








-
The African tulip tree (Spathodea campanulata P. Beauv.), is a famous evergreen ornamental tree with flaming red corollas in the plant family Bignoniaceae (Fig. 1a). It originates from Africa[1] and is now widely cultivated in tropical and subtropical areas such as India, Sri Lanka, China, and other regions[2]. The African tulip tree is the sole species within the monotypic genus Spathodea[3]. It is a species with late-acting self-incompatibility[4], and grows at an optimal temperature range of 23 to 30 °C with a minimum temperature of 10 °C.
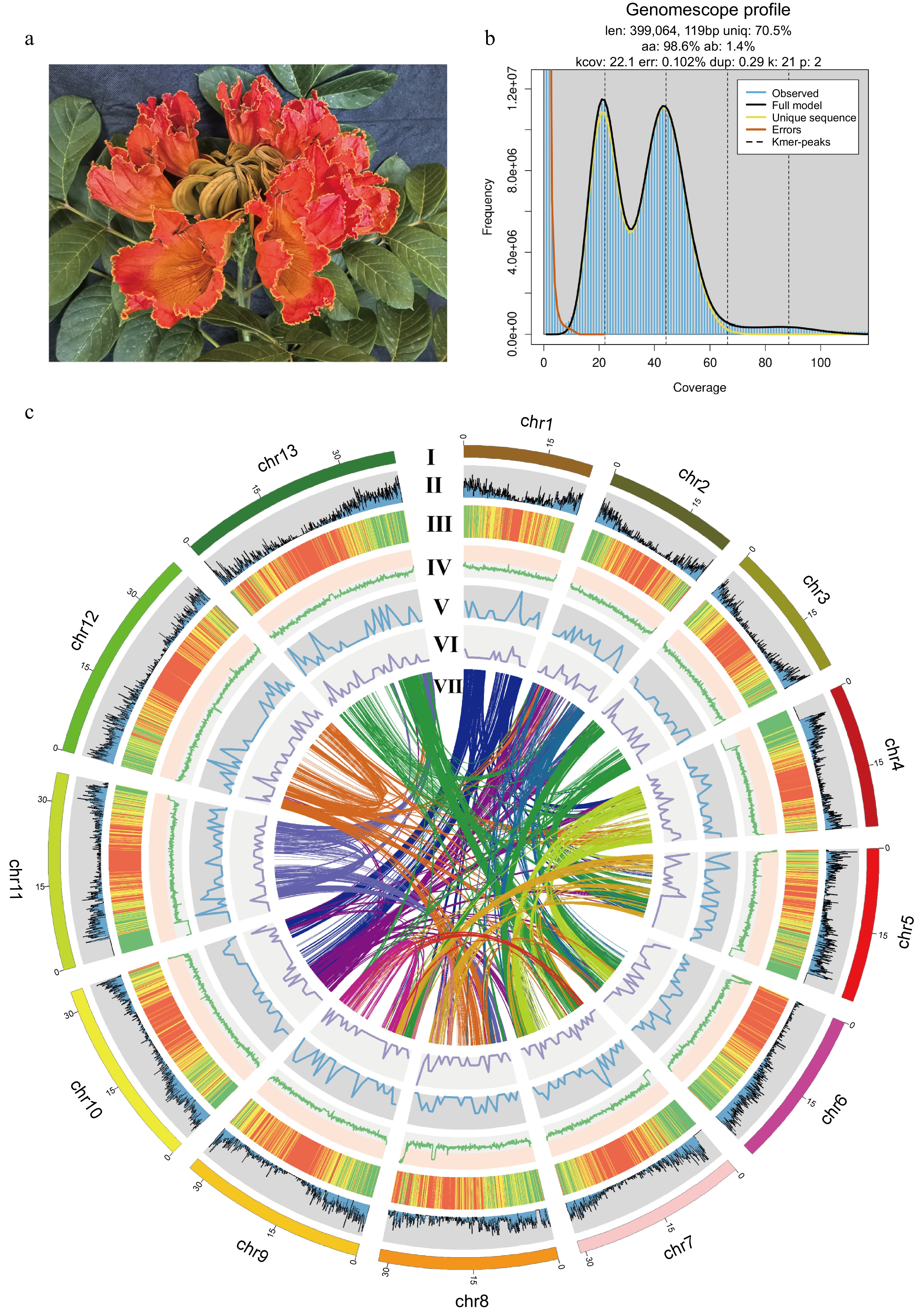
Figure 1.
