






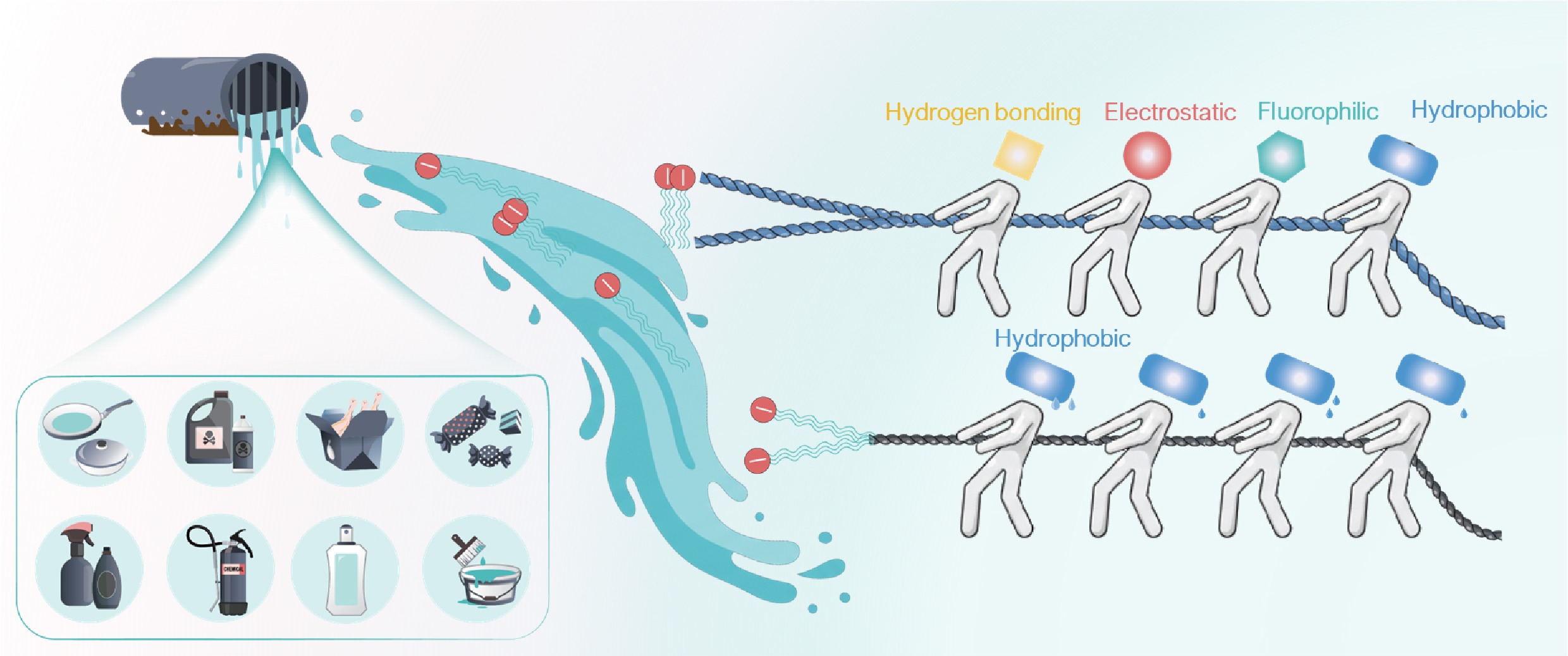



-
Per- and poly-fluoroalkyl substances (PFAS) constitute a large family of synthetic, highly persistent organic compounds[1−3]. Their exceptional chemical stability, amphiphobicity, and surfactant properties have driven widespread use across industrial and consumer applications since the 1940s (Fig. 1a)[1,4,5]. These same features, however, render PFAS extremely resistant to degradation, and have been linked to toxicological and epidemiological evidence that prompts increasingly stringent regulatory controls of PFAS in environmental compartments[6,7]. Regulatory attention has recently expanded beyond a small number of legacy long-chain PFAS to encompass a much broader suite of short-chain perfluoroalkyl acids (below the conventional long-chain cutoffs, i.e., PFCA < C8, and PFSA < C6), and emerging ether-based analogues[8]. In several jurisdictions, proposed or enacted drinking-water standards for individual PFAS or PFAS sums have reached the ng L−1 range[6,9]. This regulatory shift fundamentally alters the technical bottleneck in conventional water treatment technologies[10].
