









-
Panax notoginseng is a valuable medicinal plant with high economic importance[1]. However, its cultivation and industrial development are severely threatened by pathogenic fungi[2]. Among these diseases, root rot is particularly destructive, often resulting in severe yield losses or even complete crop failure, thereby imposing substantial economic burdens on the P. notoginseng industry[3]. These studies have stated that Fusarium fungi are the main cause of the disease[4,5]. Although the main treatment of root rot is chemical methods, using too much of these chemicals will seriously damage the environmental balance and endanger agricultural security[6]. In response to the growing awareness of these ecological risks, biological control agents (BCAs) have gained increasing attention as promising and eco-friendly alternatives for managing soilborne diseases[7]. Beneficial microorganisms inhibit harmful pathogens and promote plant growth by increasing soil microbial diversity and changing the interactions among microorganisms, ultimately making the microbial community around the root zone stronger and more resistant[8]. Trichoderma is widely recognized as having important biological control capabilities[9]. However, the molecular mechanisms underlying their antifungal activities and their precise contributions to disease suppression remain insufficiently understood, limiting their broader application in sustainable agriculture. Therefore, identifying and studying microbial opponents that specifically attack harmful soil pathogens is important for us to develop strong and lasting biological control strategies.
Among the various microorganisms used for biological control, strains of the genus Trichoderma have attracted special attention because they are so powerful that they can suppress soilborne diseases and help plants grow better[10]. Trichoderma hamatum, a representative member of the family Hypocreaceae, is particularly noted for its broad-spectrum biocontrol efficacy and plant growth-promoting capabilities[11]. In addition to fighting pathogens, T. hamatum has a host of other powerful skills. It can be antibacterial[11], be antioxidant[12], drive away insects[13], inhibit weed growth[14], and help plants grow better[15]. Members of the genus Trichoderma are ubiquitously distributed in diverse ecological niches, particularly in rhizosphere soils, agricultural environments, and habitats enriched with decomposing plant residues or organic matter[16]. Trichoderma species are widely used as biocontrol agents because they can suppress or antagonize plant pathogens. They use several methods to deal with these threats, such as taking nutrients and territory, infecting other fungi, releasing biologically active chemicals, and strengthening the defense system of the host plant[17]. There is a key process called mycoparasitism, which means that Trichoderma's mycelium actively finds harmful fungi and eats them. During this process, hydrolytic enzymes such as chitinases and β-1,3-glucanases are produced, which degrade fungal cell walls and show strong activity against Ascomycota species[18]. In addition to enzymatic degradation, Trichoderma produces various antimicrobial secondary metabolites that contribute to its biocontrol activity. For instance, T. harzianum synthesizes sesquiterpenes and aspinolides via polyketide pathways, which exhibit strong antifungal effects[19]. In addition, transcriptome analysis found that during the parasitic stage, genes responsible for producing cell wall-degrading enzymes and membrane transporters are expressed in large quantities, revealing the molecular mechanism of fungal parasitic interactions[20]. Niche competition also represents an important antagonistic strategy used by Trichoderma. By rapidly colonizing infection sites, exploiting available nutrients, and occupying ecological niches, Trichoderma effectively restricts the proliferation of soilborne pathogens[16]. Collectively, these diverse antagonistic mechanisms highlight the ecological versatility of Trichoderma and underscore its potential as an effective and sustainable biocontrol agent in agricultural systems.
Different Trichoderma strains have shown strong biological control capabilities when dealing with plant diseases caused by Fusarium. Certain Trichoderma strains, including T. asperellum, T. harzianum, and T. atroviride, have been shown to suppress pathogens caused by Fusarium species such as F. solani, F. oxysporum, F. graminearum, and F. sporotrichioides[21,22]. However, the considerable variability in antagonistic efficacy among different Trichoderma strains underscores the importance of strain-specific screening to ensure effective biocontrol outcomes. For example, in vitro assays have shown that certain Trichoderma strains can significantly suppress the mycelial growth of Fusarium through direct antagonistic interactions such as hyphal overgrowth and enzymatic degradation, achieving inhibition rates as high as 82.62%[23]. Despite these encouraging findings, the antimicrobial mechanisms of Trichoderma against F. oxysporum remain incompletely understood, largely because of genetic variation and mechanistic diversity among strains. This knowledge gap highlights the need for systematic investigations that integrate genomics, metabolomics, and functional analyses to unravel strain-specific antifungal strategies. This study highlights the practical importance of Trichoderma in crop production, demonstrating its potential as an environmentally friendly and effective approach for pest management.
The genus Trichoderma exhibits remarkable genetic diversity, which is primarily reflected in extensive variations and polymorphisms within its genome sequences. Comparative genomic studies have revealed notable differences in genome size among Trichoderma strains. For instance, T. harzianum CBMAI-0711 possesses a genome of approximately 32 Mb, whereas other strains such as Th3844 (40 Mb), Th0179 (39 Mb), and T. atroviride CBMAI-00020 (36 Mb) show substantial variation, underscoring both intra- and interspecific genomic diversity[24]. Genome mining has further demonstrated structural variation at the level of biosynthetic gene clusters (BGCs). For example, the recently described species T. agriamazonicum contains 33 BGCs, of which 27 remain uncharacterized[25]. These clusters encode polyketides and other metabolites associated with cell wall degradation, including proteins involved in lignin and chitin breakdown. Such genetic variation provides a molecular basis for the metabolic flexibility and phenotypic plasticity that enable Trichoderma species to adapt to diverse ecological niches and exert broad-spectrum antagonistic effects.
Therefore, studying the genetic structure of Trichoderma can help us better understand why it suppresses Fusarium species, the main antagonists responsible for root rot in P. notoginseng. For this research, we specifically obtained a strain called T. hamatum from the soil around the roots of a particularly well-growing P. notoginseng. Its antagonistic potential against F. oxysporum was evaluated using a five-point dual culture assay, and physical interactions between the two fungi were further characterized by scanning electron microscopy (SEM). To gain deeper mechanistic insights, we adopted a metabologenomics approach, integrating whole-genome sequencing and assembly with untargeted metabolomic profiling[26−29]. Comparative genomic annotation was subsequently performed to identify biosynthetic gene clusters and secondary metabolites associated with antifungal activity.
The study wanted to see if T. hamatum could deal with Fusarium spp., the chief culprit in causing P. notoginseng to rot. It is also necessary to understand why it can suppress these bacteria and find the reasons from a genetic and biochemical perspective. By broadening the genomic understanding of Trichoderma species, this study deepens current insights into their biology and provides a foundation for developing sustainable biocontrol strategies against root rot in P. notoginseng. Moreover, this work offers valuable data for phylogenetic and functional genomic studies of T. hamatum, thereby advancing our understanding of its biocontrol mechanisms and evolutionary adaptations.
-
The T. hamatum strain Z32, isolated from the rhizosphere soil of healthy P. notoginseng, is preserved in the Southwest Microbial Germplasm Resource Bank at Yunnan Agricultural University. For culture revival, the strain was first grown on potato dextrose agar (PDA) plates at 28 °C for approximately one week to allow the formation of dense mycelial mats. Agar plugs from these plates were then transferred into potato dextrose broth (PDB) and incubated under shaking at 200 rpm for about seven days to promote sporulation. The resulting culture was filtered to collect spores, which were flash-frozen in liquid nitrogen and stored at −80 °C for subsequent experiments.
Dual culture assay
-
To evaluate whether T. hamatum Z32 could inhibit the growth of F. oxysporum, both strains were simultaneously inoculated onto PDA plates in a dual culture confrontation assay. The extent of inhibition was assessed using a five-level rating scale. In each plate, a single plug of F. oxysporum was placed at the center, and five agar plugs containing the strain Z32 were distributed evenly around it. The control (CK) contained only the pathogen and no antagonistic fungus. To ensure experimental reliability, three biologically independent assays were conducted, each comprising three parallel technical replicates. After incubation at 28 °C for approximately 72 hours, the antagonistic activity of T. hamatum Z32 was evaluated by measuring the radial growth of F. oxysporum and the width of the clear inhibition zone formed between the two colonies. The inhibition ratio (%) was calculated according to the following formula:
$ \text{Inhibition rate}=\dfrac{\left(R_0-R_1\right)}{R_0}\times100{\text{%}} $ In the inhibition rate formula, R0 corresponds to the mean radial growth of F. oxysporum colonies under control conditions, and R1 refers to the radius of the colony obtained when F. oxysporum was cultured with the antagonistic strain Z32.