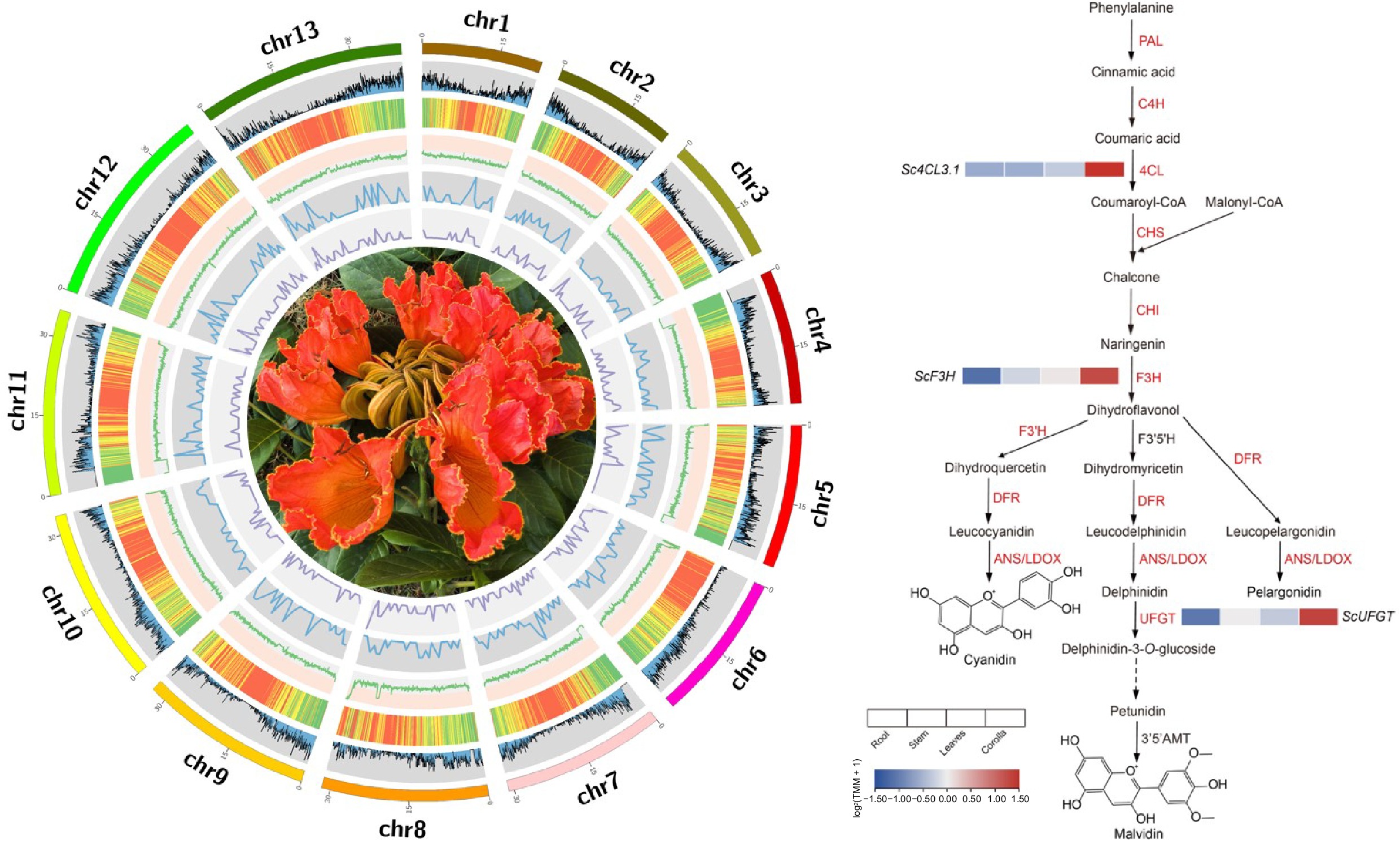
The genome of the African tulip tree. (a) Flower of the African tulip tree. (b) Distribution profiles of 21-mer analysis of short reads. (c) Circos plot showing the genome details. Labels I-VII indicate: (I) 13 pseudo-chromosomes of S. campanulata; (II) gene density; (III) repeat sequence content; (IV) GC content density; (V) density of Copia LTR-RTs; (VI) density of Gypsy LTR-RTs; (VII) syntenic blocks (all window sizes = 50 kb).
The African tulip tree also has certain medicinal properties and is traditionally used to treat and prevent various diseases. It is used in traditional herbal medicine for the treatment of ulcers, filariasis, gonorrhea, diarrhea, and fever[5−7]. The stems of the African tulip tree exhibit hypoglycemic, anticomplement, and anti-HIV activities[8]. In traditional Cameroonian medicine, it is believed to promote the healing of burn wounds[9]. An ethnobotanical survey also found that the fresh leaves of African tulip trees are often used to treat and manage cancer in Kakamega County, Kenya[10]. The leaves of African tulip trees have anticonvulsant activity, protecting mice from pentetrazole, strychnine, and electroshock-induced convulsions without exhibiting sedative effects, antipsychotic properties, or affecting motor coordination[11].
The flowers of African tulip trees are bell-shaped with a contracted base, scarlet or orange-red with bright-yellow margin and throat, large and showy, often in a corymbose raceme. The blooming canopy of the tree resembles burning flames, making it highly ornamental. This vibrant display is largely attributed to the presence of anthocyanins, a class of water-soluble flavonoid compounds widely found in plants. Anthocyanins are primarily located in the leaves, petals, and fruits, imparting rich colors such as red, blue, and purple[12]. Over 700 distinct anthocyanins have been characterized across diverse plant species[13], with cyanidin, pelargonidin, delphinidin, peonidin, petunidin, and malvidin being the most abundant[14]. Anthocyanins assist plants in adapting to diverse environmental conditions, attracting pollinators and seed dispersers, and protecting them from ultraviolet radiation, pest and disease infestations, herbivory, and low temperature stress[15−17]. Additionally, as powerful antioxidants and free radical scavengers, anthocyanins benefit human health by exhibiting anti-inflammatory activity, anticancer properties, and preventing cardiovascular and neurodegenerative diseases[18].