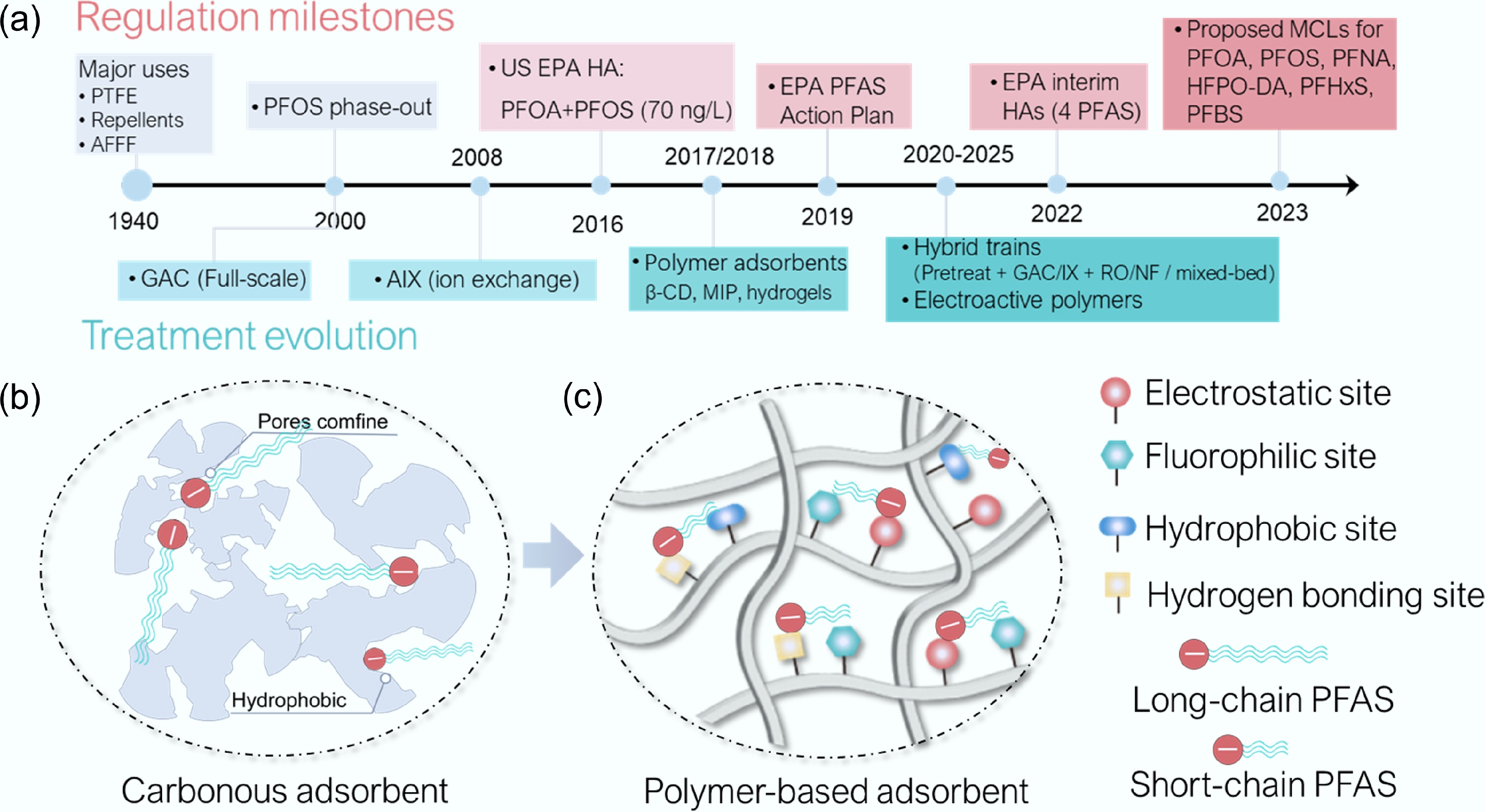
Figure 1.
Novel polymer-based adsorbents for PFAS remediation: history and concept. (a) Timeline highlighting key milestones in the development and applications of polymer-based adsorbents for PFAS removal, aligned with major US Safe Drinking Water Act (SDWA) regulatory actions and advisories that are progressively tightening PFAS limits (Supplementary Table S1). (b) Carbonaceous, and (c) polymer-based PFAS adsorption processes.
Granular activated carbon (GAC) and related carbonaceous media remain the dominant baseline technologies for PFAS removal owing to their maturity, regulatory acceptance, and compatibility with existing treatment infrastructure[11,12]. However, their performance is intrinsically decreased toward short-chain PFAS. Full-scale surveys have reported earlier breakthrough of short-chain PFAS from GAC beds (e.g., ~20,000 bed volumes for PFHxA vs ~30,000 for PFOA/PFOS)[13]. This is because short-chain congeners exhibit weaker hydrophobic interactions, stronger hydration shells, and reduced benefit from pore confinement[8,14], resulting in diminished adsorption affinity and rapid displacement under realistic water matrices containing competing ions and dissolved organic matter[15,16]. These physicochemical limitations increasingly render surface-area-driven adsorption insufficient for sustained short-chain PFAS control.
Consequently, the core solution in next-generation PFAS remediation is potentially engineering molecular-level recognition that is effective under competitive, environmentally relevant conditions[8,15,16]. Polymer-based adsorbents offer a uniquely programmable platform to meet this challenge. Through deliberate selection of monomers, crosslinkers, and architectures[17], polymers can spatially co-localize complementary interaction motifs, including electrostatic headgroup anchoring, partial desolvation control, and fluorinated tail accommodation, within confined microenvironments[15,18,19]. When integrated within a single domain, these interactions act cooperatively rather than individually, enabling affinity and selectivity toward short-chain PFAS that are unattainable with single-interaction strategies[8,15,20−23].
In this mini-review, we emphasize recent advances in polymer-based adsorbents for short-chain PFAS removal through the unifying concept of co-localized cooperative binding microenvironments and assess their life-cycle footprint. Representative polymer families include cyclodextrin-based networks, molecularly imprinted polymers, hydrogels, and electroactive polymers. These materials are evaluated according to how effectively they implement this cooperative binding principle, and how such designs translate into performance under realistic water matrices and regeneration constraints. Importantly, life-cycle assessment is integrated to examine whether gains in selectivity justify associated synthesis and operational burdens, thereby linking molecular design to net environmental benefit. By framing polymer sorbent development within both mechanistic and sustainability constraints, this review aims to provide actionable design guidance for deployable short-chain PFAS remediation systems.
-
To capture the evolving landscape of PFAS adsorbent research, a structured literature survey covering the period of 2015 to 2025 was conducted, with emphasis on publications from the past three years. Relevant studies were retrieved from Scopus, Web of Science, and Google Scholar using keywords such as 'PFAS', 'short-chain', 'adsorption', 'polymer', and specific material classes (e.g., 'cyclodextrin polymer', 'fluorogel', 'redox-active polymer', 'electroactive polymer', 'conductive polymer'). Only peer-reviewed, English-language studies reporting quantitative adsorption performance (e.g., equilibrium capacity or removal efficiency) for at least one short-chain PFAS in water were included.
Both batch and column experiments were considered, with preference given to those evaluating realistic conditions (natural water matrices, presence of dissolved organic matter [DOM], competitive anions, and variable ionic strengths). From each study, equilibrium adsorption capacities (qe) or sorption coefficients, kinetic data, effects of solution chemistry (pH, ionic strength, DOM concentration), and any reported regeneration efficiencies or reuse cycles were extracted. Synthetic methods and material characteristics were also documented to link performance with adsorbent properties. This integrative approach allows the identification of performance trends, mechanistic insights, and critical knowledge gaps across different polymer classes.