Scanning electron microscopy observation of the antagonistic region
-
The morphological interactions between T. hamatum strain Z32 and F. oxysporum were examined using SEM. For the control treatment (CK), only F. oxysporum was cultured on the PDA medium. In contrast, the experimental plates were co-inoculated with both fungi to enable direct mycelial contact. Small circular agar sections (approximately 0.5 cm in diameter) were cut from the region where the two colonies met and used for microscopic preparation.
Samples for SEM were prepared following standard fixation and dehydration procedures. Agar blocks were washed three times with 0.1 M phosphate-buffered saline (PBS) (pH 7.4) for 15 min per wash, then fixed with 1% osmium tetroxide (OsO4) in PBS for 1–2 hours at room temperature in the dark. After fixation, samples were washed again in PBS and dehydrated in a graded ethanol series (30%–100%), followed by immersion in isoamyl acetate and critical-point drying. The dried specimens were mounted on aluminum stubs with conductive carbon tape, sputter-coated with gold (~30 s), and examined using a ZEISS Gemini Sigma 300 SEM (Jena, Germany).
Molecular characterization of T. hamatum Z32
-
Genomic DNA of T. hamatum Z32 was extracted via the cetrimonium bromide (CTAB) method. The internal transcribed spacer (ITS) region was amplified using the primers ITS1/ITS4 with polymerase chain reaction (PCR) conditions of 94 °C for 10 min, then 35 cycles of 94 °C for 30 s, 54 °C for 30 s, and 72 °C for 30 s, followed by a final 10-min extension at 72 °C. Finally, the sample was heated at 72 °C for another 10 min to allow all the DNA to replicate. After purifying the PCR products, we performed Sanger sequencing using the primer pair ITS1/ITS4 (produced by Shanghai Shenggong Biotechnology Co., Ltd. completed). In order to determine the species of the fungi, we compared the detected sequence data with the NCBI ITS database using a BLAST search.
Genomic analysis: sequencing and assembly procedures
-
For genome assembly, T. hamatum Z32 was analyzed using a hybrid sequencing approach that merged high-fidelity short reads with long-read data to ensure both accuracy and continuity in the final assembly. For short-read sequencing, a reference database was first built according to the procedure outlined in[30]. We sequenced the T. hamatum Z32 genome using the DNBSEQ-T7 system (MGI, China), generating paired-end reads of 150 nucleotides (PE150).
For long-read sequencing, we used the PacBio Revio platform and prepared libraries following the SMRTbell Express Template Prep Kit 2.0 protocol (Pacific Biosciences). About 15 μg of high-quality genomic DNA was first treated to remove single-stranded overhangs, repair damage, polish ends, and add A-tails. The processed fragments were then ligated with T-tailed SMRTbell adapters at 20 °C for roughly 60 min.
We purified the ligated DNA library and assessed its fragment length and concentration using the FEMTO Pulse and Qubit instruments. Roughly 3 μg of the library was then size-selected on a BluePippin device to remove fragments smaller than 15 kb, followed by an additional round of purification and quantification. The final library was annealed with sequencing primers, mixed with Sequel II DNA polymerase at a concentration of 120 pM, and loaded onto an 8 M SMRT Cell via diffusion. We carried out sequencing on the Sequel II system for a runtime of about 1,800 min, generating long reads of 15–20 kilobases. Frasergen Bioinformatics (Wuhan, China) handled all library preparation and sequencing procedures.
Genome assembly was performed at the contig level using Hifiasm. The assembled genome was analyzed to determine the nucleotide composition (A, G, C, T, N) and overall GC content. To evaluate the genome's completeness and coverage consistency, sequencing reads generated from both short- and long-read technologies were aligned back to the assembly with BWA and Minimap2. Metrics including the mapping rate, genome coverage, and depth distribution were calculated. Coverage depth was computed for each genomic position, and a nonoverlapping 1,000-bp sliding window (or the full length for shorter contigs) was used to calculate the average sequencing depth and GC content per window. These data were visualized in density plots of GC content versus sequencing depth to evaluate GC bias and potential contamination.
The genome's completeness was further assessed using Benchmarking sets of Universal Single-Copy Orthologs (BUSCO) with the fungi_odb10 database, reporting metrics for completeness, fragmentation, and gene loss based on single-copy orthologs from OrthoDB. We used Merqury to estimate the assembly's quality value (QV) by comparing k-mer profiles derived from the genome assembly and the sequencing reads. We reported the genome size along with other assembly quality indicators and submitted the raw sequencing datasets to NCBI, accessible via BioProject PRJNA1163295 and BioSample SAMN43845003.
Prediction of the genome's composition
-
To annotate transfer RNA genes, the T. hamatum Z32 genome was analyzed with tRNAscan-SE. Ribosomal RNA (rRNA) genes were predicted with RNAmmer (v1.2). In addition, microRNA (miRNA) and small nuclear RNA (snRNA) sequences were predicted using covariance models from the Rfam database (
rfam.org ) via the INFERNAL software included in Rfam. We evaluated the completeness of the genome assembly with BUSCO, using single-copy orthologs derived from OrthoDB. At the same time, antiSMASH (version 7.0) was applied to detect and characterize gene clusters associated with secondary metabolite production.Comparative genomic analysis
-
To investigate genetic differences between T. hamatum Z32 and seven other Trichoderma strains, and to confirm the taxonomic identity of Z32, protein sequences from all eight strains were clustered according to sequence similarity using the OrthoFinder2 pipeline. Pairwise protein sequence similarities were computed with DIAMOND, and orthologous groups were identified by OrthoFinder2. Sequences from each single-copy homologous gene family were first aligned using MUSCLE. The resulting alignments were subsequently concatenated to generate a comprehensive supergene alignment in PHYLIP format.
We used RaxML's maximum likelihood method to infer the kinship between species. To determine when they were separated, we used r8s and MCMCtrees in the PAML suite to calibrate the evolutionary tree on the basis of fossil data and timelines published in studies such as TimeTree. The CAFE tool helped us analyze changes in gene family size according to orthogroup classification and identify those groups with significant changes. These groups were functionally annotated through Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment, and statistical significance was adjusted with the false discovery rate (FDR) to avoid errors caused by multiple comparisons.
We analyzed each single-copy homologous gene family shared across species with the branch-site model in codeml (PAML suite) to detect the signatures of positive selection.
Functional annotation and genomic analysis
-
We annotated the protein-coding genes of T. hamatum Z32 by comparing their sequences with the NR, KEGG, Clusters of Orthologous Groups (COG), GO, and Swiss-Prot databases. Functional predictions were performed by aligning proteins with BLASTp in Diamond (version 0.9.12.113). For GO classification, InterProScan (version 5.50-84.0) was used to assign relevant annotations.
Carbohydrate-active enzymes (CAZymes) were analyzed using the dbCAN2 process, which combines dbCAN-HMMdb V7 and HMMER (version 3.1b2). CAZymes were analyzed following the standard dbCAN2 workflow. A stringent E-value of 1 × 10−15 and a minimum coverage of 0.35 were applied to filter the sequences. Antimicrobial resistance proteins were then identified by aligning the predicted sequences to the comprehensive antibiotic resistance database (CARD) database (v3.0.1) with Diamond BLASTp, retaining only the top-scoring alignment for each query. Virulence factors were annotated by alignment against the database of fungal virulence factors (DFVF) and pathogen–host interactions database (PHI) protein databases (E-value ≤ 1 × 10–5), with the highest scoring hits retained.
Signal peptides indicative of secreted proteins were predicted using SignalP (v4.1), and transmembrane helices were identified using TMHMM (v2.0c), which applies a hidden Markov model to detect transmembrane regions.
