









-
The kiwifruit, botanically known as members of the Actinidia Lindl., traces its origins back to China. This genus encompasses around 54 species of perennial deciduous fruit crop genus. The wide array of crops are characterized by their extensive genetic and morphological traits[1]. Notably, despite such diversity, the flesh of the color of most cultivated varieties of kiwifruit cultivars tend to be green, yellow, or red when harvested. The fruit serves as a rich source of various sugars, organic acids, vitamins, and other metabolites[2−4]. Owing to its exceptionally superior nutritional value profile and appealing taste, the kiwifruit has achieved considerable economic significance over the last three decades, driving a marked expansion in its worldwide cultivation[5,6]. By 2022, the annual production of kiwifruit amounted to 4.5 million metric tons (
www.fao.org/statistics/data-releases/en ).Primary metabolites including sugars, organic acids, and free amino acids, play a pivotal role in shaping the taste and nutritional value of kiwifruit[7]. Within the Actinidia genus, there exists significant natural variation in vitamin C content and sweetness[8,9]. Among 54 recognized species, Actinidia latifolia and Actinidia eriantha stand out for having the highest levels of vitamin C[10]. In contrast, Actinidia chinensis is significantly sweeter.
Recently, the critical genes and their regulatory mechanisms have been characterized. For instance, GDP-L-galactose phosphorylase 3 (GGP3), a key enzyme in L-ascorbic acid (AsA) biosynthesis, is highly expressed in A. eriantha fruit. The promoter of GGP3 can bind to AeMYBS, which activates its expression. Additionally, the bZIP transcription factor AceGBF3 was confirmed to interact with AceMYBS1, and together they synergistically enhance AceGGP3 expression, leading to increased AsA accumulation[11]. The AcSWEET9b gene has been demonstrated to have a positive correlation with high sucrose levels in fruit, in contrast to wild varieties and other Actinidia species, the promoter sequence of AcSWEET9b was selected in red-fleshed A. chinensis resulting in increasing fruit sucrose content. This provides a plausible explanation for the increased sweetness observed in red-fleshed kiwifruits[12].
Amino acids are crucial not only for the ripening and development of fruits, but also as essential taste and nutrient elements[13,14]. Research has shown that the levels of the sweet amino acids proline and threonine increase progressively during the ripening of kiwifruit, potentially contributing more significantly to the fruit's sweetness compared to other amino acids[13]. Furthermore, aromatic amino acids (AAAs), are fundamental components of proteins. They function as precursors for a wide range of secondary metabolites, such as flavonoids, ligin, vitamin E, and others[15,16]. The primary synthesis pathway for AAAs occurs in plant plastids, via the shikimic acid pathway[17]. Precise regulation of this synthesis process is essential for maintaining optimal levels of AAAs, which is vital for promoting normal plant growth, reproduction, and enhancing their resilience to environmental stressors. Studies have indicated that aminotransferases in Arabidopsis participate in the synthesis and metabolism of AAAs, helping to maintain plant amino acid balance[18]. However, the metabolism of AAAs in kiwifruit remains poorly understood.
Despite the growing number of studies investigating the biological roles of metabolites and genes during kiwifruit development, there remains a paucity of research comparing metabolites across different kiwifruit species. In this study, we selected two commercially popular kiwifruit species that exhibit distinct characteristics and flavors: A. eriantha, which has green flesh and a higher vitamin C content, and A. chinensis, which features red flesh and a sweeter taste. We employed liquid chromatography-mass spectrometry (LC-MS) and RNA sequencing to quantify the accumulation of various metabolites during the ripening process, with a specific focus on the regulatory mechanisms underlying significant differences in the AAAs pathway. This research aims to deepen our understanding of nutrient metabolism during kiwifruit ripening.
-
Fruits of A. eriantha 'White', and A. chinensis 'Hongyang' were harvested at DAF180 from three independent trees. A total of 30 fruits for each species were sent to the laboratory shortly after harvesting. The seedless flesh was sliced into small pieces and promptly frozen using liquid nitrogen, then stored in a −80 °C refrigerator.
Sample preparation and metabolite extraction
-
The pulp was ground into a powder (MM 400, Retsch). Next, 100 mg of the resultant powder underwent overnight extraction at 4 °C to obtain either lipid-soluble or water-soluble metabolites. The metabolomics analysis was conducted using an LC/MS system. The raw data, acquired using MassLynx V4.2, undergoes processing by Progenesis QI software for tasks such as peak extraction and alignment. Identification is based on the online METLIN database within Progenesis QI and Biomark's internally developed library, with theoretical fragment identification and mass deviation falling within a 100 ppm range.
Differential metabolites analysis
-
Using the R package PHEATMAP, hierarchical cluster analysis was performed on metabolites in various samples, and the outcomes were presented through heatmaps accompanied by dendrograms.
The color gradients in the heatmaps represented the normalized signal intensities of metabolites, which were scaled to unit variance. Metabolites classified as significantly differentially accumulated metabolites (DAMs) had p-values below 0.05, variable importance in projection (VIP) scores exceeding 1, and fold changes greater than 2 or less than 0.5. The DAMs were annotated through the KEGG compound database, available at
www.kegg.jp/kegg/compound/ . Subsequently, these metabolites were aligned with the KEGG Pathway database atwww.kegg.jp/kegg/pathway.html . Pathways that included significantly regulated metabolites were subjected to metabolite set enrichment analysis, and the relevance of these pathways was assessed using p-values calculated from the hypergeometric test.RNA extraction and sequencing
-
For total RNA extraction, the modified CTAB method was used as described in a previous study[19]. One μg RNA per fruit sample was utilized, and NEBNext UltraTM RNA Library Prep Kit for Illumina (manufactured by NEB, USA) was used to create sequencing libraries according to the manufacturer's guidelines. The library preparations were sequenced on an Illumina sequencing platform, yielding 150 bp paired-end reads.