The biosynthesis and regulation of anthocyanins in plants is a complex process. Phenylalanines serve as precursors, undergoing a series of conversions catalyzed by enzymes such as phenylalanine ammonia-lyase (PAL), cinnamic acid 4-hydroxylase (C4H), 4-coumarate CoA ligase (4CL), chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), dihydroflavonol 4-reductase (DFR), and anthocyanidin synthase (ANS/LDOX). These enzymes lead to the formation of colorless dihydroflavonol and ultimately colored anthocyanidins. Transcription factors also play a crucial role in regulating anthocyanin biosynthesis. For instance, the R2R3-MYB transcription factor positively regulates the accumulation of anthocyanins and proanthocyanidins in strawberry by trans-activating specific genes[19]. The complex formed by PpBBX16 and PpHY5 stimulates the activity of the PpMYB10 promoter, enhancing light-induced anthocyanin accumulation. Overexpression of PpBBX16 also promotes anthocyanin accumulation in pear fruit peels[20]. MdNAC52 binds to the promoters of MdMYB9 and MdMYB11, promoting anthocyanin biosynthesis in apples[21]. Plant hormones also interact with these transcription factors, influencing the regulation of anthocyanin accumulation. In apples, it has been demonstrated that MdABI5 interacts with MdbHLH3, enhancing its transcriptional activity on MdDFR and MdUF3GT, and promoting anthocyanin biosynthesis under abscisic acid (ABA) treatment[22]. VvWRKY5 can directly bind to the W-box element in the promoter of VvLOX, a gene associated with jasmonic acid (JA) biosynthesis, thereby enhancing its activity and promoting the synthesis of anthocyanins in grapes[23].
Significant progress has been achieved in the research of anthocyanins in plants such as Arabidopsis, apples, and grapes, uncovering diverse molecular mechanisms that regulate their synthesis. However, in contrast, research on the anthocyanins of the African tulip tree remains relatively scarce. Despite existing reports indicating that the pigment components of the African tulip tree's corolla primarily consist of cyanidin, pelargonidin, and malvidin[24], no comprehensive analysis of the related metabolic pathways has been conducted from the genome and transcriptomic perspectives.
The plant family Bignoniaceae comprises 86 genera and 852 species of shrubs, lianas, and trees[25]. Its diversity, coupled with high similarity in reproductive morphology, once posed significant challenges in taxonomy[26]. Despite the publication of genome sequences for four species within the family: Jacaranda mimosifolia, Handroanthus impetiginosus, Jacaranda copaia, and Handroanthus guayacan, the overall lack of genomic data for this family still limits phylogenetic inference, which primarily rely on plastid markers (such as matK, ndhF, rbcL) or a limited number of nuclear regions, such as the multi-copy nuclear ribosomal internal transcribed spacer (ITS) or the low-copy gene PepC, at present[27−29]. While these markers provide a framework for phylogenetic studies at the family and infra-familial levels, the limited number of informative sites within the DNA regions traditionally used for phylogenetic reconstruction hinders a thorough understanding of phylogenetic relationships.
More genome sequences of Bignoniaceae are therefore needed to elucidate the phylogenetic relationships and evolutionary history of this family. Genome sequences are the cornerstone of genetic research and molecular breeding. As of now, there is no available genome sequence for the African tulip tree. The lack of a genome for the African tulip tree has hindered our ability to reveal the phylogenetic relationships and evolutionary history within the family Bignoniaceae, impeding in-depth understanding of the origin, evolution, and ornamental trait formation of the African tulip tree. Here, we report the first genome of the African tulip tree and explore the relationship between anthocyanin metabolism and the coloration of its flowers. Our goal was to contribute essential genome sequence data and features to foundational research on the African tulip tree because genome sequences are the foundation of genetics.
-
The material of the African tulip tree originates from the College of Breeding and Multiplication in Sanya, Hainan Province (China). Fresh tender leaves of the African tulip tree were collected for genome sequencing. Additionally, fresh roots, stems, leaves, and flowers of the African tulip tree were collected for transcriptome sequencing to prepare for genome annotation. Immediately after collection, the samples were snap-frozen in liquid nitrogen and stored in an ultra-low temperature freezer at −80 °C for subsequent nucleic acid extraction. High-quality genomic DNA was isolated from the young leaves using the CTAB method[30]. The genomic DNA was randomly fragmented, and a short-read library for the African tulip tree was constructed according to the TruSeq DNA Sample Preparation Guide, which was then run on the NovaSeq 6000 sequencing platform for paired-end sequencing, yielding 150 bp sequence reads. Furthermore, the HiFi library was constructed following the Revio platform protocol. The library was prepared for sequencing using the PacBio Binding kit, where primers and polymerase were attached to the library. The final reaction product was purified using AMpure PB Beads and then sequenced on the Revio system sequencer. Additionally, a Hi-C library was sequenced on the Illumina NovaSeq platform with 150 bp paired-end reads. Total RNA was extracted using CTAB, and RNA-seq libraries were constructed and sequenced on the Illumina NovaSeq platform with 150 bp paired-end reads on both sides. All sequencing data were filtered using fastp v0.23.2[31] software to obtain clean data for subsequent analysis.