Life cycle assessment (LCA) procedures
-
Life cycle assessment (LCA) was performed to quantify and compare the environmental burdens of polymer-based adsorbents with conventional GAC, and anion-exchange resins for PFAS removal. The functional unit is the treatment of 1 m3 of influent water that lowers target PFAS from their initial concentrations to the relevant regulatory limits. The results were additionally normalized to the mass of PFAS removed[24]. The system boundary covered a gate-to-grave life cycle, including monomer/precursor production, polymerization/activation, use-phase regeneration, and end-of-life disposal or recycling. Environmental impacts were characterized using ReCiPe 2016 Midpoint (H) (including GWP100, human toxicity [non-cancer], terrestrial ecotoxicity, marine eutrophication, particulate matter formation, and fossil resource scarcity), and aggregated into ReCiPe Endpoint (H/A) damage categories (human health, ecosystems, resources). In parallel, USEtox was applied for human/ecotoxicity comparison (Supplementary File Section 2)[25,26].
-
Global regulatory actions have profoundly reshaped PFAS production, use, and environmental occurrence. The listing of PFOS under Annex B (2009), and PFOA under Annex A (2019) of the Stockholm Convention accelerated the phaseout of legacy long-chain PFAS and drove a shift toward short-chain, and ether-based alternatives (Fig. 2)[3,12]. Across major PFAS families including perfluoroalkyl carboxylic acids (PFCA), perfluoroalkyl sulfonic acids (PFSA), fluorotelomer sulfonates (FTS), fluorinated amine sulfonamides (FASA), and emerging ether derivatives such as hexafluoropropylene oxide dimer acid (GenX), the defining structural trend is a reduction in the perfluoroalkyl segment length (n ≈ 1–10) while retaining the chemically robust C–F backbone[3,8,17].
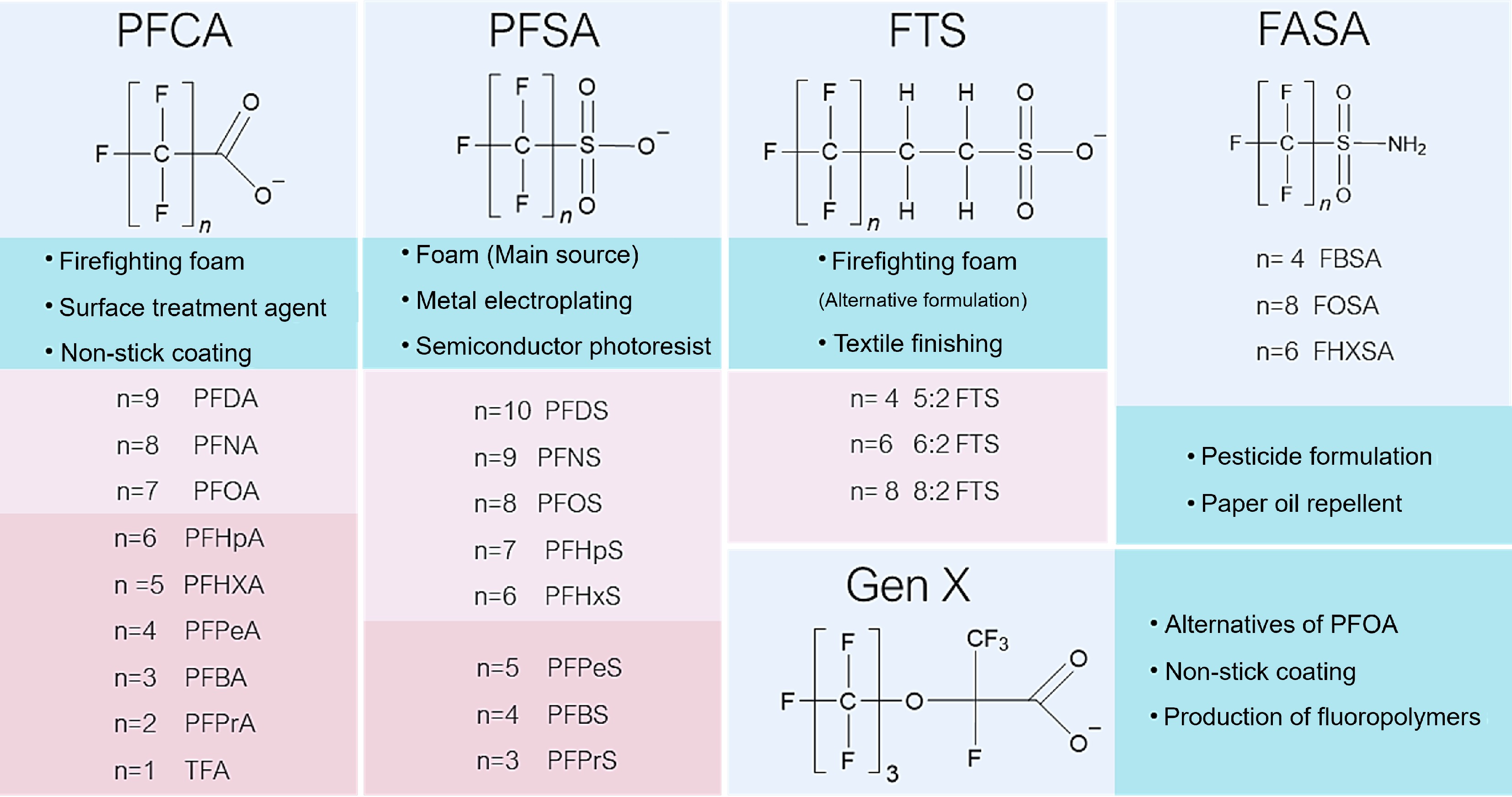
Figure 2.
Chemical structures of widely explored PFAS compounds. n represents individual C−F chain lengths.