Transcriptome data analysis
-
We filtered out low-quality reads using Trimmomatic (v0.39)[20] and mapped clean reads to A. chinensis (
http://actinidiabase.moilab.net/download/genome/362501/ ), and A. eriantha (www.ncbi.nlm.nih.gov/datasets/genome/GCF_019202715.1 ) reference genome using STAR (v2.7.0)[21] with default parameters, respectively. The reads uniquely aligned to each gene were quantified using featureCounts[22] and then normalized to transcripts per million (TPM). For the comparison of gene expression patterns between two species, orthologue genes were pinpointed using the reciprocal blast function in TBtools[23]. Differential expression analysis was conducted on the orthologue genes of two species with the DESeq2 R package[24]. Significant differential expression was defined by a padj value less than 0.05 and an absolute fold change of at least 2. Functional annotations for the differentially expressed genes (DEGs) were carried out using eggNOG5[25]. Furthermore, the clusterProfiler R package[26] was employed to detect enriched Gene Ontology (GO) terms and KEGG pathways.Correlation analysis and visualization
-
The correlation coefficients for the contents of 428 metabolites found in ripening fruits of A. chinensis and A. erinatha were calculated. A Raincloud plot was created utilizing R. After excluding genes with relatively low expression (average TPM less than 1), a correlation analysis was conducted between DEGs and differentially accumulated metabolites (DAMs). Additionally, transcription factors were predicted among all DEGs. A correlation network was constructed by selecting Pearson correlation coefficients with an absolute value (|PCC|) greater than 0.95 and a p-value less than 0.01. These networks were visualized by Cytoscape[27].
-
The metabolite profiles of ripening fruits from A. chinensis and A. eriantha were characterized using a widely targeted metabolomics approach. A total of 428 compounds were identified, of which 109 were annotated in the KEGG database (Supplementary Table S1). The principal component analysis (PCA) of all samples distinctly separated the two species, showing tight clustering of the biological replicates within each species (Fig. 1a). Hierarchical heatmap clustering analysis, based on metabolite concentration data, further confirmed that all biological replicates were grouped appropriately. These findings underscore the high reliability of our data (Fig. 1b). To assess differences in metabolite composition between A. chinensis and A. eriantha, we calculated the correlation coefficients for the 109 common compounds present in both species (Fig. 1d; Supplementary Table S2). Compounds with the highest correlation coefficients are involved in carbohydrate and energy metabolism, suggesting a significant similarity in metabolic patterns for these metabolites between the two kiwifruit species. However, differences in amino acid metabolism, as well as in the metabolism of terpenoids and polyketides, exist between the species, contributing to variations in taste, flavor, and nutritional properties.
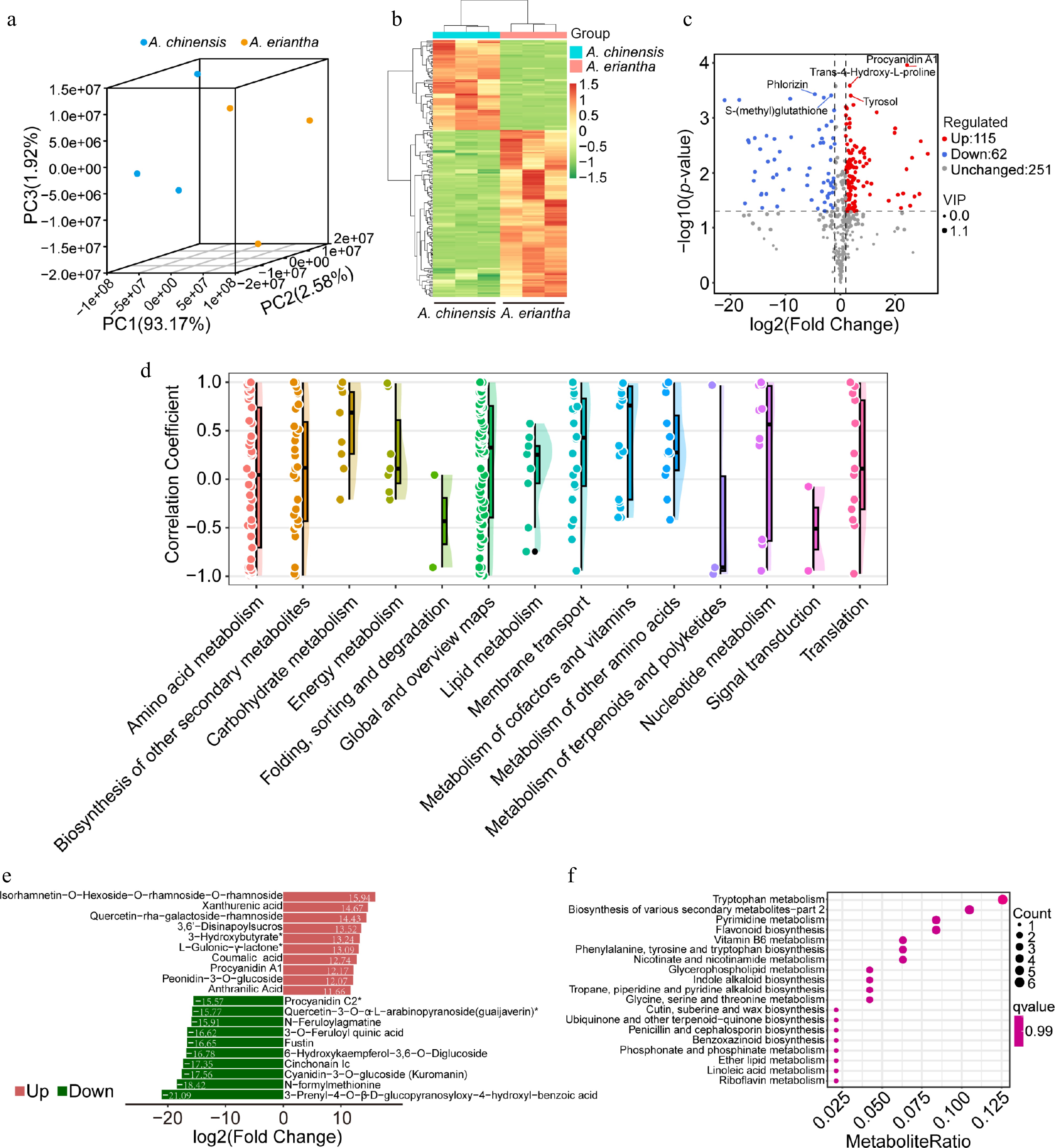
Figure 1.