Estimation of genome size by k-mer analysis
-
The k-mer analysis was conducted for the genome survey. To obtain clean reads, the raw data were filtered by removing low-quality reads, short reads, adapter sequences, and polyG tails. Subsequently, Jellyfish v2.2.10[32] was utilized to perform k-mer = 21 frequency distribution analysis. The genome size, heterozygosity, and duplication rate were estimated using GenomeScope v2.0[33]. The estimated genome size of the African tulip tree was 399.1 Mb with a heterozygosity of 1.4%.
Chromosome-level genome assembly
-
Firstly, the HIFI raw data were converted to fastq format using samtools v1.21[34]. To obtain clean HIFI and HiC data, fastp v0.23.2 was used to filter out low-quality reads, short reads, adapter sequences, and polyG tails from both the HIFI and HiC data. The filtered HIFI data were then assembled into contigs using hifiasm 0.16.1[35] with default parameters. Subsequently, the clean HiC data were aligned to the final assembled contigs using juicer v1.6[36] to obtain the interaction matrix. The contigs were then ordered and anchored using 3D de novo assembly v180114[37]. Finally, Juicebox v1.11.08[38] was used to manually inspect the HiC contact map of the final assembly. We performed a de novo assembly of the African tulip tree genome at the chromosome level, resulting in an assembled genome size of 407.9 Mb with a contig N50 of 31.7 Mb. Among them, 395.5 Mb were anchored onto 13 pseudo-chromosomes, representing a fixation rate of 97.1%.
Gene prediction and annotation
-
RepeatModeler v2.0.3[39] assisted in clustering the repeats by building a de novo repeat library. Then RepeatMasker v4.1.2[40] was applied to identify the repetitive sequences. A total of 239.3 Mb repeat elements were identified, which accounted for 58.7% of the African tulip tree genome. Utilize HISAT2 v2.1.0[41] to align all transcriptome data to the genome. Subsequently, employ BRAKER v3.0.3[42] to automatically undertake the training of species-specific parameter models and the annotation of gene structures, leveraging both the alignment results of the transcriptome data and protein evidence. Overall, a total of 23,695 protein-coding genes were predicted, with 97.5% of the complete BUSCO genes covered. For functional annotation, we queried our predicted protein-coding genes against the eggNOG database[43].
Orthologue and phylogenetic analyses
-
Protein sequences of nine other species (Oryza sativa, Vitis vinifera, Olea europaea, Utricularia gibba, Salvia bowleyana, Striga asiatica, Erythranthe guttata, Avicennia marina, and Handroanthus impetiginosus) were extracted from the public database HortDB V1.0[44] (
https://bioinformatics.hainanu.edu.cn/hortdb/ ) and PLAZA 5.0[45] (https://bioinformatics.psb.ugent.be/plaza/versions/plaza_v5_dicots/ ). Only the longest transcript of each gene representing the coding gene was retained to construct the phylogenetic tree. Orthologs and paralogs were identified using OrthoFinder v2.5.5[46] with default parameters, using O. sativa as the outgroup. The maximum likelihood tree was constructed using IQ-TREE v2.2.3[47] with 1,000 ultrafast bootstrap replicates. Divergence times among different species were estimated using R8s v1.81[48] based on fossil records from the TimeTree database[49] (www.timetree.org ), employing the approximate likelihood method. The fossil calibration points used were O. sativa vs V. vinifera at 142.1−163.5 million years ago (MYA), O. europaea vs U. gibba at 63.6−80.0 MYA, and H. impetiginosus vs A. marina at 38.5−74.3 MYA. CAFE v5.1.0[50] software was used to assess the changes in the size of gene families, examining both contractions and expansions. The gene family clustering of S. campanulata, S. asiatica, E. guttata, A. marina, and H. impetiginosus was clustered using jvenn[51] website (www.bioinformatics.com.cn/static/others/jvenn/example.html ).Syntenic analysis
-
The JCVI[52] tool, configured with its default settings, was utilized to detect collinear blocks. Utilizing protein names as search queries, we scoured the genomes of various plant species to locate the most compatible matches. Each aligned block served as an indicator of homologous pairs that originated from a shared ancestor. To compute the synonymous substitution rates (Ks) among homologs within these collinear regions, we employed the Nei-Gojobori method, which is integrated within the PAML (Phylogenetic Analysis by Maximum Likelihood) software package[53]. The average Ks value was taken as a representative of the entire collinear region.
Anthocyanin biosynthesis and flower pigmentation
-
Paired-end clean reads were aligned to the reference genome using HISAT2 v2.1.0. featureCounts[54] was used to count the read numbers mapped to each gene. Gene expression levels were then estimated as fragments per kilobase of transcript per million mapped fragments (TPM). All the protein sequences related to anthocyanins in Arabidopsis thaliana were downloaded from BRAD[55], the anthocyanin protein sequences were merged, and uBLAST 2.16.0+ was used to construct an anthocyanin protein sequence database. The protein sequences of genes annotated in the African tulip tree that are related to the anthocyanin metabolic pathway were compared with those in the anthocyanin protein sequence database to identify candidate genes with sequence similarities to anthocyanin-related proteins. An expression heatmap was constructed based on the expression matrix.