Short-chain PFAS have replaced their long-chain counterparts in firefighting foams, metal electroplating, textile finishing, semiconductor photoresists, and non-stick coatings because they preserve surfactant properties and thermal stability while lowering bioaccumulation potential[17,21]. However, shortening the perfluoroalkyl tail fundamentally alters aqueous behavior: short-chain congeners exhibit increased polarity, stronger hydration shells, and reduced steric bulk[27]. As a result, compounds such as PFHxA, PFBA, and PFBS exhibit greater mobility and persistence in aquatic environments, shifting the dominant environmental risk from bioaccumulation toward widespread, chronic low-level exposure[9,28]. From a treatment perspective, the overall performance becomes increasingly governed by the weakest-retained species. These trends underscore the need to rethink adsorption design principles beyond those developed for hydrophobic organic contaminants or long-chain PFAS, particularly under high ionic strength or DOM-rich conditions.
Carbonaceous adsorbents as the engineering baseline
-
Carbonaceous adsorbents, including GAC[29], powdered activated carbon (PAC)[29,30], carbon nanotubes (CNT), and graphene-based materials[31,32], remain the most established technologies for PFAS removal. Their appeal lies in high surface area, mature manufacturing, and regulatory familiarity.
GAC is widely implemented in municipal drinking water, groundwater remediation, and industrial polishing due to its reliability and ease of retrofitting[33]. For legacy long-chain PFAS (e.g., PFOA and PFOS), commercial GAC filters can typically achieve > 95% removal, yielding effluent concentration in the order of 10 ng L−1. However, when faced with short-chain PFAS (e.g., PFBA, PFPeA, and GenX), the adsorbent performance decreases dramatically[34]. A global survey of 23 drinking water treatment plants found that C4–C6 short-chain PFAS broke through 2−4 times faster than long-chain species under comparable conditions[11]. Likewise, a two-year field study tracking 15 PFAS species found that although initially achieving 92%–100% removal, short-chain compounds showed early breakthrough after approximately one year of continuous operation (~29,300 bed volumes)[13]. This short-chain limitation mainly arises from coupled deficits in interaction strength and adsorption selectivity[8,14]. Adsorption on GAC is dominated by hydrophobic interactions and micropore confinement. However, these same mechanisms are less effective for short-chain PFAS[10,35]. In addition, GAC lacks headgroup-specific recognition motifs, so competitive adsorption follows an affinity ranking: short-chain PFAS are readily displaced by longer-chain PFAS, and coexisting inorganic ions and DOM can further suppress uptake, reducing adsorption performance by up to 60%[11,13].
PAC is typically used for short-term or emergency PFAS control during contamination incidents or regulatory exceedances[36]. Comparative studies showed that, with a 10 min contact time, PAC removed 60%–90% of PFOA and PFOS, compared with 20%–40% for GAC under identical conditions[37]. The main difference between PAC and GAC lies in morphology and dynamics (mass transfer), while both share the same mechanistic limitations for short-chain PFAS removal[27,38]. Accordingly, PAC mainly improves kinetics for PFAS removal but does not reverse the chain-length-dependent affinity hierarchy that governs short-chain breakthrough.
CNT and graphene-based materials represent next-generation carbon adsorbents offering ultrahigh surface areas and tunable surface chemistries[12,39]. Commercial CNT and graphene derivatives can achieve long-chain PFAS adsorption capacities exceeding 200 mg g−1[12,35]. However, their deployment is constrained by cost, aggregation under elevated ionic strength, and limited robustness in complex waters[31,40]. For example, as Cl− concentration increases from 0.001 to 0.05 M, competitive effects are observed at low concentrations (0.001–0.01 M), where Cl− reduces PFAS uptake; once the electrolyte concentration exceeds the CNT critical coagulation concentration (0.037 M), CNTs tend to aggregate, thereby decreasing accessible surface area and adsorption capacity. Moreover, simple CNT modifications that primarily enhance electrostatic interactions are often unsatisfactory because adsorption becomes pH-sensitive, with capacities decreasing as pH increases[40]. Consequently, many recent works incorporate CNT in electrostatically driven adsorption, while the contribution to intrinsic PFAS selectivity is limited[20,41]. Nonetheless, their physicochemical properties still make CNTs attractive in building PFAS removal systems, and challenges in selectivity and water-matrix adaptability should be carefully addressed.
Cooperative intermolecular interactions for short-chain PFAS capture
-
From a structural perspective, PFAS offers two addressable interaction motifs, namely an anionic headgroup (–COO–/–SO3–, and ether oxygen atoms in some species), and a shortened perfluorinated tail (–(CF2)n–CF3 or –O–CF2–). Effective capture therefore requires sorbents that simultaneously engage both cationic motifs to anchor the headgroup, and hydrophobic/fluorous domains to accommodate the tail and resist displacement in realistic matrices[8,14,27].
Four interaction classes that are particularly relevant have been described (Table 1)[8,15,20−23]. Electrostatic binding targets the anionic headgroups (–COO–/–SO3–) through cationic motifs such as –NR4+, –NH3+/–NH2+–, imidazolium+/pyridinium+, and guanidinium, but is susceptible to screening and competition from background anions[21,23]. Hydrophobic/dispersion interactions mainly engage the perfluorinated tail (–[CF2]n–CF3) via sp2-carbon/aromatic or –(CH2)n– domains; for short-chain PFAS, these contributions are weak unless amplified by micropore/nanoconfinement[42]. Fluorophilic recognition strengthens tail accommodation by introducing fluorous segments (e.g., –[CF2]n–/–CF3-rich units or –C[F]x– surfaces) that stabilize perfluoroalkyl/perfluoroether motifs[22]. Hydrogen bonding provides directional stabilization through donors/acceptors (e.g., –NH–, –OH, –CONH–, –NHCONH–, C=O), typically as a cooperative 'secondary handle' adjacent to electrostatic sites to stabilize partially dehydrated headgroups[42,43]. When these interactions are integrated within the same binding domain, headgroup anchoring can modulate desolvation, while tail accommodation reduces susceptibility to competitive displacement. This cooperative-binding paradigm provides a unifying framework for understanding why certain polymer-based adsorbents outperform conventional materials under realistic conditions. In the following section, representative polymer sorbents are categorized according to how effectively their architectures encode and spatially organize these complementary interactions, and how this organization translates into short-chain PFAS removal selectivity, regenerability, and environmental robustness.