Analysis of metabolites in A. chinensis and A. eriantha fruits. (a) Clustering of all fruit samples using Principal Component Analysis (PCA). (b) Hierarchical cluster-based heatmap of differential metabolites detected in fruit samples. (c) Volcano plot illustrating the identified differentially accumulated metabolites (DAMs). (d) Correlation coefficients of metabolites content patterns in ripening fruit shared by A. chinensis and A. eriantha. (e) Top 10 upregulated and downregulated metabolites based on log2(Fold Change). (f) Enriched components based on KEGG analysis of all DEGs.
Identification of the differentially accumulated metabolites in kiwifruits
-
We determined the DAMs based on the criteria mentioned in the methods. In total, 115 metabolites were classified as differentially upregulated, 62 as downregulated, and 251 as unchanged when comparing A. eriantha to A. chinensis (Fig. 1c). According to the −log10 (p-value), Procyanidin A1, Trans-4-Hydroxy-Lproline, Tyrosol were recognized as top three upregulated metabolites. On the contrary, Phlorizin and S-(methyl)glutathione were the two most downregulated metabolites. Based on the log2FC value, the top 10 upregulated and downregulated metabolites were listed (Fig. 1e). The contents of rhamnoside and xanthurenic acid were significantly higher in A. eriantha compared to A. chinensis. Conversely, A. eriantha accumulated lower concentrations of 3-prenyl-4-O-β-D-glucopyranosyloxy-4-hydroxyl-benzoic acid, N-formylmethionine, and kuromanin compared to A. chinensis. These results suggest that A. eriantha is less sweet and has higher antioxidant activity. The top six enriched KEGG terms associated with DAMs were involved in tryptophan metabolism, biosynthesis of secondary metabolites, pyrimidine metabolism, flavonoid biosynthesis, vitamin B6 metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis (Fig. 1f). These metabolites are primarily associated with nutritional components and antioxidant agents.
Difference in contents of AAAs related metabolisms
-
Given the distinct accumulation of AAAs as core metabolites, we isolated DAMs involved in the tryptophan metabolism pathway to understand the patterns of metabolite accumulation differences between two kiwifruit species. Serotonin (Fig. 2e), tryptamine (Fig. 2f), shikimic acid (Fig. 2k), and 5-hydroxyindole-3-acetic acid (Fig. 2l) were found in higher amounts in A. chinensis compared to A. eriantha. Notably, the contents of serotonin and tryptamine are extremely low in A. eriantha. In contrast, several other metabolites, including indole (Fig. 2a), L-phenylalanine (Fig. 2b), L-tryptophan (Fig. 2c), methoxyindoleacetic acid (Fig. 2d), indole-3-carboxaldehyde (Fig. 2i), and L-(-)-cystine (Fig. 2j), exhibited higher concentrations in A. eriantha. Additionally, only trace amounts of xanthurenic acid (Fig. 2g), and anthranilic acid (Fig. 2h) were detected in A. chinensis.
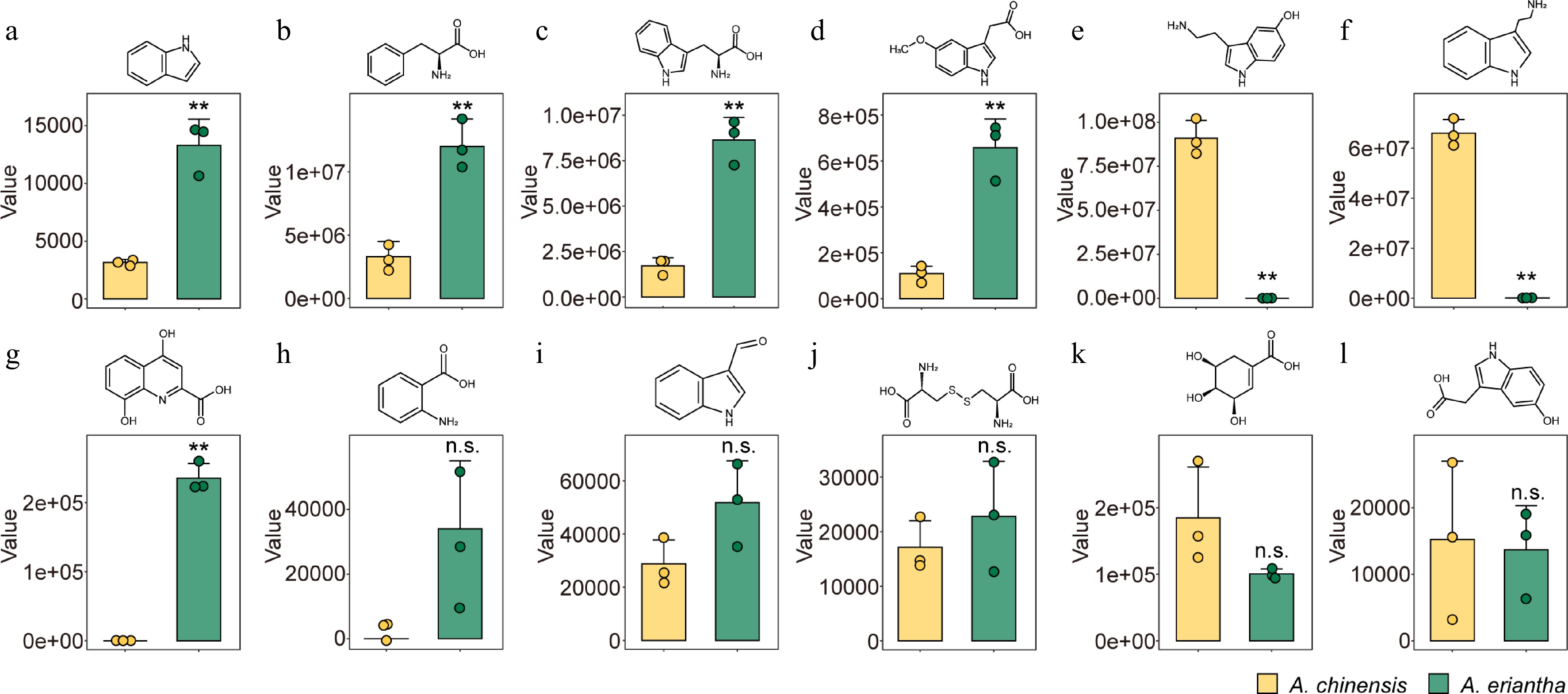
Figure 2.
