGs can be introduced to bacterial hosts, leading to increased bacterial fitness and promoted associations between mushroom-forming fungi and bacteria (Auxiliary recruitment hypothesis). In case of mushroom-forming mycorrhizal fungi, bacterial lysis can lead to decreased mycorrhizal formation or diminished mycorrhizal activity (phage attenuation hypothesis). These putative phages' roles imply the necessity for researching phage communities in mushroom microbiomes, although thorough experimental approaches should be performed to prove or disprove the suggested hypotheses. Please note that the figure describing the phage-associated hypotheses (left panel) is modified from the artwork in by Berrios[115]. Please see this latest insightful forum article for more information on mycorrhizal fungi-bacteria-phage interactions. Another experimental approach is MCMM, a powerful tool for examining metabolic associations among microbes at the community level (right panel). From metagenome data, genome-scale metabolic models (GEMs) of each metagenome-assembled genome (MAG) can be reconstructed. Using these GEMs, potential cross-feeding interactions and transporter-mediated metabolic exchanges can be inferred, enabling the identification of putative key metabolites required for mushroom-forming fungi. Furthermore, community-scale metabolic gap-filling of GEMs, including host fungi, enables a mechanistic understanding of the observed microbial associations by revealing essential metabolic interdependencies and complementary pathways in the examined mushroom microbiomes. Finally, metatranscriptomics and metabolomics can help researchers assess whether the predicted metabolic interactions are biologically active and whether the predicted key metabolites are present in the samples. For more information of MCMM, please see a cutting-edge review article addressed by Quinn-Bohmann et al.[119]. The figure was created in BioRender.
-
The rise of high-throughput meta-omics, along with scalable bioinformatics pipelines and databases, has accelerated research on microbiomes in natural environments and host organisms. Recent advancements in understanding the associations between mycorrhizal fungi, plants, and microbiomes show that the traditional binary view of symbiosis needs to be expanded to consider community- and ecosystem-level interactions. Although there are practical challenges in studying mushroom-forming fungi and their microbial partners, this review suggests complementary approaches that will help researchers gain a better understanding of these fungi and their associated microbiomes. Achieving this agenda will require integrated designs: compartment-resolved, longitudinal sampling of the hyphosphere and fruiting bodies; strain-resolved meta-omics connected to metabolomics; isolation and synthetic community experiments; and iterative model-experiment loops in which MCMM predictions are validated and adjusted. Ultimately, a thorough understanding of mushroom microbiomes can transition the field from mere description to practical intervention. This knowledge can inform the design of stable, productive, and resilient microbial consortia for sustainable mushroom cultivation, especially in the context of climate variability, while also advancing our understanding of symbiosis and ecosystem functions.
-
Not applicable.
-
The authors confirm their contributions to the paper as follows: systematic search for Supplementary Tables S1 and S2: Bae IH, Kim H; figure configuration: Kim H; draft manuscript preparation: Bae IH, Kim H, Lee YH. All authors reviewed and approved the final version of the manuscript.
-
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
-
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIT) (RS-2025-00512558 and RS-2023-00275965 to Lee YH and RS-2022-NR072199 to Kim H). Bae IH is grateful for a graduate fellowship through the Brain Korea 21 Plus Program.
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: In Hyup Bae, Hyun Kim
- Supplementary Table S1 Dominant taxa in the mycosphere microbiomes of mushroom-forming fungi.
- Supplementary Table S2 Dominant taxa in the sporocarp (fruiting body) microbiomes of mushroom-forming fungi.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Jilin Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Bae IH, Kim H, Lee YH. 2026. Microbiomes in mushroom-forming fungi. Panfungi 1: e002 doi: 10.48130/panfungi-0025-0003 

Microbiomes in mushroom-forming fungi
- Received: 03 November 2025
- Accepted: 17 December 2025
- Published online: 28 January 2026
Abstract: The microbiome is the integrated information on the composition, functional potential, and environmental determinants of a microbial community within a defined ecosystem. High-throughput omics technologies, cutting-edge bioinformatic tools, and extensive databases have accelerated microbiome research in diverse environments and host organisms. Over the past decade, microbial ecologists have explored microbiomes associated with macrofungi (including mushroom-forming fungi) as well as arbuscular mycorrhizal fungi. In this review, current knowledge on the taxonomic composition and functional properties of microbiomes associated with mushroom-forming fungi are summarized. Future research directions to deepen and expand our knowledge of mushroom fungi-microbiome interactions are further discussed. Collectively, this review provides a conceptual and empirical foundation for advancing mushroom microbiome studies, and for developing microbiome-based strategies and platforms to enhance mushroom production and manage environments.
-
Key words:
- Mushroom /
- Hyphosphere /
- Sporocarp /
- Microbiome