-
A genome survey using k-mer analysis has estimated the genome size of the African tulip tree to be approximately 399.1 Mb, with a heterozygosity rate of 1.4% (Fig. 1b). Using HiFi sequencing and HiC reads for assembly, the genome of the African tulip tree was anchored onto 13 pseudo-chromosomes (Fig. 1c), with a total length of 407.9 Mb and contig N50 length of 31.7 Mb. The total genomic GC content reached 33.9%. The genomic Benchmarking Universal Single Copy Orthologs (BUSCO) value was 98.1% [C: 98.1% (S: 95.7%, D: 2.4%), F: 1.0%, M: 0.9%, n: 1614], suggesting a reference-scale genome for comparative analyses and gene mining.
A total of 239.3 Mb repeat elements were identified, which accounted for 58.7% of the assembled genome. The most abundant repetitive elements were long terminal repeat (LTR) elements (123.8 Mb; Ty1/Copia: 45.2 Mb; Gypsy/DIRS1: 65.6 Mb; Retroviral: 0.4 Mb), followed by DNA transposons (22.3 Mb), with an additional 76.4 Mb of unclassified repetitive sequences. Through genome annotation, we predicted a total of 23,695 protein-coding genes in the African tulip tree. The protein sequence obtained through annotation achieved a BUSCO completeness of 97.5% (Table 1).
Table 1. Genomic statistics of Spathodea campanulata.
Genome Spathodea campanulata Ploidy 2n = 26 Estimated genome size (Mb) by k-mer 399.1 Assembled genome size (Mb) 407.9 Genomic heterozygosity (%) 1.4 Contig N50 (Mb) 31.7 Number of contigs 164 Number of pseudochromosomes 13 Repeat sequence content (%) 58.7 GC content (%) 33.9 Number of gene models 23,695 Genome BUSCOs (%) 98.1 Gene BUSCO (%) 97.5 Comparative genomics unlocked the evolution of species within the family Bignoniaceae
-
By comparison of the protein sequences of S. campanulata with those of nine other species sequenced in near families, we tried to unlock the evolutionary relationship among them. They are H. impetiginosus, O. europaea, U. gibba, S. bowleyana, E. guttata, S. asiatica, A. marina, V. vinifera, and O. sativa. Phylogenetic analysis revealed that the differentiation between Acanthaceae and Bignoniaceae occurred approximately 56.7 million years ago. The African tulip tree diverged from its relative H. impetiginosus around 31.4 million years ago. Expansion or contraction of the gene family is an important feature in selective evolution. Compared with H. impetiginosus of the same family, African tulip tree evolution involved new genes and gene families, but their evolution occurred independently with a different degree of gene families being lost for each species. The African tulip tree exhibited an expansion in 726 gene families and a contraction in 933 gene families (Fig. 2a). Gene family clustering was performed, and the results were visualized using jvenn. In the African tulip tree, there are 23,695 genes grouped into 16,187 gene families, of which 2,264 are single-copy gene families. In contrast to H. impetiginosus, the evolution of the African tulip tree encompassed the arising of novel genes and gene families. However, these evolutionary