Contents of differentially accumulated metabolites (DAMs) associated with the tryptophan metabolism pathway in A. chinensis and A. eriantha fruits. (a) Indole, (b) L-phenylalanine, (c) L-tryptophan, (d) methoxyindoleacetic acid, (e) serotonin, (f) tryptamine, (g) xanthurenic acid, (h) anthranilic acid, (i) indole-3-carboxaldehyde, (j) L-(-)-cystine, (k) shikimic acid, and (l) 5-hydroxyindole-3-acetic acid. ** p < 0.01, n.s. non-significance.
Transcriptome profiles of two kiwifruit species
-
We conducted an in-depth examination of the variations in gene expression profiles between the two kiwifruit species. Through transcriptome sequencing with three biological replicates, we obtained a combined total of 20.60 Gb of clean reads for A. chinensis and 18.58 Gb of clean reads for A. eriantha. PCA was applied to the samples using transcripts per million (TPM) values of DEGs, all biological replicates grouped together, attesting to the high reliability of our sequencing data (Fig. 3a). The PC1 accounted for the majority of the data variance (97%), reflecting the distinction between the two kiwifruit species. Specifically, utilizing the criteria of |log2FC| > 1 and padj < 0.05, we identified 3,168 upregulated genes and 3,174 downregulated genes (Fig. 3b). In a heatmap that included all DEGs, a clear distinction between the two species was observed, similar to the findings in the metabolome analysis (Fig. 3c). This indicated that the changes in metabolite accumulation during the ripening process are closely regulated by differential gene expression. To gain insights into the functional implications of the DEGs, we conducted a Gene Ontology (GO) enrichment analysis (Fig. 3d). Notably, the biological process category showed significant enrichment in terms related to AAAs family catabolic process and photosynthesis, strongly hinting at the involvement of these enriched genes in AAAs metabolism.
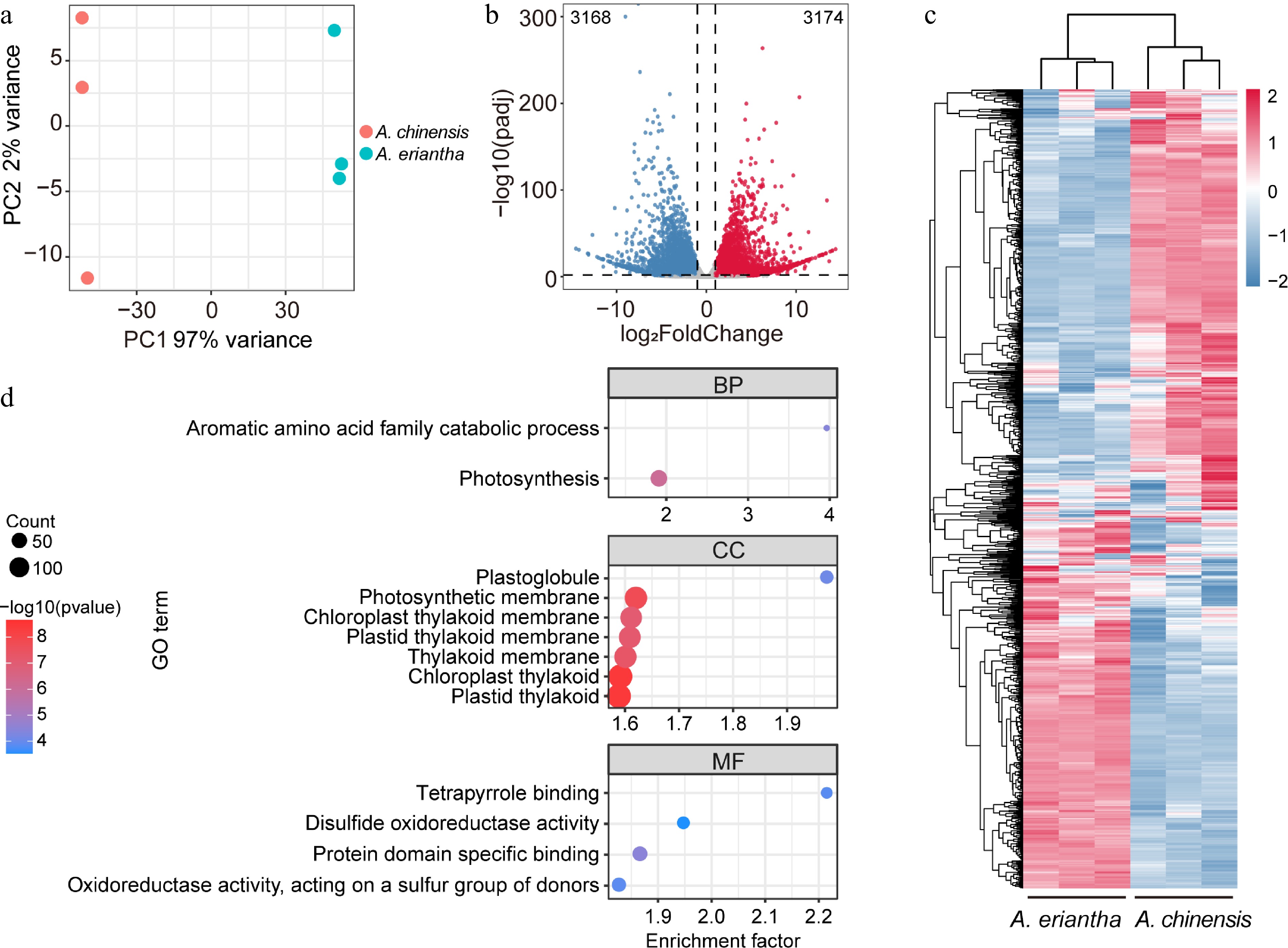
Figure 3.