Table 1. Summary of intermolecular interactions for short-chain PFAS capture
Binding type Electrostatic Hydrophobic Fluorophilic Hydrogen bonding Schematic 



Target PFAS moiety Anionic headgroups:
–COO–, –SO3–Fluorinated tail:
–(CF2)n–CF3Fluorinated tail:
–(CF2)n–, –O–CF2–Headgroup O atoms(–COO–/–SO3–) and
ether O (–O–)Sorbent binding site Cationic sites: –NR4+, –NH3+/–NH2+–, imidazolium+/pyridinium+, guanidinium (–C[=NH2+]NH2) Hydrophobic domains: sp2-C (graphitic/aromatic), –(CH2)n–; micropores (geometric confinement) Fluorous domains: –(CF2)n–/–CF3, fluorinated surfaces –C(F)x–, fluorinated pore walls/cavities Donors: –NH–, –OH, –CONH–, –NHCONH–; Acceptors: C=O, –N: (paired with PFAS O atoms) -
Polymers provide an intrinsically programmable platform in which monomers, crosslinkers, and architectures (e.g., block, graft, network) can be selected to co-localize complementary forces within tailored microenvironments (Table 1). Here, four representative polymer families are highlighted that operationalize distinct combinations of these forces: β-cyclodextrin (β-CD) polymers[44,45], molecularly imprinted polymers (MIPs)[46,47], hydrogels[16,18], and electroactive polymers (Fig. 3)[41].
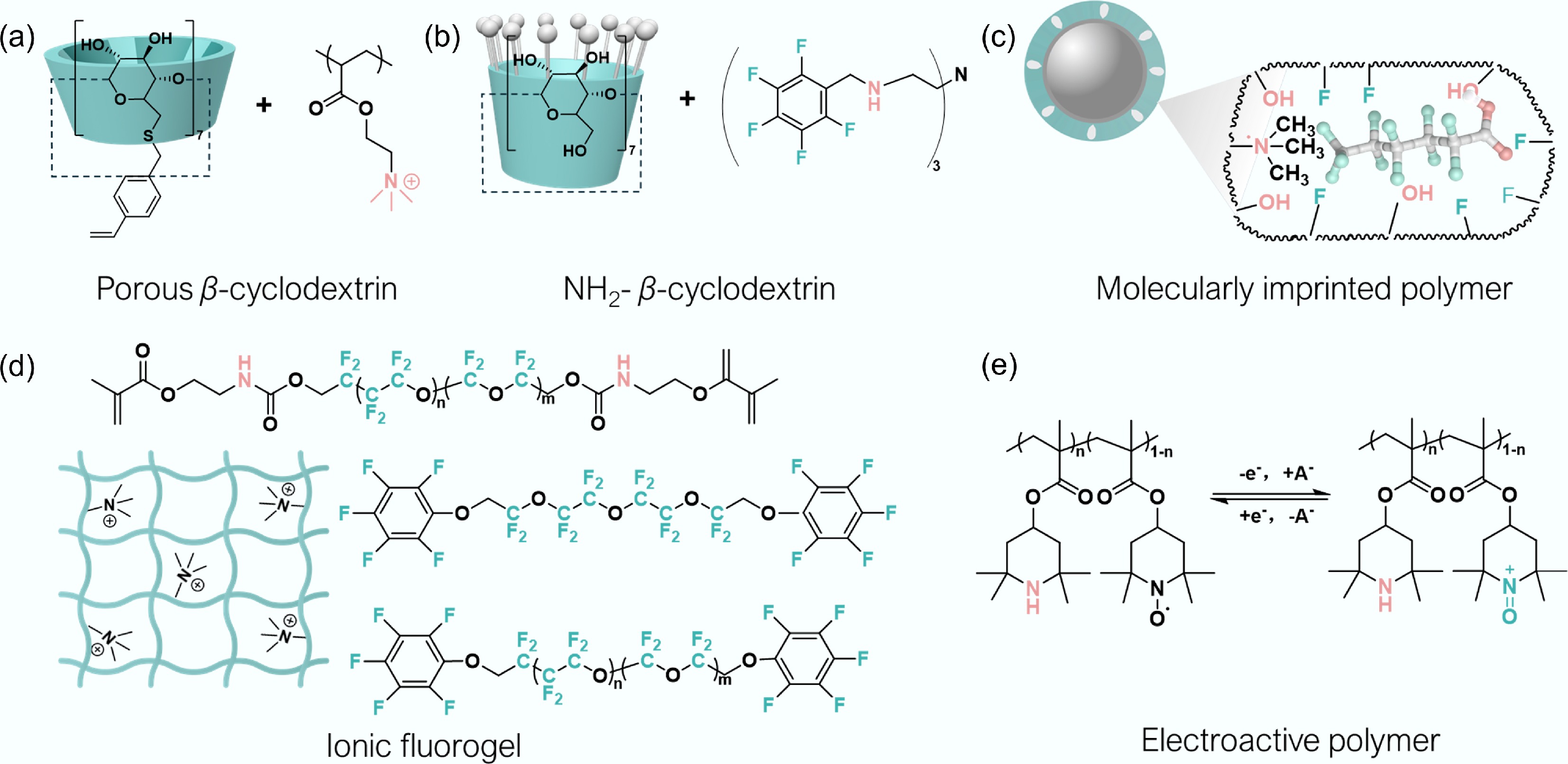
Figure 3.