trajectories unfolded independently, with each species enduring differing levels of gene family reduction. We have characterized the orthogroups shared by the genome of S. campanulata and its related species. Specifically, 8,656 orthogroups were identified as common to five species. Notably, S. campanulata and H. impetiginosus share 14,820 orthogroups, which to some extent reveal their phylogenetically related yet distinct relationships. Compared to other species, S. campanulata harbors 100 unique orthogroups, whereas H. impetiginosus possesses 515 unique orthogroups (Fig. 2b).
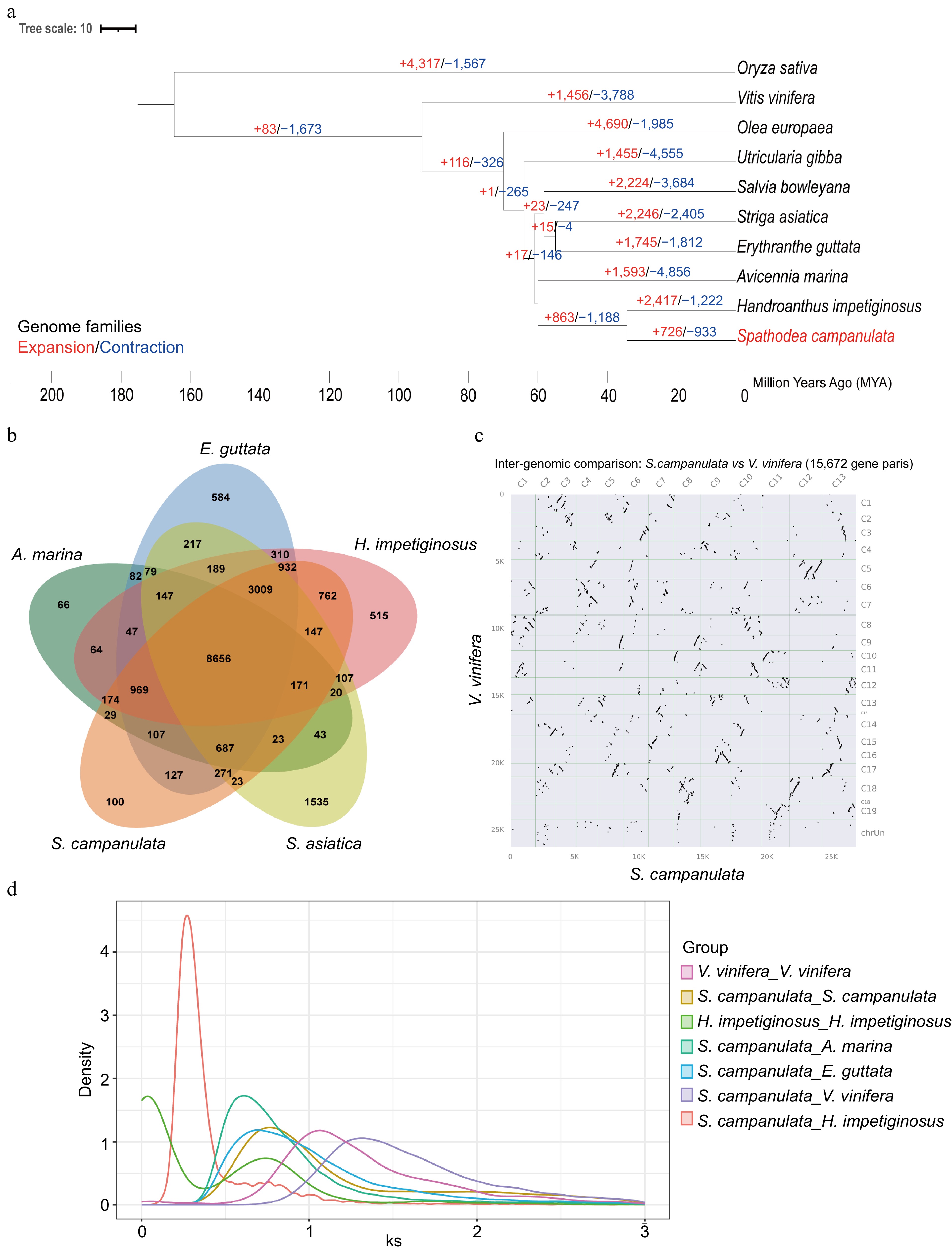
Figure 2.
Comparative genomic analysis of the African tulip tree. (a) Expansion and loss of gene orthogroups leading to the current African tulip tree genome. (b) Venn diagram showing orthogroups shared by the African tulip tree and related genomes. (c) Syntenic dot plot between S. campanulata and V. vinifera. (d) The Ks distribution map within and between species.
Through the analysis of collinearity dot plots, we have identified a notable 1:2 relationship in the genomic architecture between the grape and the African tulip tree (Fig. 2c). When this finding is integrated with the examination of Ks distribution plots both intra- and inter-specifically, an intriguing pattern emerges. Both the grape and the African tulip tree exhibit a shared peak, suggesting a common evolutionary history or a conserved genomic segment between the two species. Notably, subsequent to this shared peak, each species demonstrates distinct individual peaks (Fig. 2d). This observation suggests that following their divergence, the African tulip tree experienced a whole-genome duplication event at some juncture in its evolutionary trajectory.
Anthocyanin metabolism and its gene differential expression in the corolla of the African tulip tree
-
We investigated the different expression of genes for the anthocyanin metabolic pathway by comparative transcriptomic analysis of flower and other tissues of the African tulip tree. A total of nine genes in the phenylalanine metabolic pathway involved in anthocyanin synthesis were highly expressed in the corolla. There are genes of 4-coumarate: CoA ligase 3 (Sc4CL3.1), chalcone synthase (ScCHS2 and ScCHS3), chalcone isomerase (ScCHI1 and ScCHI2), flavanone 3-hydroxylase (ScF3H), those determine the ability of corolla pigment precursor synthesis. Twenty three candidate genes were identified that were potentially linked to pigment deposition, which contribute to anthocyanin accumulation and the formation of pigment complexes. These genes are all involved in anthocyanin biosynthesis, modification, and transport. Also the gene of flavonoid 3'-hydroxylase (ScF3'H1 and SCF3'H2), flavonoid 3-O-glucosyltransferase (ScUFGT) which directly catalyze synthesis and accumulation of cyanidin and delphinidin. However, higher expression of the dihydroflavonol-4-reductase (DFR) and leucoantho-cyanidin dioxygenase (ANS/LDOX) were not found in the corolla of the African tulip tree (Fig. 3).