Secondary metabolite analysis and identification
-
For metabolomic profiling, 50 μL of each sample was mixed with 150 μL of an extraction solvent containing an internal standard (acetonitrile–methanol, 1:4, v/v). The mixture was vortexed for 3 min and centrifuged at 12,000 rpm and 4 °C for 10 min. A 150-μL aliquot of the supernatant was transferred to a fresh tube, frozen at −20 °C for 30 min, and centrifuged again under the same conditions for 3 min. Subsequently, 120 μL of the clarified supernatant was loaded into autosampler vial inserts for liquid chromatography–mass spectrometry (LC-MS) analysis, with all steps performed on ice to preserve the metabolites' integrity.
Chromatographic separation was conducted using a Waters ACQUITY Premier HSS T3 column (1.8 μm, 2.1 × 100 mm) with Solvent A (0.1% formic acid in water) and Solvent B (0.1% formic acid in acetonitrile). The column temperature was maintained at 40 °C, with a flow rate of 0.4 mL/min and an injection volume of 4 μL. Analyses were performed in both positive and negative ionization modes. The elution gradient started at 5% B, increased to 20% B over 2 min, then to 60% B within 3 min; after that, it increased further to 99% B in 1 minute (held for 1.5 min), returned to 5% B in 0.1 min, and equilibrated for 2.4 min.
Mass spectrometry was carried out on a Sciex system using Analyst TF software (v1.7.1) in information-dependent acquisition (IDA) mode. The source conditions were nebulizer gas 1 (GAS1) = 50 psi, heater gas 2 (GAS2) = 50 psi, curtain gas (CUR) = 25 psi, and temperature (TEM) = 550 °C. Declustering voltages were set at 60 V (positive) and −60 V (negative), whereas the ion spray voltages were 5,000 V and −4,000 V, respectively. Time-of-flight mass spectrocopy (TOF MS) scans covered m/z 50–1,000 with 200 ms of accumulation time, and product ion scans ranged from m/z 25–1,000 with 40 ms of accumulation time. The collision energy was ±30 V with a 15-V spread, unit resolution, a charge state of 1, and an intensity threshold of 100 cps. Isotopes within 4 Da were excluded, the mass tolerance was 50 ppm, and the number of candidate ions per cycle was limited. Raw data were converted to mzXML format using ProteoWizard.
Data were processed using XCMS for peak detection, alignment, and correction of retention time. Peaks missing in over 50% of samples were removed, and the remaining missing values were imputed via the k-nearest neighbors (KNN) method. Peak areas were normalized using support vector regression (SVR). Metabolites were annotated according to internal and public databases, prediction libraries, and metDNA technology. Quality control (QC) samples with coefficient of variation (CV) < 0.3 were used to assess data quality, and only metabolites with identification scores of > 0.5 and CV < 0.3 in the QC samples were retained. When combining positive and negative ion datasets, the metabolites with the lowest CVs were prioritized to ensure reliability.
Statistical analysis
-
We calculated the mean and standard deviation for each measurement and carried out statistical analyses using SPSS software (version 27.0, IBM, Armonk, NY, USA).
-
In the five-point dual culture assay, T. hamatum Z32 effectively inhibited F. oxysporum, producing a clearly visible inhibition zone with diffuse yet well-defined edges (Fig. 1c). Quantitative analysis showed an inhibition rate of 62% (Supplementary Table S1). The results showed that strain Z32 has strong antifungal properties, and it may have used several methods to prevent the growth of fungi, such as releasing biologically active secondary metabolites and seizing their common living space.
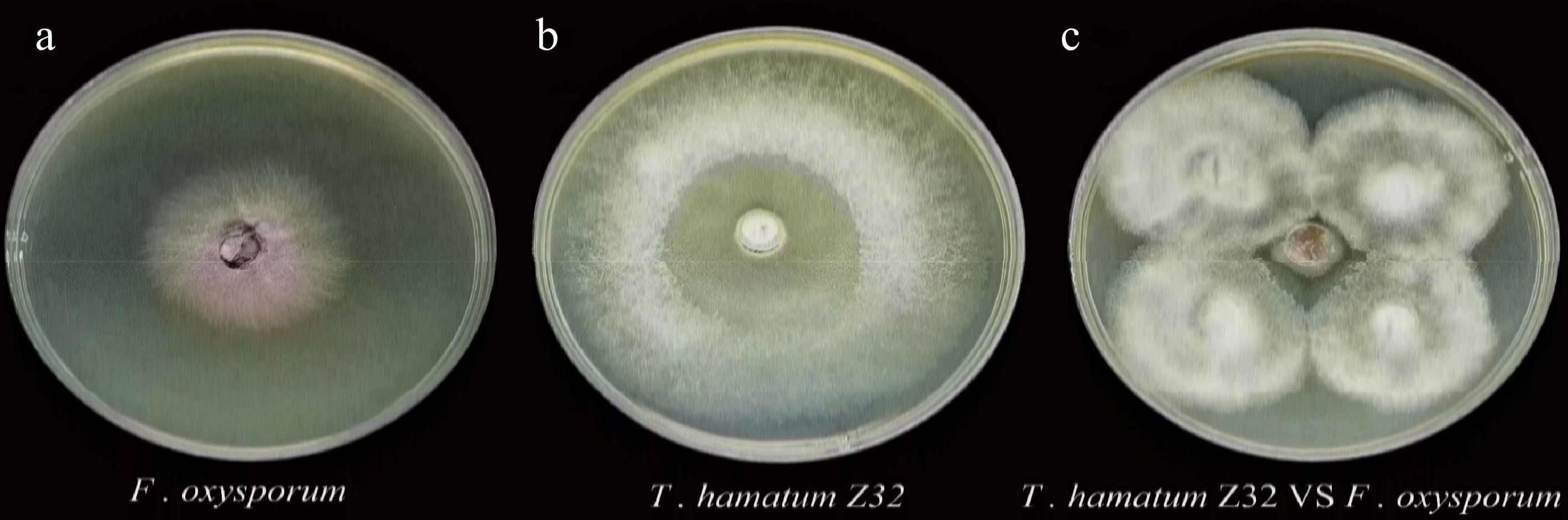
Figure 1.
Individual cultures and dual culture assay of T. hamatum Z32 and F. oxysporum.
(a) F. oxysporum cultured alone (pathogen control); (b) T. hamatum Z32 cultured alone (antagonist control); (c) dual culture plate showing F. oxysporum inoculated at the center and T. hamatum Z32 inoculated at four peripheral points.The SEM examination found that after F. oxysporum grew together with T. hamatum Z32, its physical structure changed significantly. Under monoculture conditions, the hyphae of both F. oxysporum and T. hamatum Z32 exhibited intact structures with smooth surfaces (Fig. 2a, b). In contrast, co-culture conditions induced pronounced hyphal shrinkage and surface wrinkling in F. oxysporum (Fig. 2c). These structural deformations suggest that strain Z32 exerts antagonistic effects, likely through mechanisms such as nutrient competition and occupation of ecological niches. Collectively, the SEM observations provide further evidence that T. hamatum Z32 effectively suppresses the growth of F. oxysporum.

Figure 2.
Electron microscope images taken using scanning technology were recorded and used to show the surface and structural characteristics of the antimicrobial species.
(a) F. oxysporum; (b) T. hamatum Z32; (c) co-culture of F. oxysporum and T. hamatum Z32. The arrows mark where T hamatum Z32 interacts with harmful fungi, and we can see that T hamatum Z32's mycelium entangles the pathogen. This entanglement can cause the pathogen's mycelium to wither and shrink, and can also cause structural damage through resistance.Genomic features of T. hamatum Z32
-
High-precision (HiFi) sequencing of T. hamatum strain Z32 generated 7.35 Gb of raw data, with an average sequencing depth of 172.2 times the genome size. After quality control, a total of 376,147 high-quality reads were retained (Supplementary Table S2). By integrating HiFi reads with Hi-C data, the T. hamatum Z32 genome was reconstructed into 20 contigs totaling 42.69 Mb, with an N50 of 6.74 Mb and a GC content of 46.26% (Fig. 3; Supplementary Table S3). Mapping the sequencing reads back to the assembly yielded a high alignment rate of 98.36% and genome coverage of 99.96%, demonstrating the robustness of the assembly. BUSCO analysis indicated that 98.6% of the core conserved genes were fully represented, confirming the high quality and reliability of the genome assembly and providing a solid basis for subsequent functional and comparative genomic studies (Supplementary Table S4).