Summary of chemical structures of the latest representative PFAS polymer-adsorbents. (a) Porous β-cyclodextrin polymers. Adapted from Zhang & Wang[48], with copyright © 2022 American Chemical Society. (b) NH2-β-cyclodextrin polymers. Adapted from Ippolito et al.[49], with copyright © 2024 American Chemical Society. (c) Molecularly imprinted polymer. Adapted from Yang et al.[50], with copyright © 2018, Elsevier. (d) Ionic fluorogel adsorbent. Adapted from Liu et al. and Chang et al. [51,52], with copyright © 2018 and 2022, American Chemical Society, Wiley-VCH. (e) Conducting polymers. Adapted from Qiu et al.[41], with copyright © 2020 and 2024, Wiley-VCH, Elsevier[29].
β-Cyclodextrin polymers
-
β-Cyclodextrin (β-CD) is a cyclic oligosaccharide consisting of seven glucose units forming a hydrophobic inner cavity and hydrophilic exterior (Fig. 3a and b). When polymerized or crosslinked into solid networks, these cavities act as fixed host–guest pockets capable of selectively binding organic molecules[53]. Early β-CD adsorbents crosslinked with epichlorohydrin displayed limited affinity for PFAS, particularly short-chain compounds[44]. Subsequent efforts improved performance by introducing aromatic crosslinkers such as tetrafluoroterephthalonitrile (TFN), which impart fluorine-rich surfaces, and by post-functionalizing networks with cationic amines to enhance electrostatic anchoring of anionic PFAS[54]. Dichtel and coworkers first demonstrated sub-ppb removal with β-CD polymers, decreasing PFOA from 1 to < 10 ng L−1 under environmentally relevant conditions[44]. Recent β-CD polymer variants illustrate the benefits of combining intermolecular interactions. β-CD-COFs modified with amines have achieved adsorption capacities of 0.33–1.51 mmol g−1 toward a panel of short- and long-chain PFAS, outperforming commercial ion-exchange resins and activated carbon[55]. Styrenated β-CD copolymers containing quaternary ammonium groups demonstrated > 99% removal of PFBA and PFPeA at ~1 µg L−1 concentration[48]. Note that β-CD sorbents also exhibit fast kinetics and high affinity for short-chain species. Distribution coefficients (Kd) for congeners with < 4 fluorinated carbons reached 103–107 L kg−1, compared with < 106 L kg−1 for activated carbon and biochar analogues[56].
A key advantage of β-CD materials is facile regeneration. Spent cyclodextrin networks can be eluted with mild solvents (water–alcohol mixtures or dilute base) to desorb > 50%–95% of bound PFAS within minutes[57]. In contrast to activated carbon, β-CD polymers maintain adsorption in the presence of DOM; their nanoporous structure limits humic fouling[58]. Xiao et al.[44] reported > 90% removal of PFHxA, PFOA, and PFOS from lake water (ng L−1 spikes), even with 10 mg L–1 humic acid. Collectively, β-CD polymers offer rapid, selective short-chain PFAS removal (Fig. 4), with straightforward regeneration and tolerance to natural water matrices[54,58]. To translate these promising results into robust field performance, several gaps still need to be addressed. Most reported performance relies on µg L−1 batch spikes and simplified matrices, so adsorption, breakthrough, and regeneration at ng L−1 levels, and under continuous-flow conditions remain insufficiently explored. Moreover, the individual contributions of cavity inclusion, cationic anchoring, and fluorophilic microdomains are not fully understood in crosslinked networks, and regeneration using solvent/base eluents warrants further optimization.
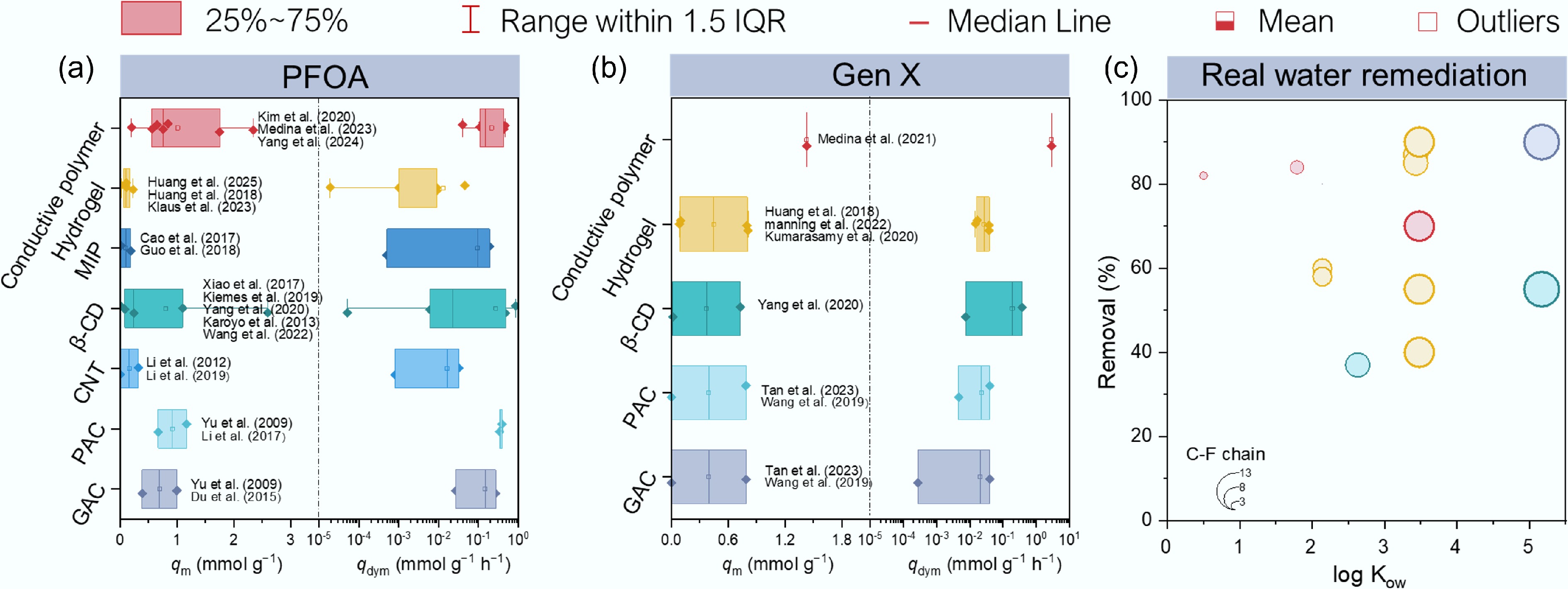
Figure 4.