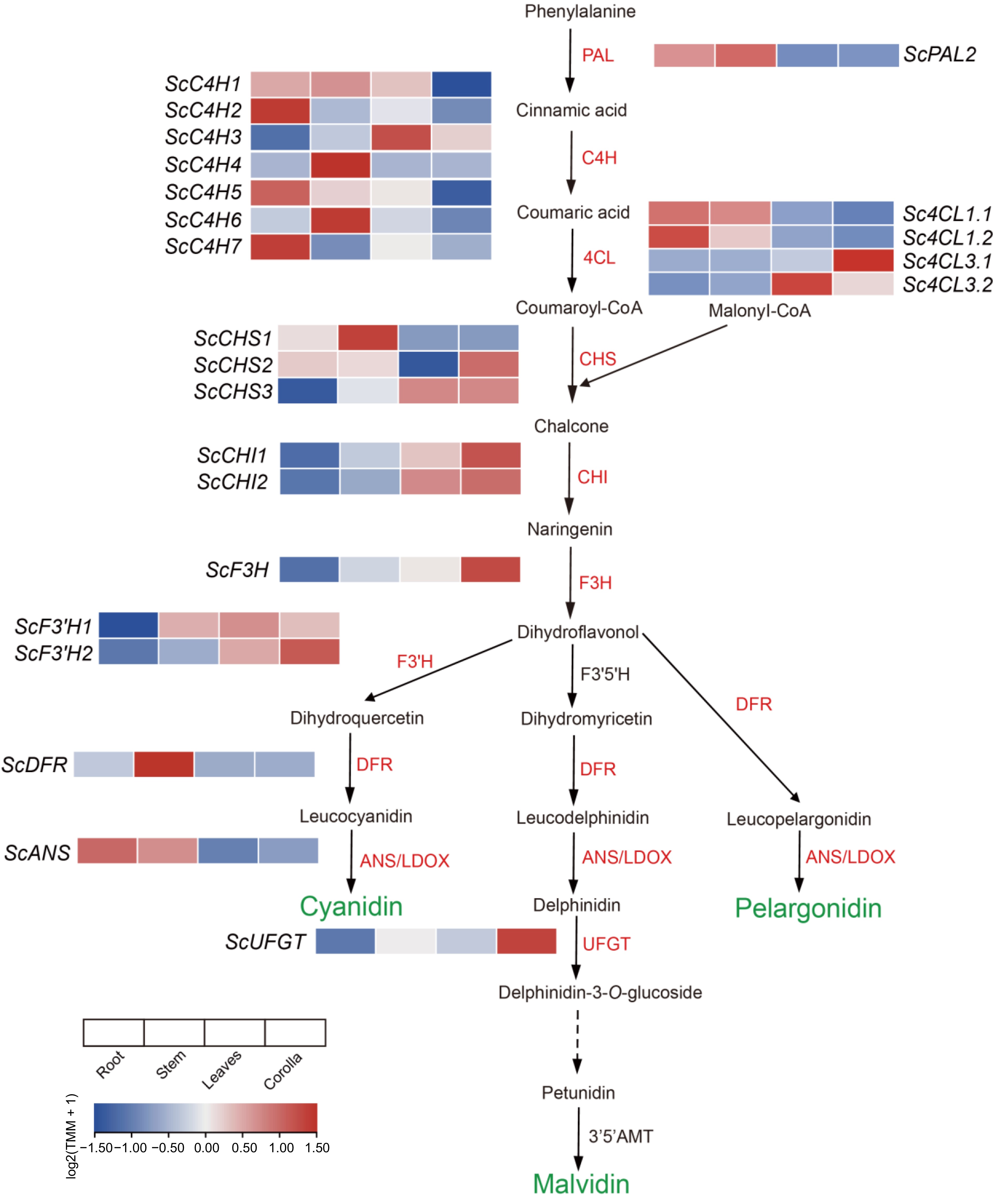
Figure 3.
Preferentially expressed genes in the phenylalanine metabolism pathway in the corolla of the African tulip tree (modified from Berardi et al.[56]) .
-
The African tulip tree, a species known for its ornamental value and medicinal properties, has long lacked genomic resources, hindering efforts to understand its evolutionary history and underlying genetic mechanisms. By generating a reference-scale genome, we aim to address these gaps and contribute essential data for further genetic and functional studies. The genome assembly of the African tulip tree has a total length of 407.9 Mb, a contig N50 of 31.7 Mb, and a GC content of 33.9%. These metrics suggest that the genome assembly is highly contiguous and representative of the species' overall genomic structure. The high BUSCO completeness (98.1%) indicates that the assembly is near-complete and suitable for downstream comparative genomics and gene function analysis. The identification of 239.3 Mb of repeat elements, accounting for 58.7% of the genome, aligns with similar genome analyses in other plant species, where repetitive elements contribute significantly to genome size and structural complexity. The abundance of LTR elements, in particular, suggests a dynamic history of retrotransposon activity, which has likely influenced the genome's architecture and its evolutionary trajectory[57]. Through genome annotation, we predicted 23,695 protein-coding genes, with a BUSCO completeness of 97.5% in the annotated gene set. We not only provide a comprehensive catalog of the protein-coding genes in the African tulip tree but also lay the groundwork for identifying genes involved in important biological processes such as secondary metabolism, stress response, and flower development.
The expansion of gene families may result in gene dosage effects and enhance certain phenotypes, which plays an important role in species evolution and environmental adaptation. The comparative genomics analysis reveals important insights into the evolutionary history of the African tulip tree. The comparative genomics analysis reveals important insights into the evolutionary history of the African tulip tree. Acanthaceae and Bignoniaceae occurred approximately 56.7 million years ago. Furthermore, within the Bignoniaceae family, Spathodea stands out as a pivotal genus, with the recently assembled genome of S. campanulata offering fresh perspectives on its unique evolutionary trajectory. Of particular note, the divergence between S. campanulata and its closely related species H. impetiginosus is estimated to have occurred some 31.4 million years ago. The gene family clustering results showed that the African tulip tree shares 14,820 orthogroups with H. impetiginosus, its closest relative, but also exhibits significant gene family expansion and contraction. Specifically, 726 gene families have expanded, while 933 gene families have contracted in the African tulip tree. These findings reflect selective evolutionary pressures and lineage-specific adaptations, with some gene families likely playing important roles in the plant's ecological success and ornamental traits.