Transcriptome dynamics of fruit samples for A. chinensis and A. eriantha. (a) Clustering of all fruit samples subjected to transcriptome analysis using Principal Component Analysis (PCA). (b) A volcano plot displaying the up-and downregulated differentially expressed genes (DEGs) when comparing fruit samples between A. chinensis and A. eriantha. (c) Heatmap with scaled expression value of DEGs in fruit samples of A. chinensis and A. eriantha. (d) Enriched components according to GO analysis.
Integrated transcriptome and metabolome analyses
-
To get an insight into the variations of gene expression related to AAAs pathway between the two distinguished kiwifruit species, we focused on the phenylalanine, tryptophan, and tyrosine pathways. Our transcriptional and metabolic enrichment analyses both highlighted major alterations in these pathways, which may reflect the differing nutritional attributes of the two species. In the present study, we uncovered several structural genes whose orthologs in A. chinensis and A. eriantha exhibit distinct expression patterns (Supplementary Table S3). For example, higher content of tryptophan observed in A. eriantha might result from upregulated TSA1 and TSB2 compared to A. chinensis (Fig. 4a). However, more accumulated phenylalanine in A. eriantha can be explained by the reduced transcription of the TAA enzyme (Fig. 4a), which enables it to degrade such substrate, and much more 3-phenyl-2-oxopropanoate storing in A. chinensis, metabolism profiles supported this point (Supplementary Table S3). Similarly, the elevated transcriptional ability of the TAT enzyme in A. eriantha correlates with the lower levels of tyrosine in this species (Fig. 4b). Metabolic analysis of tyrosine accumulation in A. chinensis, further validates these observations (Supplementary Table S4).
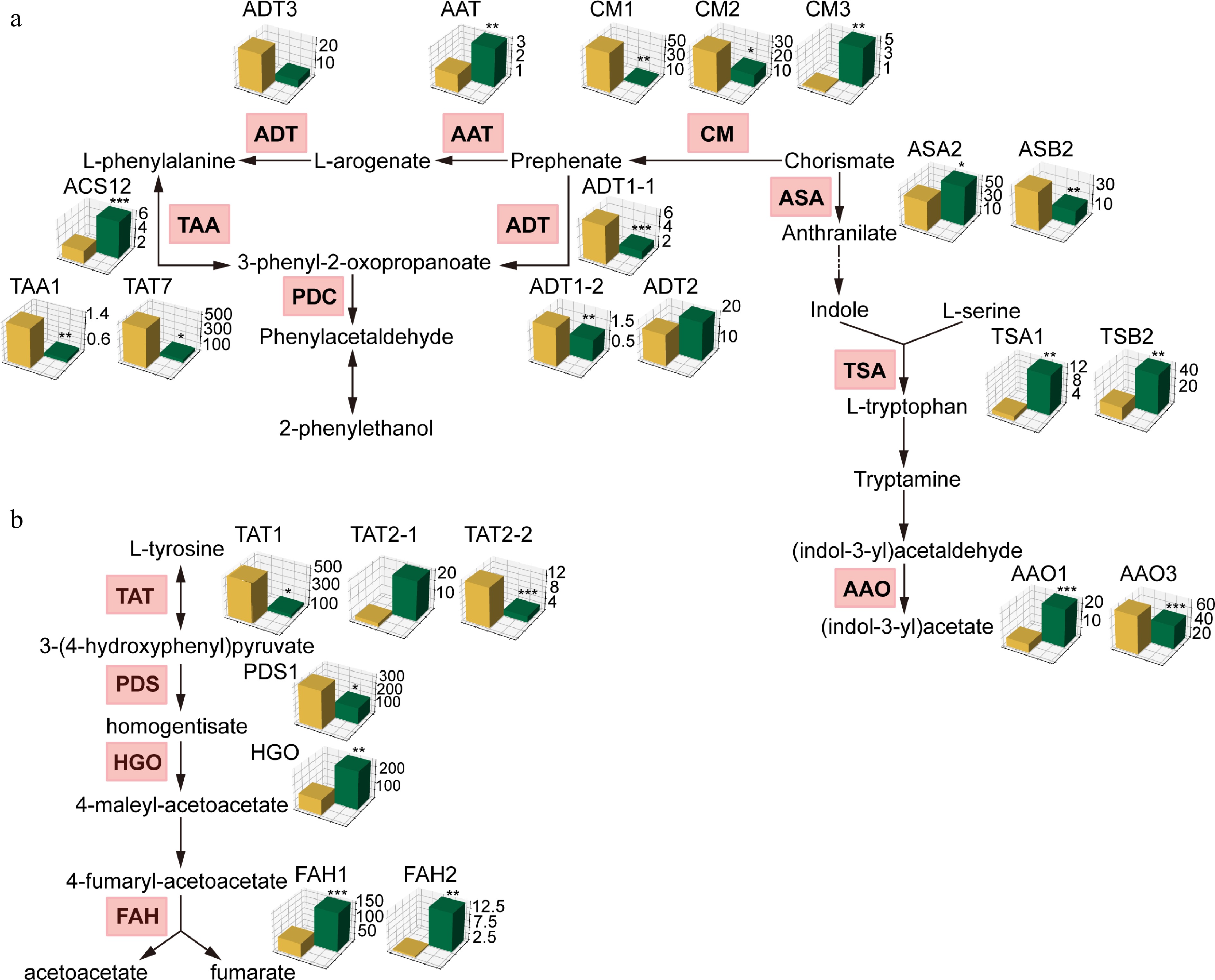
Figure 4.