Comparison of carbon-based and polymer-based adsorbents for PFAS remediation. (a), (b) Comparison of qm and dynamic adsorption capacity (qdyn, mmol g−1 h−1) value for (a) PFOA (n = 18), (b) Gen X (n = 18) in reported literature (listed in Supplementary Tables S2 and S3). The centerline, upper limits and the lower limits of the box, upper whiskers and lower whiskers, and the point represents the median, 25% of the maximum and minimum, the maximum and minimum, and the average of qm or qdyn in different adsorbents. (c) Comparison of the short-chain PFAS removal efficiency of different polymer-adsorbents (listed in Supplementary Tables S4).
Molecularly imprinted polymers
-
Molecularly imprinted polymers (MIP) leverage templating strategies to create binding sites that are complementary to a target molecule in shape and functionality (Fig. 3c)[46,59]. Template removal yields cavities that recognize PFAS via cooperative shape matching, hydrogen bonding, and ionic pairing[50,60].
Early MIPs imprinted against PFOA or PFOS showed high affinity toward long-chain templates (> 600 mg g−1) but limited uptake of short-chain PFAS (PFBA capacity ~100 to 200 mg g−1; just 14%–50% removal for short-chain species)[61,62]. Mismatches between cavity geometry, and the shorter fluorinated tails accounted for much of this selectivity gap[62]. Performance improvements have come from several strategies. First, direct imprinting with short-chain templates (e.g., GenX/HFPO-DA) increases selectivity, albeit with some decrease in long-chain capacity[63]. Second, functional-monomer tuning, for example, introducing sulfonamide, or other polar monomers, enhances recognition of short-chain sulfonates. Advances have produced porous MIPs with capacities of ~230 mg g−1 for PFBA, and ~380 mg g−1 for PFOA, narrowing the short- to long-chain performance gap to ~60%[62,64].
The selectivity of MIP can also be engineered using multi-template and composite designs. Introducing a secondary template yields binding sites spanning different chain lengths: a biochar-supported MIP prepared with PFOS + PFBA templates achieved 30%–50% higher PFBA and PFHxA uptake than single-template PFOS MIPs[65]. MIPs can also incorporate fluorophilic and cationic monomers (e.g., trifluoromethylated acrylates, vinylbenzyl amines) to emulate PFAS's dual binding requirements. An ultrathin imprinted film from a perfluorinated styrene derivative paired with a urea H-bond donor exhibited six-fold selectivity for PFAS over co-ions[60].
Regeneration of MIP is generally straightforward because imprinting interactions are non-covalent, allowing PFAS to be desorbed under relatively mild conditions. Most PFAS-MIPs are regenerated by washing with methanol or acetonitrile, often with 1%–2% NaOH or NH4OH to disrupt ionic interactions[58]. Similarly, PFOS-MIPs have been reused repeatedly in SPE cartridges with MeOH/0.1% NaOH elution[60].
While most applications have targeted analytical enrichment, several studies demonstrate real-water performance. A commercial Supel MIP® PFOS extracted trace PFAS from groundwater and river water following pre-rinsing to manage natural organic matter interference[60,66]. A biochar-MIP composite removed up to 70% of PFBS, PFHxA, and other short-chain PFAS from airport runoff, vs < 20% removal by non-imprinted controls[67]. These results confirm that MIPs can function in complex matrices. Despite clear gains in selectivity, further work is needed to translate PFAS-MIPs into practical adsorbents beyond analytical enrichment[62,67,68]. Reported performance is often derived from template-matched systems and cartridge/batch tests, whereas long-term capture and breakthrough under mixed-PFAS, NOM/ion-rich matrices, and continuous-flow operation are underexplored. In addition, imprinting designs would benefit from approaches that balance cavity specificity with compositional breadth to maintain coverage across structurally diverse PFAS, and regeneration protocols warrant optimization to minimize secondary eluents while preserving selectivity and cycling stability.
Hydrogel
-
Hydrogel-based adsorbents with water-swollen, cross-linked polymer networks are attracting interest for short-chain PFAS removal because their hydrated matrices facilitate rapid mass transfer and mitigate fouling more effectively than dry resins (Fig. 3d). Tailored hydrogel chemistries enable co-localization of ionic, hydrophobic, and fluorophilic binding domains while maintaining accessible diffusion pathways.
Among hydrogel systems, ionic fluorogels are among the most extensively studied. These include styrene–divinylbenzene networks bearing fluoroalkyl pendants and quaternary ammonium groups, as well as alginate matrices incorporating amine-functionalized sorbents[16,51,52]. The cooperative binding motif is central to their performance: fluorinated microdomains solvate the perfluoroalkyl tail, while cationic sites anchor the anionic headgroups. In municipal water, ionic fluorogels achieved near-quantitative removal of a 21-PFAS mixture (including PFBA, PFBS, PFHxA, and GenX) at μg L−1 levels using only 1–10 mg L−1 adsorbent, with equilibrium reached within < 30 min, and minimal interference from natural solutes[51]. Hybrid systems illustrate the modularity of hydrogel design. An acrylamide hydrogel composite embedded with PAC achieved > 95% removal of PFOA/PFOS at ng L−1, combining the hydrogel's swelling-enhanced mass transfer with PAC's adsorption sites[69].