Figure 3.
Circos plot displaying the genome of T. hamatum Z32.
From outermost to innermost, the concentric rings represent contigs, gene density, repeat sequences, noncoding RNAs (ncRNAs), and GC content. Although the genome assembly includes 20 contigs, only the 7 longest are shown, as the shorter contigs contribute little or no gene information.Genome feature identification and annotation
-
The T. hamatum Z32 genome contains 12,137 protein-coding genes, with an average gene length of 2,589 bp and an average coding sequence (CDS) length of 1,242 bp. Each of these genes has an average of 2.46 exons, and the average lengths of the exons and introns are 505.15 and 923.51 bp, respectively (Supplementary Table S5). The genome of T. hamatum Z32 contains 450 noncoding RNAs (ncRNAs), including 221 transfer RNAs (tRNAs), 204 ribosomal RNAs (rRNAs), and 25 small nuclear RNAs (snRNAs) (Table 1).
Table 1. ncRNA statistics and composition in T. hamatum Z32.
Type Copy Average length (bp) Total length (bp) % of genome miRNA 0 0.00 0 0.00 tRNA 221 87.54 19,346 0.05 rRNA 204 2,209.75 450,790 1.06 18S 75 1,794.55 134,591 0.32 28S 79 3,929.13 310,401 0.73 5S 50 115.96 5,798 0.01 snRNA 25 138.20 3,455 0.01 CD-box 14 129.14 1,808 0.00 hypermethylated allele-coxA associated (HACA)-box 3 170.33 511 0.00 splicing 8 142.00 1,136 0.00 scaRNA 0 0.00 0 0.00 The function annotation process used databases such as InterPro, GO, KEGG, SwissProt, TrEMBL and NR, and the specific information is presented in Table S6. Among the predicted genes in T. hamatum Z32, 7,943 (about 73.3%) were classified into the three principal GO categories: Biological processes, cellular components, and molecular functions. Biological processes were the most abundant, with genes predominantly involved in metabolic activities, cellular functions, and processes occurring in single organisms (Supplementary Fig. S1). KEGG pathway analysis revealed that 4,157 genes (38.36%) belong to the five major pathway classes, with metabolism-related pathways forming the largest group, particularly those associated with carbohydrate, amino acid, and lipid metabolism (Supplementary Table S6; Supplementary Fig. S2).
Comparative genomic analysis
-
By conducting a phylogenetic analysis of 1,884 single-copy orthologically conserved genes, we inferred the evolutionary relationships among the eight Trichoderma species and one outgroup strain, as shown in Fig. 4. The analysis estimated the divergence time from the outgroup, Saccharomyces cerevisiae S288C, at approximately 424 million years ago (MYA). Within the Trichoderma genus, T. hamatum diverged from T. asperelloides and T. asperellum CBS433.97 about 6 MYA, whereas T. asperelloides and T. asperellum CBS433.97 separated from each other around 2 MYA (Fig. 4).
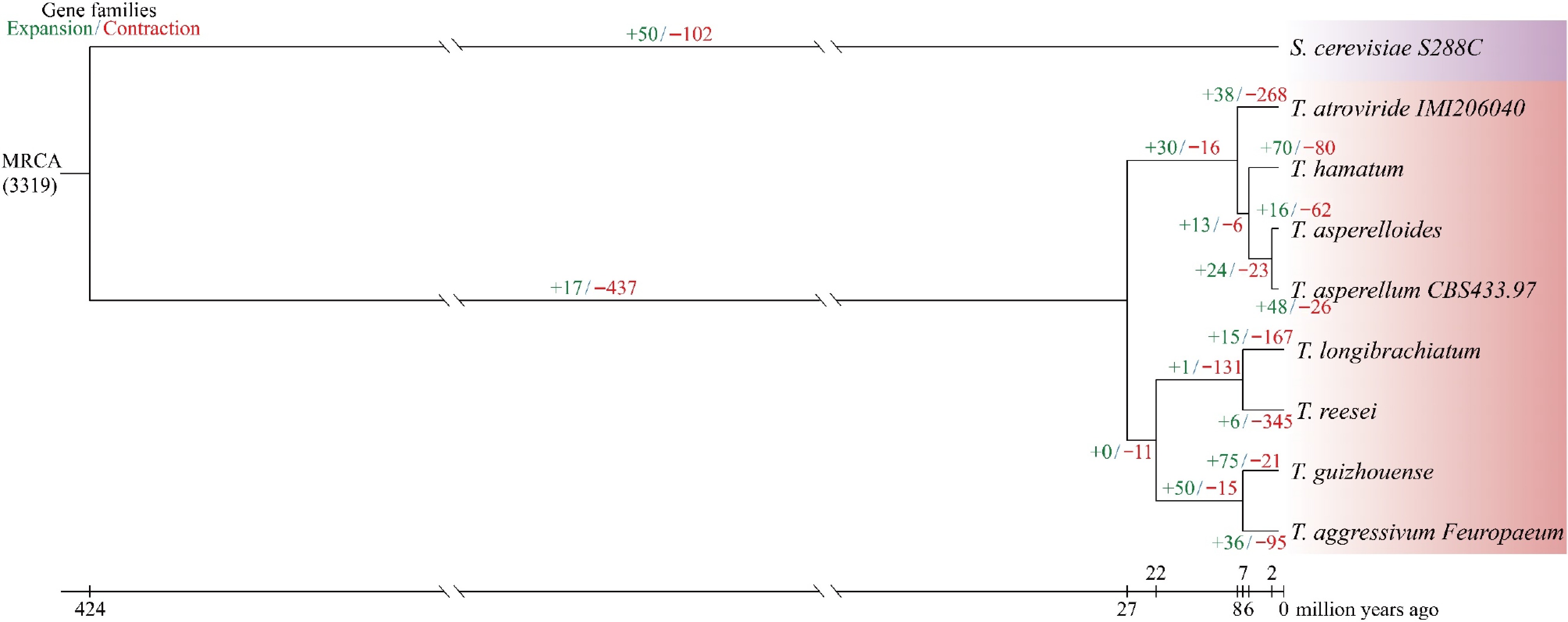
Figure 4.
A maximum likelihood phylogenetic tree was generated using 1,884 single-copy orthologs from eight Trichoderma strains and one outgroup.
All nodes were supported with 100% bootstrap values, and branch lengths indicate the estimated divergence times in millions of years ago (MYA). For each species, green and red numbers denote gene family expansions and contractions, respectively, while the background color reflects the corresponding taxonomic group.Evolutionary analysis of the nine fungal species showed that gene family contractions were more frequent than expansions (Fig. 4). In Trichoderma, T. hamatum, T. asperelloides, and T. asperellum CBS433.97 exhibited expansions in 70, 16, and 48 gene families, respectively, whereas contractions were observed in 80, 62, and 26 gene families, respectively.
Analysis of enzyme production potential in T. hamatum Z32
-
CAZymes are essential for diverse biological processes in Trichoderma species, particularly their mycoparasitic lifestyle. Genomic analysis of T hamatum Z32 with the dbCANseq database found that it has 451 genes responsible for encoding CAZymes. The predicted genome of T. hamatum Z32 contains genes assigned to six major CAZyme families: 252 glycoside hydrolases (GHs), 13 carbohydrate-binding modules (CBMs), 73 auxiliary activity enzymes (AAs), 25 carbohydrate esterases (CEs), 78 glycosyltransferases (GTs), and 10 polysaccharide lyases (PLs). Specific data can be seen in Fig. 5. Among these, GHs constituted the largest CAZyme family.