The synteny analysis and Ks distribution plots between the grape and African tulip tree genomes suggest that the duplication occurred early in the species' evolutionary history, leading to a significant structural and functional diversification of gene families. WGD is known to contribute to the complexity of plant genomes, enabling the development of new traits and facilitating adaptation to different environments[58]. The African tulip tree's WGD could explain the species' extensive gene family expansions, particularly those related to secondary metabolism, which could have implications for the plant's medicinal properties and flower pigmentation.
Our comparative transcriptomic analysis of the African tulip tree revealed nine highly expressed genes in the phenylalanine metabolic pathway, including Sc4CL3.1, ScCHS2, ScCHS3, ScCHI1, ScCHI2, and ScF3H, highlighting their essential roles in anthocyanin precursor synthesis and contributing to the vivid pigmentation of the corolla. Additionally, 23 candidate genes linked to pigment deposition were identified, such as ScF3'H1, ScF3'H2, and ScUFGT. UFGT is critical for stabilizing anthocyanins and enhancing pigment accumulation. For example, UFGT is directly responsible for anthocyanin color transformation as well as anthocyanin accumulation during apple-fruit ripening[59]. VvUFGT catalyzes the glycosylation of anthocyanins, thereby accelerating the accumulation of anthocyanins in grapes[60]. DFR and ANS, typically crucial in later biosynthetic stages, showed low expression in the corolla, suggesting tissue-specific regulatory mechanisms or alternative pathways. These results enhance our understanding of the genetic and metabolic basis of pigmentation in African tulip trees.
Overall, the genome sequence of the African tulip tree represents a significant step forward in the study within the family Bignoniaceae and ornamental plants in general. The genome of the African tulip tree provides a foundational resource for future research into the genetics of this species, including investigations into its ornamental traits, medicinal properties, and evolutionary history. The insights into gene family evolution and whole-genome duplication in the African tulip tree may offer broader implications for understanding the role of WGD in plant adaptation and diversification. Moreover, the identification of genes involved in anthocyanin biosynthesis opens up possibilities for manipulating flower color in breeding programs.
This work was supported by the National Natural Science Foundation of China (32172614) and supported by the Project of National Key Laboratory for Tropical Crop Breeding (No. NKLTCB202337), Hainan Province Science and Technology Special Fund (ZDYF2023XDNY050), and the Hainan Provincial Natural Science Foundation of China (324RC452).
-
The authors confirm contribution to the paper as follows: study conception and design: Wang W, Chen F; plant samples collection: Zhang J, Guo G, Wang S, Chen F; experimentsconducted, analyses performed, and draft manuscript preparation: Wang S. All authors reviewed the results and approved the final version of the manuscript.
-
Raw data High-Fidelity sequencing, Illumina, Hi-C data are available online at the National Genomics Data Center (https://ngdc.cncb.ac.cn) with the project ID PRJCA031779 . The genome, coding sequences, proteins, and gff files can be found at FigShare (https://doi.org/10.6084/m9.figshare.27798735) .
-
The authors declare that they have no conflict of interest.
-
Received 5 December 2024; Accepted 5 February 2025; Published online 7 May 2025
-
The chromosome-level genome of the African tulip tree has been assembled into 13 pseudo-chromosomes.
Protein sequences of the African tulip tree were compared with nine other species to evaluate gene family expansion and contraction.
Acanthaceae and Bignoniaceae originated about 56.7 million years ago. The African tulip tree diverged from H. impetiginosus around 31.4 million years ago.
Using the assembled genome, 23 genes involved in anthocyanin biosynthesis were identified, nine of these genes potentially related to cyanidin and malvidin accumulation showed high expression in the corolla.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang S, Guo G, Zhang J, Wang W, Chen F. 2025. Chromosome-level genome of Spathodea campanulata revealed genetic characteristics of malvidin and cyanidin accumulation in the corolla. Tropical Plants 4: e018 doi: 10.48130/tp-0025-0004 

Chromosome-level genome of Spathodea campanulata revealed genetic characteristics of malvidin and cyanidin accumulation in the corolla
- Received: 05 December 2024
- Revised: 23 January 2025
- Accepted: 05 February 2025
- Published online: 07 May 2025
Abstract: The African tulip tree (Spathodea campanulata) is a rare tropical ornamental species, renowned for its large, scarlet flame-bubble corolla and medicinal properties. However, its evolutionary and genetic characteristics, particularly concerning economically significant traits, have been poorly understood due to the absence of a sequenced genome. Here, we present the first chromosome-level assembled genome for this species, organized into 13 pseudo-chromosomes, with a total size of 407.9 Mb and 23,695 predicted protein-coding genes. Comparative genomic analysis reveals that the African tulip tree diverged from Handroanthus impetiginosus approximately 31.4 million years ago, accompanied by significant changes in gene families and a whole-genome duplication event. Additionally, we identified 23 genes involved in the anthocyanin biosynthesis pathway, with nine showing high expression levels in the corolla, potentially linked to the accumulation of cyanidin and malvidin, which likely contribute to its vibrant coloration. This study significantly enhances our understanding of the evolutionary and genetic dynamics of this species and its family.
-
Key words:
- Spathodea campanulata /
- Genome assembly /
- Evolution /
- Anthocyanin biosynthesis /
- Malvidin