Comparative transcriptomic analysis of genes involved in phenylalanine, tryptophan, and tyrosine metabolism pathways between A. chinensis and A. eriantha. Barplots of orthologous genes were generated using TPM value. * p < 0.05, ** p < 0.01, *** p < 0.001.
Transcriptional modulation of AAAs pathway
-
To gain insights into the AAAs metabolism pathway during fruit ripening, we first screened for potential differentially expressed transcription factors (TF). A total of 506 DEGs encoding TFs were identified, predominantly belonging to the MYB and MYB_related families, bHLH family, ERF family, and C2H2 family (Supplementary Table S5). Next, we calculated the correlation using differential expressed TFs and DAMs, subset with correlation coefficient absolute values of > 0.9 and p-value < 0.05 was generated (Supplementary Table S5). Additionally, correlation networks targeting L-Phenylalanine and L-Tryptophan were visualized. Interestingly, AcNAC100 was positively correlated with L-Tryptophan in A. chinensis (Fig. 5a), while AeNAC100 was positively associated with L-Phenylalanine in A. eriantha (Fig. 5d). Similarly, AcAPRR2 and AeAPRR2, were both negatively correlated with L-Phenylalanine in A. chinensis (Fig. 5b) and L-Tryptophan in A. eriantha (Fig. 5c), respectively.
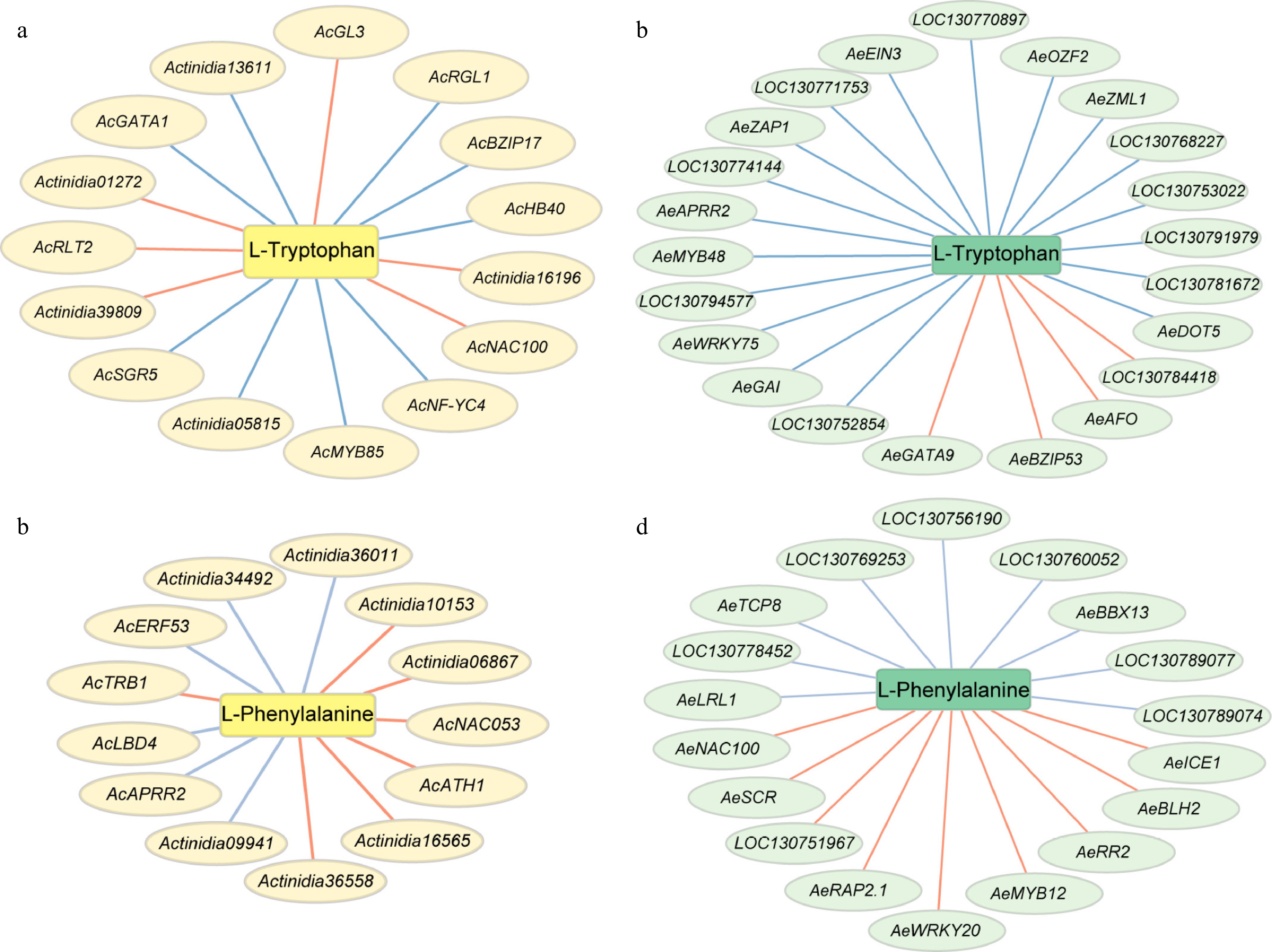
Figure 5.