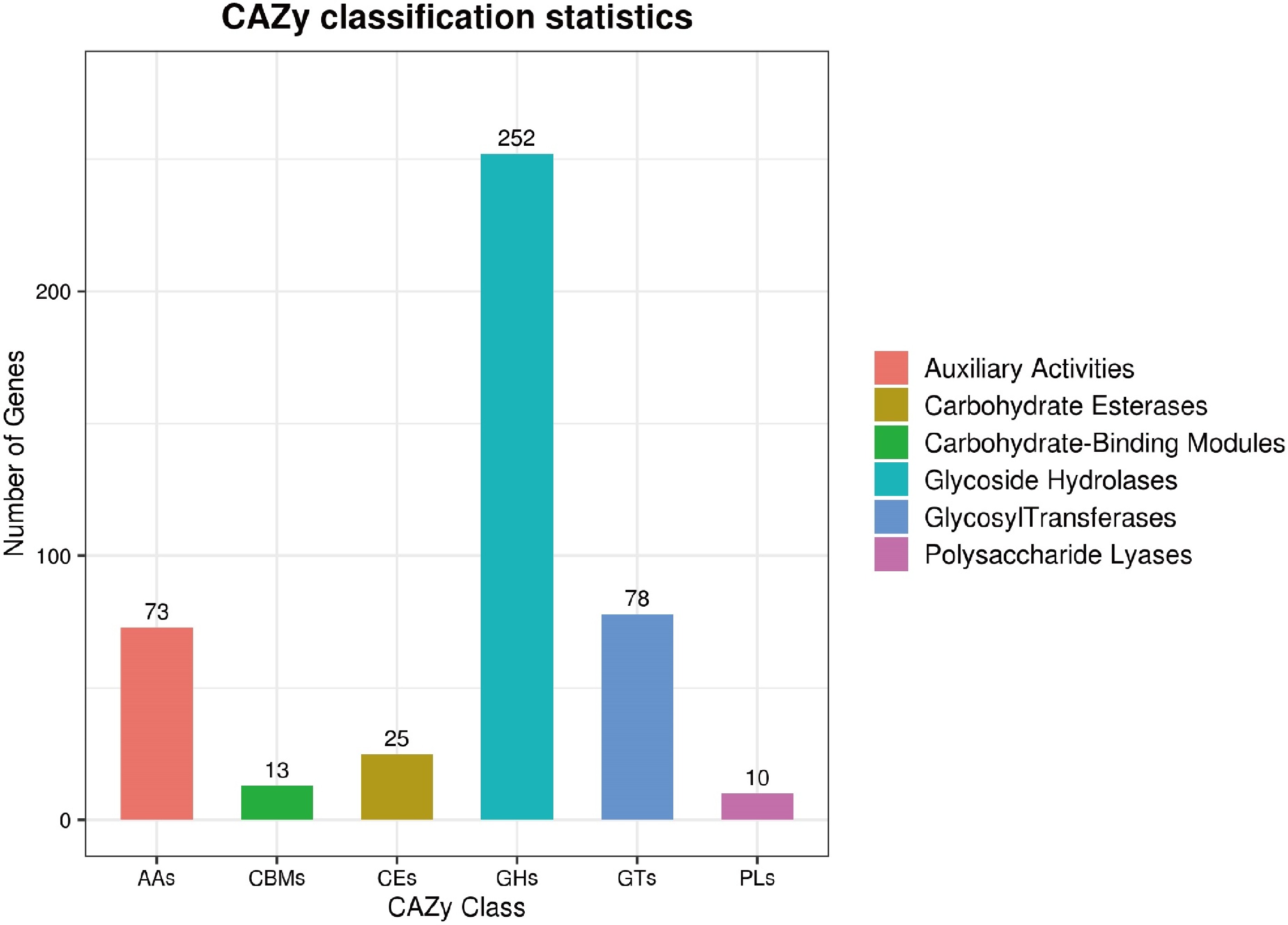
Figure 5.
The predicted CAZyme genes in T. hamatum Z32 were grouped into different families.
In the T. hamatum Z32 genome, the identified genes were categorized into six main CAZyme families: auxiliary activities (AAs), carbohydrate esterases (CEs), carbohydrate-binding modules (CBMs), glycoside hydrolases (GHs), glycosyltransferases (GTs), and polysaccharide lyases (PLs).In addition, annotation against the PHI-base database revealed 4,706 genes associated with pathogen–host interactions (Supplementary Table S7). Most of these genes are associated with weaker toxicity (2,086 genes), followed by genes that do not affect pathogenicity (1,953 genes), and finally those that lead to loss of pathogenicity (264 genes) (Fig. 6; Supplementary Table S7).
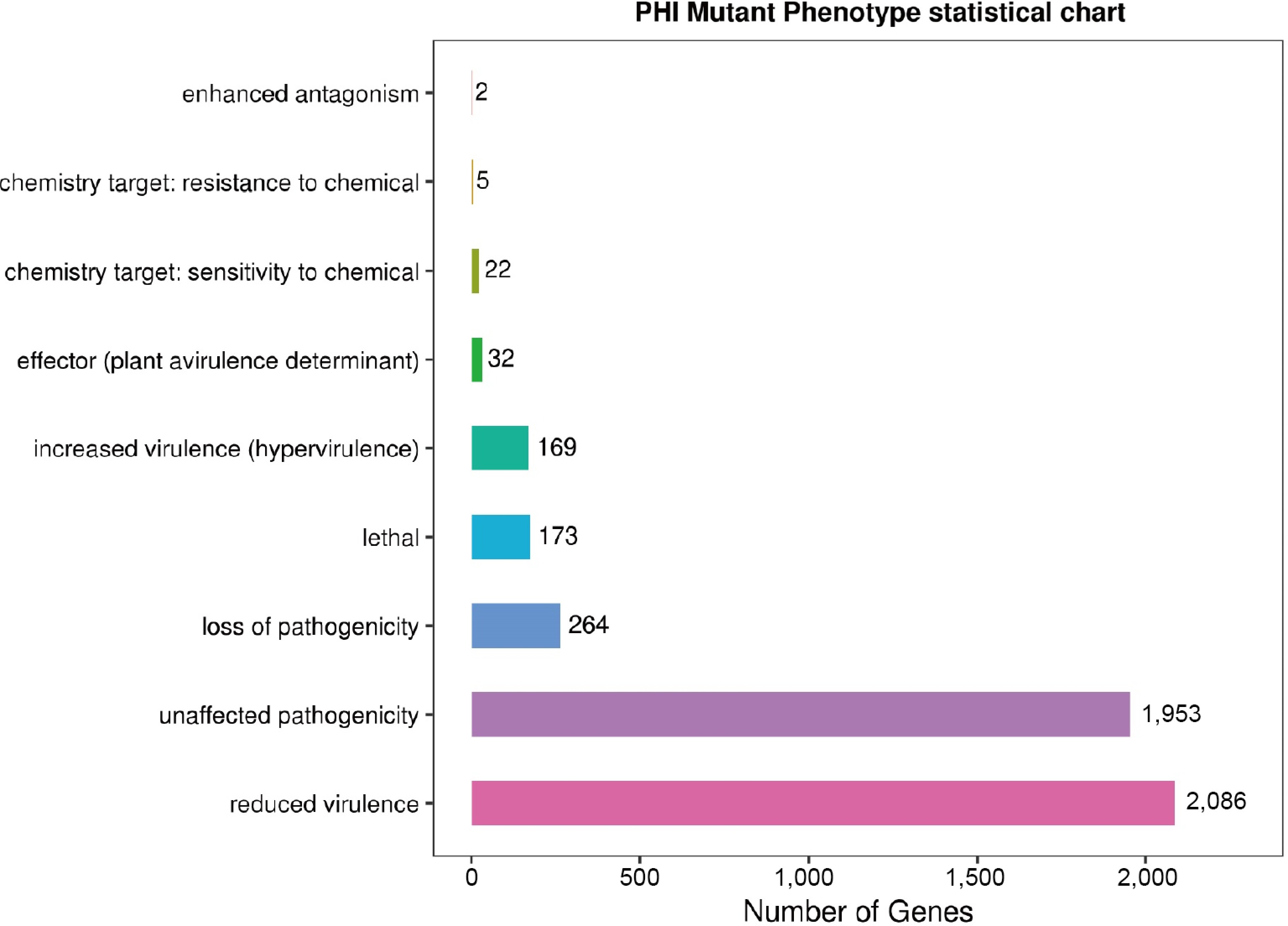
Figure 6.
The figure shows the distribution of pathogen–host interaction (PHI) mutation phenotypes, with nine mutation types along the vertical axis and the corresponding number of annotated genes on the horizontal axis.
Examining the gene assembly for producing secondary metabolites in T. hamatum Z32
-
We used antiSMASH software (v7.1.0) to analyze the genome of T. hamatum Z32 to find genes that produce secondary metabolites. It was found that 29 such gene clusters were distributed across nine scaffolds (Supplementary Table S8). The T. hamatum Z32 genome contains 15 biosynthetic gene clusters (BGCs) related to nonribosomal peptide synthetases (NRPSs) and NRPS-like variants, 2 clusters corresponding to Type I polyketide synthases, and 7 clusters associated with terpene synthesis. To evaluate the strain's biocontrol potential, 10 of these BGCs were linked to previously characterized secondary metabolites, with their predicted chemical structures and genomic locations summarized in Fig. 7.
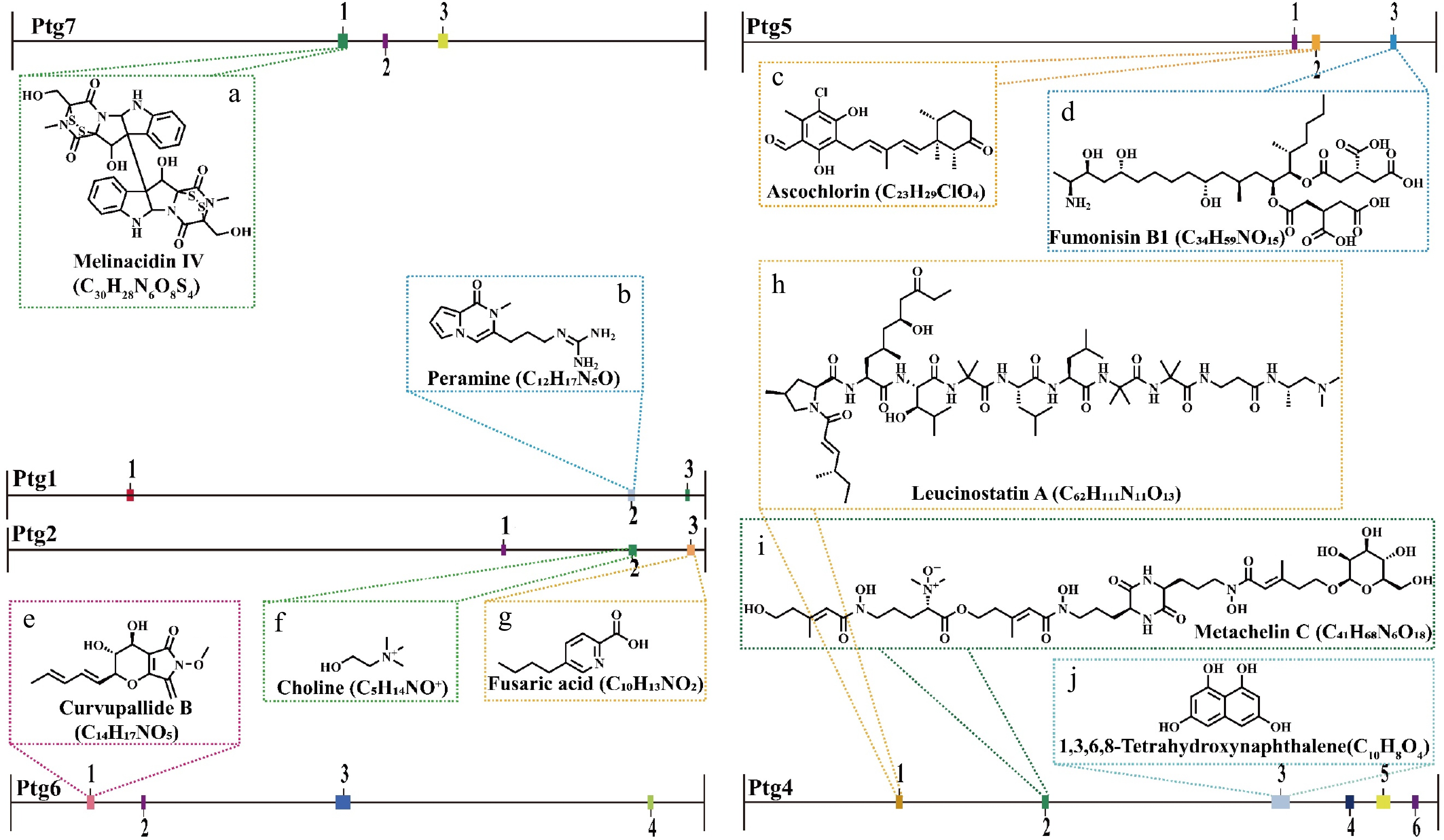
Figure 7.
The secondary metabolite biosynthetic gene clusters identified in the T. hamatum Z32 genome using antiSMASH (version 7.0).
The predicted compounds associated with each cluster are: (a) Melinacidin IV (C30H38N4O5S4), (b) peramine (C12H17N5O), (c) ascochlorin (C23H29ClO4), (d) fumonisin B1 (C34H59NO15), (e) curvupallide B (C14H17NO5), (f) choline (C5H14NO+), (g) fusaric acid (C10H13NO2), (h) leucinostatin A (C62H111N11O13), (i) metachelin C (C41H68N6O18), and (j) 1,3,6,8-tetrahydroxynaphthalene (C10H8O4).Among them, three BGCs exhibited 100% amino acid sequence homology with characterized clusters: Region 1.2 of Ptg1 encodes an NRPS cluster responsible for peramine biosynthesis (Fig. 7b), Region 2.2 of Ptg2 encodes an NRPS-like cluster related to choline biosynthesis (Fig. 7f), and Region 4.3 of Ptg4 harbors NRPS and T1PKS clusters involved in 1,3,6,8-tetrahydroxynaphthalene biosynthesis (Fig. 7j). Additionally, the cluster located in Region 7.1 (Pgt7, NRPS) showed 75% sequence similarity to the melinacidin IV biosynthetic cluster (Fig. 7a). Other identified clusters were predicted to participate in the biosynthesis of ascochlorin (Fig. 7c), fumonisin B1 (Fig. 7d), curvupallide-B (Fig. 7e), fusaric acid (Fig. 7g), leucinostatin A (Fig. 7h), and metachelin C (Fig. 7i).
Secondary metabolite analysis of T. hamatum Z32
-
To validate the predicted secondary metabolites via BGC analysis, non-targeted metabolomic profiling was performed on the fermentation broth of T. hamatum Z32. In total, 5,883 secondary metabolites were detected (Supplementary Table S9), with amino acids and their derivatives accounting for the largest fraction (26.99%), followed by benzene and its substituted derivatives, which comprised 15.84% of the identified compounds (Supplementary Fig. S3). In addition, diverse compound classes were detected, including indole derivatives (e.g., indole-3-carbinol, naltrindole), choline-related compounds (e.g., choline, dehydrocorydaline), terpenoids, and phenolic acids (e.g., ferulic acid).
Principal component analysis (PCA) was performed to assess the overall metabolic variation among T. hamatum Z32 samples. The first two principal components, PC1 and PC2, explained 66.08% and 18.52% of the total variance, respectively, capturing the major metabolic differences. The QC samples are closely clustered together in the two-dimensional score plot, proving that this analytical method is both stable and reliable. Within the treatment group, two of the three biological replicates clustered closely, whereas one replicate showed a slight deviation in its principal component space, potentially reflecting inherent metabolic variation or differential treatment responses. Nevertheless, the overall within-group variation was markedly smaller than the between-group variation, supporting the reliability and interpretability of the metabolomic dataset (Supplementary Fig. S4).