Putative transcriptional regulatory network involved in phenylalanine, and tryptophan metabolism. Red lines indicate positive correlation, blue lines indicate negative correlation. (a) and (b) present regulatory network in A. chinensis, (c) and (d) present regulatory network in A. eriantha.
-
Fruit quality and nutritional value are critical factors that influence consumer preference[28]. Many previous studies have highlighted the abundance of vitamin C in commercial kiwifruit[29,30] and have delved into the mechanisms behind its accumulation[11,31,32]. Our research aimed to investigate the varying metabolites linked to nutritional properties in different kiwifruit species and to uncover the potential regulatory mechanism through multi-omics approach. In this study, we employed a widely-targeted metabolomics method to investigate metabolite differences between two key kiwifruit species: green flesh A. eriantha and red flesh A. chinensis. We identified 428 compounds, which provided a more comprehensive landscape of the metabolites contributing to specific flavors and nutrients of ripening kiwifruits. The analysis of detected metabolites in A. eriantha and A. chinensis showed that flavonoid biosynthesis and the citrate cycle have high correlation coefficients, while amino acid metabolism, terpenoids, and polyketides exhibit distinct patterns. Similar results were observed in a comparison between A. chinensis and A. latifolia[12], as A. latifolia and A. eriantha are both green-fleshed kiwifruit species with high VC content.
Kiwifruit, renowned for its nutrient-richness and unique flavor, boasts not only a high content of vitamins, minerals, and pectin but also an abundance of various amino acids, with AAAs being particularly prominent[33]. The AAAs in fruit are a crucial component of its nutritional content, significantly influencing its flavor and nutritional value[34,35]. These amino acids also play key roles in various biochemical processes within the fruit. For example, phenylalanine in kiwifruit can be converted into aroma compounds such as styrene through a series of enzymatic reactions, contributing significantly to the fruits' aroma components of kiwifruit. Meanwhile, tyrosine, another important amino acid in kiwifruit, is involved in protein synthesis and metabolism within the fruit, playing a vital role in maintaining fruits' normal physiological functions. Furthermore, the AAAs in fruits possess various biological activities. For instance, phenylalanine exhibits antioxidant properties, playing an important role in protecting cells from oxidative stress damage. The amino acid content of fruit and fruit-derived foods has been studied intensely because of their contribution to nutritional value, aroma, taste, and health-promoting effects[36]. Additionally, amino acid concentration was determined in strawberries[37], and was measured to evaluate the microclimate effects on grapes[38], and used for authenticity purposes in citrus fruit juices[39].
Recently, the contents of free amino acids in different kiwifruit cultivars have been extensively studied. Ma et al.[40] analyzed various cultivars belonging to A. deliciosa and A. chinensis and found that glutamic acid was the most abundant amino acid, whereas tryptophan was the least abundant. Additionally, correlations between flesh color and free amino acids were observed. A. chinensis Hongyang, has a higher abundance of total free amino acids and non-essential amino acids but lower total essential amino acids including AAAs. The analogous results were obtained in our study, indole, L-phenylalanine, L-tryptophan, methoxyindoleacetic acid, indole-3-carboxaldehyde, L-(-)-cystine, xanthurenic acid, and anthranilic acid have higher contents in A. eriantha than A. chinensis (Fig. 2). In this study, we further confirmed that nutritional differences existed between A. eriantha and A. chinensis. Our report contrasts with the findings of a previous study that asserted that the contents of AAAs including phenylalanine, tryptophan, and tyrosine are higher in commercial maturity A. chinensis than in A. deliciosa.
During the cultivation and processing of kiwifruit, the content and proportion of AAAs can be affected by various factors, such as variety, growth environment, harvest time, storage conditions, and processing methods[40]. To maintain the quality and nutritional value of kiwifruit, it is essential to control these factors effectively to optimize the content and proportion of AAAs in fruits. Although AAAs are an important nutritional component in kiwifruit, research on their metabolic mechanisms remains limited. The metabolism of AAAs is regulated by specific structural genes. In our study, we identified homologous genes in A. chinensis based on Arabidopsis genes involved in the metabolism pathway of AAAs. We discovered that the majority of the structural genes in the L-phenylalanine, L-tryptophan metabolism pathway, and L-tyrosine degradation pathway show different expression patterns. L-phenylalanine and L-tyrosine can be degraded by tyrosine aminotransferase (TAT)[41]. The higher expression levels of TAT7 and TAT1 were found in A. chinensis (Fig. 4a), which potentially leads to lower L-phenylalanine and L-tyrosine contents in fruit compared to A. eriantha. Meanwhile, a higher expression level of tryptophan synthase (TSA1/TSB2) in A. eriantha, results in more accumulation of L-tryptophan in fruit compared to A. chinensis. Hence, we suggest that altering the activity of essential structural genes involved in AAAs metabolism could serve as a viable strategy to sustain optimal amino acid levels and fruit nutritional value.
To understand the regulatory mechanisms of the AAA metabolism pathway in kiwifruit, we aimed to identify differentially expressed TFs through correlation analysis. TFs belonging to the NAC, MYB, WRKY, and bZIP families, which are differentially expressed, show a high correlation with L-Phenylalanine and L-Tryptophan. The consequence suggested that the metabolism of these two AAAs are likely mediated by these common TFs. Our results are supported by previous studies. For instance, MYB transcription factors including MYB20, MYB42, MYB43, and MYB85, are known to directly activate lignin biosynthesis genes and phenylalanine biosynthesis genes during secondary wall formation in Arabidopsis[16]. Additionally, TaWRKY24 has been reported to integrate tryptophan metabolism pathways to participate in defense against Fusarium crown rot in wheat[42].
PgMYB308-like transcription factors in pomegranate were reported to enable to enhance the level of shikimate, aromatic amino acids, and lignins, but represses the synthesis of flavonoids and hydrolyzable tannins[43]. Consistent with these findings, we believe that such TFs play similar roles in the biosynthesis and degradation of AAAs in kiwifruit. Notably, these TFs show distinct expression patterns during AAAs metabolism, which contributes to the varied accumulation of these amino acids in fruits.
-
In summary, different components of metabolites in two commonly planted kiwifruit species A. chinensis and A. eriantha were clarified through widely-targeted metabolomics approach using ripening fruit. Except for known differences occurring in sugar and VC accumulation, contents of AAAs were investigated. Accordingly, structural genes involved in L-phenylalanine, L-tryptophan pathway were mainly focused. Based on the joint analysis between metabolome and transcriptome, differentially expressed TFs containing NAC, MYB, WRKY, and bZIP were recognized as potential transcriptional regulators participating in the metabolic pathways of AAAs. Manipulation of these structural genes or related transcription factors through gene editing may be an effective way to alter the AAAs accumulation in kiwifruit. Overall, this study provides more scientific instruction on kiwifruit breeding in the future.
This research was funded by the Zhejiang Provincial Natural Science Foundation of China (No. LQ23C150003), and the Zhejiang A&F University R&D Fund Talent Startup Project (No. 2022LFR015). The funding organizations had no involvement in the study design, data collection and analysis, decision to publish, or manuscript preparation.
-
The authors confirm contribution to the paper as follows: conception and design of the study: Liu C; kiwifruit collection: Liu C, Qian Y; technical assistance: Zhu X, Zhu J, Zhao J; analysis and interpretation of results: Hu X, Li L, Wang X; data collection: Xiao H, Sun X; draft manuscript preparation: Hu X, Li L, Liu C. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files. The RNAseq raw data analyzed during the current study are available in the CNCB (China National Center for Bioinformation). The project number is PRJCA034092. Further enquiries can be directed to the corresponding author.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Xiaoli Hu, Lin Li, Xuanyu Wang
- Supplementary Table S1 428 metabolites detected in A. chinensis and A. eriantha fruit samples.
- Supplementary Table S2 Correlation coefficients of metabolites contents in ripening fruit of A. chinensis and A. eriantha.
- Supplementary Table S3 TPM values of genes involved in phenylalanine, tryptophan and tyrosine metabolism pathways for A. chinensis and A. eriantha.
- Supplementary Table S4 Contents of detected metabolites in A. chinensis and A. eriantha fruit samples.
- Supplementary Table S5 List of differentially expressed genes correlated with two aromatic amino acid metabolites.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Hu X, Li L, Wang X, Xiao H, Zhu X, et al. 2025. Transcriptional and metabolomic analysis reveal different aromatic amino acid production and potential regulatory mechanisms for two kiwifruit species. Fruit Research 5: e023 doi: 10.48130/frures-0025-0013 

Transcriptional and metabolomic analysis reveal different aromatic amino acid production and potential regulatory mechanisms for two kiwifruit species
- Received: 05 January 2025
- Revised: 17 March 2025
- Accepted: 09 April 2025
- Published online: 04 June 2025
Abstract: Kiwifruit, as a recently domesticated horticultural fruit crop, has substantial economic and nutritional value, particularly its high vitamin C content. In this study, we investigated the different metabolites accumulation in the ripening fruit of two mainly cultivated kiwifruit species A. chinensis and A. eriantha. A total of 428 metabolites were identified, with low correlation coefficients found in amino acid metabolism, as well as in the metabolism of terpenoids and polyketides between two species, indicating the variations in taste, flavor, and nutritional properties. Furthermore, 115 items were clarified as differentially accumulated metabolites. Based on the Kyoto Encyclopedia of Genes and Genomes pathways enrichment analysis, differentially accumulated metabolites (DAMs) were majorly involved in tryptophan metabolism, biosynthesis of secondary metabolites, pyrimidine metabolism, flavonoid biosynthesis, vitamin B6 metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis. Transcriptome analysis revealed 3,168 upregulated genes and 3,174 downregulated genes, we performed Gene Ontology (GO) enrichment analysis suggesting that differentially expressed genes were associated with the aromatic amino acids (AAAs) family catabolic process, which verified the results of metabolome analysis. Expression patterns of homologous genes of these two kiwifruits participating in AAAs metabolism pathway were compared, key structural genes including TSA1, TSB2, TAA, and TAT showed opposite expression profiles. Additionally, we built a co-expression network to identify multiple transcription factors involved in regulating the AAAs metabolism pathways, predominantly belonging to the MYB and MYB_related families, bHLH family, ERF family, and C2H2 family. Our research provides new perspectives on the accumulation of aromatic amino acids between different kiwifruit species. We proposed the potential transcription factors that regulate the AAAs metabolism pathway. This work offers theoretical support for inventing new more nutritional kiwifruit species by altering specific structural genes and corresponding regulatory elements.
-
Key words:
- Kiwifruit /
- Nutrient /
- Metabolism /
- Transcriptomics /
- Aromatic amino acids