-
F. oxysporum is the main pathogen responsible for root rot in P. notoginseng, representing a major challenge to the sustainable cultivation of this medicinal plant[31]. Currently, disease management largely depends on chemical fungicides[32]; however, their widespread use has caused unintended consequences, including pesticide residues and the emergence of fungicide-resistant microbial populations[33]. These problems show that we are in urgent need of safer and more effective alternatives than traditional chemical treatments[34]. Under such conditions, biological control strategies have attracted considerable attention because of their safety, environmental friendliness, and sustainability[35]. The expanding use of microbial inoculants, particularly fungal biocontrol strains, underscores the need for effective and dependable biocontrol agents (BCAs) within sustainable plant disease management programs[36].
There are a variety of microorganisms in the soil around plant roots, among which bacteria that help plant growth, known as plant growth-promoting rhizobacteria (PGPRs), have been the most widely studied. They can make plants grow better[37], stimulate plant defense responses[38], and suppress harmful bacteria in the soil[39]. Studies have found that using PGPRs can greatly reduce reliance on artificial fertilizers and pesticides, as these not only protect the ecological balance but also reduce harm to human health[40].
Trichoderma fungi are recognized as vital members of the PGPR community. When they establish themselves in plant roots, they can alter the microbial landscape of the rhizosphere, enrich populations of beneficial fungi, and enhance plants' tolerance of soils contaminated by heavy metals such as cadmium and mercury[41]. In addition to fostering plant growth through mechanisms like phosphate solubilization[11] and nitrogen fixation[42], these fungi produce a variety of antimicrobial compounds that help suppress pathogenic microorganisms[43]. Among them, T. hamatum stands out for its ability to generate diverse secondary metabolites that specifically inhibit plant pathogens, demonstrating considerable potential as an environmentally sustainable biocontrol agent[44].
In the case of P. notoginseng, a T. hamatum strain isolated from its rhizosphere exhibited strong antagonistic activity against root rot. Laboratory experiments showed that this strain effectively curtailed the growth of F. oxysporum, the primary pathogen behind the disease (Supplmentary Table S1). The combination of broad-spectrum antagonism and plant growth-promoting properties positions Trichoderma species as key contributors to sustainable agriculture, and they are increasingly recognized as valuable tools for eco-friendly disease management strategies[17].
To explore the genomic characteristics of the biocontrol fungus T. hamatum Z32, we performed comprehensive whole-genome sequencing followed by comparative genomic analyses. The assembled genome was subsequently compared with those of seven other Trichoderma species and one outgroup strain to elucidate its evolutionary relationships and functional attributes. According to the results of evolutionary tree analysis, strain Z32 has the closest genetic relationship with T. asperelloides and T. asperellum CBS43397. Comparative analysis revealed significant gene family dynamics, with 70 families that have expanded and 80 families that have contracted (Fig. 4). GO enrichment analysis revealed that in T. hamatum Z32, the expanded gene families were primarily enriched in metabolic pathways associated with phenylpropanoid metabolism, particularly those involved in ferulic acid conversion and cinnamic acid degradation (Supplementary Fig. S5a). As cinnamic, ferulic, and p-hydroxycinnamic acids, key intermediates in plant secondary metabolism, are derived from the phenylpropanoid pathway, the capacity of strain Z32 to convert or degrade these compounds may contribute to its plant growth-promoting effects. In contrast, the contracted gene families were mainly enriched in transmembrane transporter activity (Supplementary Fig. S5b), potentially affecting the strain's capacity to utilize environmental resources or metabolic products. KEGG pathway enrichment analysis indicated that the expanded gene families of T. hamatum Z32 were primarily associated with the lysine biosynthetic pathway (Supplementary Fig. S5c). This pathway likely plays a pivotal role in the organism's metabolism, potentially facilitating increased lysine accumulation, enhanced metabolic flexibility, and improved adaptability under diverse environmental or symbiotic conditions. Conversely, the contracted families were enriched in sulfur metabolism (Supplementary Fig. S5d), which may reduce the organism's sulfur utilization efficiency and constrain its ecological adaptability. Notably, previous studies have reported that T. guizhouense exhibits strong plant growth-promoting and biocontrol capabilities[41], and the high genomic similarity between Z32 and T. guizhouense (Supplementary Fig. S6) further supports the substantial biocontrol potential of this strain.
Trichoderma fungi can directly fight plant pathogens. They have several methods: They can parasitize other fungi, make antibacterial substances[45], release enzymes that destroy cell walls[46], and compete for territory in root-zone ecosystems. These antagonistic activities are closely linked to the functional genes encoded within Trichoderma genomes[32]. In line with this, SEM revealed that hyphal coiling by Z32 induced the shrinkage and breakage of F. oxysporum hyphae. Similar mechanisms have been widely documented. Zhang et al.[47]reported that the suppression of F. oxysporum by T. harzianum E5 primarily results from direct physical interaction. During this process, the hyphae of T. harzianum coil around the pathogen's mycelia, thereby restricting its growth and proliferation. Rees et al.[48]demonstrated that T. hamatum suppresses Armillaria mellea through hyphal coiling and coverage, completely inhibiting the pathogen's growth. Similarly, Zhang et al.[49]observed that T. hamatum exhibits parallel hyphal growth and coiling around Sclerotinia spp., leading to hyphal deformation, shrinkage, and breakage. Taken together, these findings, together with our observations, suggest that T. hamatum Z32 likely suppresses F. oxysporum through a combination of hyphal coiling and robust competitive growth.
Genomic functional annotation, antagonistic assays, and electron microscopy collectively indicated that T. hamatum possesses significant potential to suppress pathogenic fungi through mycoparasitism. On the molecular scale, secondary metabolites in plants are important for resisting pathogens because of their antibacterial ability. Genetic analysis of genes related to secondary metabolites showed that T. hamatum Z32 contains 29 BGCs and can produce five major categories of secondary metabolites. These include alkaloids (such as peramine and fusaric acid), amine derivatives (such as choline), polyketides (such as 1,3,6,8-tetrahydroxyphthalene, ascochlorin, fumonisin B1), NRPS-derived peptides (such as melinacidin IV, curvupallide-B, leucostatin A, and metachelin C), and siderophores (such as metachelin C).
Previous studies have demonstrated that alkaloids exhibit broad-spectrum antifungal activity against diverse plant pathogens, including Fusarium spp.[50]. Polyketides, such as wailupemycins M–P and bikaverin, have been reported to inhibit both phytopathogenic fungi (e.g., Fusarium spp.) and oomycetes (e.g., Phytophthora)[51]. Similarly, nonribosomal peptides, including cyclic lipopeptides and fusaric acid derivatives, display potent antifungal activity and can markedly suppress the radial growth of F. oxysporum[51]. Moreover, siderophores derived from Pseudomonas stutzeri exhibit strong antagonistic effects against F. oxysporum[52]. Taken together, these findings support the hypothesis that BGC-derived secondary metabolites produced by T. hamatum Z32 may contribute substantially to its inhibitory activity against F. oxysporum.
Through genomic analysis and verification of existing research results, we speculated that the inhibitory effect of this strain may be related to its secondary metabolites; however, direct experimental validation of their functional roles is still lacking. Thus, the current results indicate only a potential association rather than a definitive causal relationship. Moreover, given that metabolite expression may exhibit dynamic changes under different culture conditions, the predictions may be subject to certain biases. Future research will focus on the following aspects: (1) Purification and isolation of key secondary metabolites; (2) in vitro antifungal assays to verify the biological functions of individual compounds; (3) determination of whether the antifungal activity of this strain is indeed attributable to these metabolites, in conjunction with the activity of hydrolytic enzymes; and (4) integration of transcriptomic and metabolomic data to elucidate the molecular mechanisms linking metabolite biosynthesis with their functional roles, thereby providing a comprehensive understanding of their contribution to the antagonistic activity of this strain.
In T. hamatum Z32, a total of 451 carbohydrate-active enzymes (CAZymes) were identified through genome annotation. The majority were glycoside hydrolases (252), followed by glycosyltransferases (78) and auxiliary activity enzymes (73), indicating a diverse enzymatic repertoire involved in carbohydrate metabolism. This enzyme composition suggests that Z32 possesses strong potential for carbohydrate degradation and modification, which may underlie its interactions with host plants and its capacity to suppress pathogens. GHs, as components of plant defense systems, can catalyze specific reactions that contribute to pathogen resistance[53].
Integration with nontargeted metabolomic analysis of Z32 fermentation broth further revealed the presence of bioactive metabolites with antimicrobial potential, including choline derivatives, triterpenoids, and indole-related compounds. Choline compounds have been reported to participate in osmoregulation and metabolic processes, indirectly enhancing plants' stress tolerance[54]. Triterpenoids serve as key components of plant defense, providing protection either directly through inhibition of herbivorous pests or indirectly via modulation of ecological interactions[55]. Indole and its derivatives exhibit broad-spectrum bioactivities, including antifungal and insecticidal effects, and can promote plant growth and development[56]. Collectively, these genomic and metabolomic features suggest that T. hamatum Z32 employs multiple molecular mechanisms to enhance plant health and suppress pathogenic fungi.
The genomic evidence suggests that T. hamatum Z32 can generate diverse secondary metabolites through several key biosynthetic routes, including NRPS-like, terpene, NRPS, and T1PKS pathways. The diverse metabolic pathways identified in T. hamatum Z32 likely form the biochemical basis of its antagonistic activity against pathogens and may also contribute to the enhanced tolerance and vigor in the host plants. For instance, terpenoid volatiles can function as herbivore-induced plant volatiles (HIPVs), mediating tritrophic interactions among plants, herbivores, and natural enemies[57]. In addition, some plants generate structurally complex metabolites through NRPS–terpene hybrid gene clusters, potentially providing synergistic enhancement of defense capabilities[58]. The enrichment of TPS-a and TPS-b genes in T. hamatum may represent an adaptive mechanism that broadens its terpenoid biosynthetic potential and enhances chemical diversity. These include monoterpenes, sesquiterpenes, and diterpenes, allowing organisms to better cope with complex biological challenges[59]. Metabolites produced by T1PKS gene clusters, as reported in fungi such as Sarcopodium sp., appear to contribute to the activation of plant defense responses. These findings suggest that T1PKS-associated pathways could be further explored for developing environmentally sustainable biofertilizers and biological control strategies[60].
Given its rich repertoire of biosynthetic gene clusters and secondary metabolism-associated genes, T. hamatum Z32 appears to be well suited for use as a biocontrol agent against root rot in P. notoginseng. However, the present study primarily evaluated its biocontrol potential at the genomic and metabolomic levels, without field validation or direct assessment of the cytotoxic effects of compounds such as fumonisin B1 and fusaric acid on plants. Future studies should focus on comprehensive field efficacy evaluations and further advance the development and optimization of biocontrol formulations based on T. hamatum Z32.
-
We found a strain of T. hamatum called Z32, which is particularly powerful and can effectively fight the bad fungus F. oxysporum. Strain Z32 effectively inhibited the growth of F. oxysporum, the main pathogen responsible for root rot in P. notoginseng. Dual culture experiments revealed an inhibition rate of about 62%. Under microscopy, Z32 hyphae were observed to intertwine and wrap around the pathogen's filaments, a behavior consistent with mycoparasitic activity. The genome of T. hamatum Z32 (42.69 Mb; 20 contigs; N50 of 6.74 Mb; GC content of 46.26%) contained 10,837 predicted genes, including CAZymes such as glycoside hydrolases, as well as multiple secondary metabolite biosynthetic gene clusters, highlighting its potential for biocontrol. Comparative phylogenomic analysis with seven other Trichoderma species revealed 70 expanded and 80 contracted gene families. Untargeted metabolomic profiling identified 5,883 metabolites, including indole derivatives (e.g., indole-3-methanol and indole-3-lactic acid) associated with plant growth regulation, and several antifungal compounds (e.g., choline, dehydrocorydaline, and ferulic acid). Our results provide insights into the genetic and biochemical factors underlying the biocontrol activity of T. hamatum Z32 and suggest its potential for developing environmentally friendly strategies to manage root rot in P. notoginseng. As some findings are derived from in silico analyses, experimental verification is necessary to confirm its effectiveness against F. oxysporum.
We thank all authors and mentors for their valuable support in data analysis and manuscript preparation. Funding This research was funded by The Sino-Vietnamese International Joint Laboratory for Characteristic & Cash Crops Green Development of Yunnan Province (202403AP140013), the National Key Research and Development (2021YFD1601003), and Yunnan Provincial Joint Funds for Agriculture General Program(202301BD070001-198).
-
The authors confirm their contributions to the paper as follows: conceptualization and experimental design: Liu T, Yang S, Xu X, Yang N; data collection: Yang N, Zhang H, Feng Y, Shi C, Zhou Y, Jia J, Tiên LH, Tín HT; data analysis and interpretation: Yang N, Zhang H, Jiang J, Shi C, Tiên LH, Tín HT; manuscript drafting: Yang N. All authors reviewed the results and approved the final version of the manuscript.
-
All research outputs from this work are openly available. The T. hamatum Z32 genome is accessible through NCBI (BioProject: PRJNA1163295; BioSample: SAMN43845003), and the raw metabolomics data have been deposited at the China National Center for Bioinformation (CNCB) under Project No. PRJCA046955, OMIX ID OMIX012064.
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Neng Yang, Honglin Zhang
- Supplementary Table S1 Inhibition rate against pathogens.
- Supplementary Table S2 HiFi data statistics.
- Supplementary Table S3 Base composition and GC content in the genome.
- Supplementary Table S4 Sequencing, assembly metrics, and genome quality of T. hamatum.
- Supplementary Table S5 Basic statistics of gene prediction.
- Supplementary Table S6 Functional annotation statistics.
- Supplementary Table S7 Summary of PHI-base mutant phenotypes identified in the T. hamatum Z32 genome.
- Supplementary Table S8 BGCs statistics.
- Supplementary Table S9 Summary table of the number of identified metabolites.
- Supplementary Fig. S1 Statistical map of functional annotation classification based on GO database.
- Supplementary Fig. S2 KEGG Pathway Annotation of Coding Sequences in the Whole Genome of T. hamatum.
- Supplementary Fig. S3 Map of secondary metabolite content and species of strain T. hamatum Z32.
- Supplementary Fig. S4 Principal component analysis (PCA) of three biological replicates of T. hamatum Z32.
- Supplementary Fig. S5 Comparative genomic analysis of T. hamatum and eight other strains.
- Supplementary Fig. S6 GO(A,B) and KEGG(C,D) enrichment bubble plots.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Yunnan Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang N, Zhang H, Feng Y, Shi C, Jiang J, et al. 2025. Mechanistic insights into the antagonistic effects of Trichoderma hamatum Z32 against root rot pathogens revealed by genomic and metabolomic analyses. Agrobiodiversity 2(4): 78−88 doi: 10.48130/abd-0025-0011 

Mechanistic insights into the antagonistic effects of Trichoderma hamatum Z32 against root rot pathogens revealed by genomic and metabolomic analyses
- Received: 01 July 2025
- Revised: 17 October 2025
- Accepted: 07 November 2025
- Published online: 27 November 2025
Abstract: Root rot in Panax notoginseng is primarily caused by Fusarium oxysporum. A rhizosphere-derived Trichoderma hamatum strain, Z32, was isolated and evaluated for its biocontrol potential. Dual culture assays showed that Z32 inhibited F. oxysporum's growth by 62%, and microscopy revealed hyphal coiling, indicating competition-based antagonism. Genome sequencing produced a 42.69 Mb assembly with 10,837 predicted genes, including 451 carbohydrate-active enzyme (CAZyme) genes involved in cell wall degradation. Ten biosynthetic gene clusters related to antimicrobial metabolites were identified, and metabolomic profiling confirmed multiple antifungal and growth-promoting compounds. These findings reveal that T. hamatum Z32 suppresses soilborne pathogens through diverse mechanisms, providing a promising and environmentally friendly strategy for controlling root rot in P. notoginseng.
-
Key words:
- Root rot disease /
- Biological control /
- Trichoderma hamatum /
- Genomics /
- Metabolomics.