A major advantage of hydrogels is straightforward regeneration. The soft, non-covalent binding in many hydrogel systems enables mild elution conditions without thermal reactivation. Ionic fluorogels can undergo ≥ 3 reuse cycles via water : methanol (1:1) rinsing with 1 M NaCl, desorbing > 90% of PFAS per cycle with negligible network degradation[51]. Hydrogel sorbents have also been evaluated in real-water matrices. For instance, fluorogel-packed columns removed PFBA, PFBS, and GenX below detection limits from settled drinking water despite competing anions and natural organic carbon[52]. A vesicular polyacrylamide/PAC hydrogel similarly achieved > 85%–90% removal of C4–C6 PFAS from surface water, whereas conventional AC removed < 50% for the short-chain fraction[70].
Hydrogel architectures also demonstrate resilience to variable pH and DOM. A polyaniline-based hydrogel maintained > 90% PFAS removal from pH 3–10, and in the presence of 50 mg L−1 humic acid[16,18]. Benefiting from water-swollen, permeable networks that enable rapid mass transfer and allow co-localization of cooperative binding motifs, hydrogel adsorbents can achieve efficient short-chain PFAS capture and mild regeneration. Future improvements should focus on strengthening mechanical stability under repeated swelling–deswelling, minimizing functional-component degradation, and developing scalable regeneration protocols.
Electroactive polymers
-
Electroactive polymers represent an innovative class of PFAS adsorbents that leverage electrochemical control to capture and release contaminants[71,72]. Materials including polypyrrole (PPy), polyaniline, and viologen- or quinone-functionalized backbones reversibly switch among oxidation states, modulating charge and thus affinity toward anionic PFAS (Fig. 3e)[73−75].
Mechanistically, applying a potential induces electrostatic sorption of PFAS anions into the polymer matrix, often augmented by hydrophobic and fluorophilic interactions where present[20,41]. In its doped state, PPy bears positive charges and features a hydrophobic carbon-rich matrix, enabling simultaneous headgroup attraction and tail partitioning[76]. Similarly, redox polymers bearing nitroxide radicals (N–O•), which convert to cationic oxoammonium species (+N=O) under mild bias, have been shown to selectively electrosorb short-chain PFAS from water[41].
Nanostructured conductive polymers exhibit particularly high uptake across a wide spectrum of PFAS (Fig. 4a, b). A vesicle-like PPy prepared by Yu et al.[77] had an estimated sorption capacity of ~500 mg g−1 for PFBA, and ~509 mg g−1 for PFOA, removing ≥ 90% of 12 PFAS (C3–C8) from contaminated groundwater (spiked); by contrast, a commercial resin removed < 50% of short-chain congeners. Fluorinated redox-copolymers incorporating nitroxide radicals and trifluoromethyl groups demonstrated strong short-chain PFAS selectivity, adsorbing up to 205.8 mg g−1 PFBA, and 860.8 mg g−1 PFHxA[20]. More recently, Kim et al. achieved high removal of ultra-short to long-chain PFAS (e.g., trifluoroacetic acid to PFOS) by integrating redox electrodialysis with electrosorption, illustrating the versatility of electro-mediated capture[78]. Moreover, electroactive polymers can be tuned to selectively capture certain PFAS. For example, by adjusting the voltage, an electrode can selectively target PFAS below a certain chain length, effectively 'gating out' longer ones if desired[72]. This level of control makes electroactive polymers one of the most attractive emerging solutions for short-chain PFAS remediation.
Importantly, redox polymers can be regenerated in situ: applying a reverse or neutral potential releases bound PFAS, concentrating them into a low-volume brine for disposal or destruction[20,41,78]. Electro-regeneration with PFAS was directly evidenced by electron quartz crystal microbalance (EQCM) over successive cycles, with minimal performance decay upon repeated regeneration[72]. In other cases, solvent or acid rinses restore capacity for PPy and polyaniline systems. For instance, Yu et al. showed pH 1 methanol could regenerate porous PPy, enabling at least five adsorption–desorption cycles with > 95% capacity recovery[77].
Electroactive polymers also provide a pathway toward capture-and-destroy integration, wherein concentrated PFAS are directly mineralized at the electrode surface via anodic oxidation. Preliminary studies indicate that pre-concentration on redox-polymer electrodes enables more energy-efficient oxidation than in dilute solution[41]. These materials have demonstrated robust performance in complex waters, including brines and high-DOC matrices[20]. Remaining challenges include scale-up, electrode durability, and managing PFAS concentrates when destruction is not integrated[20,78]. Validating electroactive polymers under continuous-flow, ng L−1-level conditions, and quantifying mass-transfer/kinetic constraints, and energy demand across realistic conditions are not sufficient. Nevertheless, with continuing advances in polymer chemistry (e.g., robust redox-active backbones, fluorine-containing ionic liquids), and electrochemical reactor design, electroactive polymers are poised to complement (See comprehensive comparison in Fig. 4), especially for the efficient removal of short-chain PFAS under field conditions. Emphasis should also be placed on improving long-term durability (including antifouling/scaling control) and regeneration efficiency, ideally coupled with on-site destruction to minimize secondary waste streams.
-
Polymer-based adsorbents can deliver high affinity and selectivity for short-chain PFAS; however, sustainability cannot be justified by removal performance alone. Because the performance gains summarized above (Fig. 4) are achieved under matrix and regeneration constraints, the required adsorbent inventory and reuse cycles become decisive for net impacts. Therefore, the practical value of polymer sorbents depends on whether these benefits translate into net environmental advantages over incumbent media such as GAC and ion-exchange resins[15,27,79]. To evaluate these trade-offs, an LCA on a per-m3-treated basis was conducted[25,26,79,80]. To isolate synthesis-related burdens, we focused on electroactive polymers (representative of relatively energy- and reagent-intensive synthesis, Supplementary Table S5, S6, and Supplementary File Section 2.1)[41]. The functional unit was defined as the treatment of 1 m3 of PFAS-contaminated water to a common effluent target. As illustrated in Fig. 5a, the gate-to-grave boundary comprises polymerization (monomer production, solvent use, electricity), and post-modification (electricity, oxidant, solvents). Module fabrication and end-of-life were addressed in sensitivity analyses.
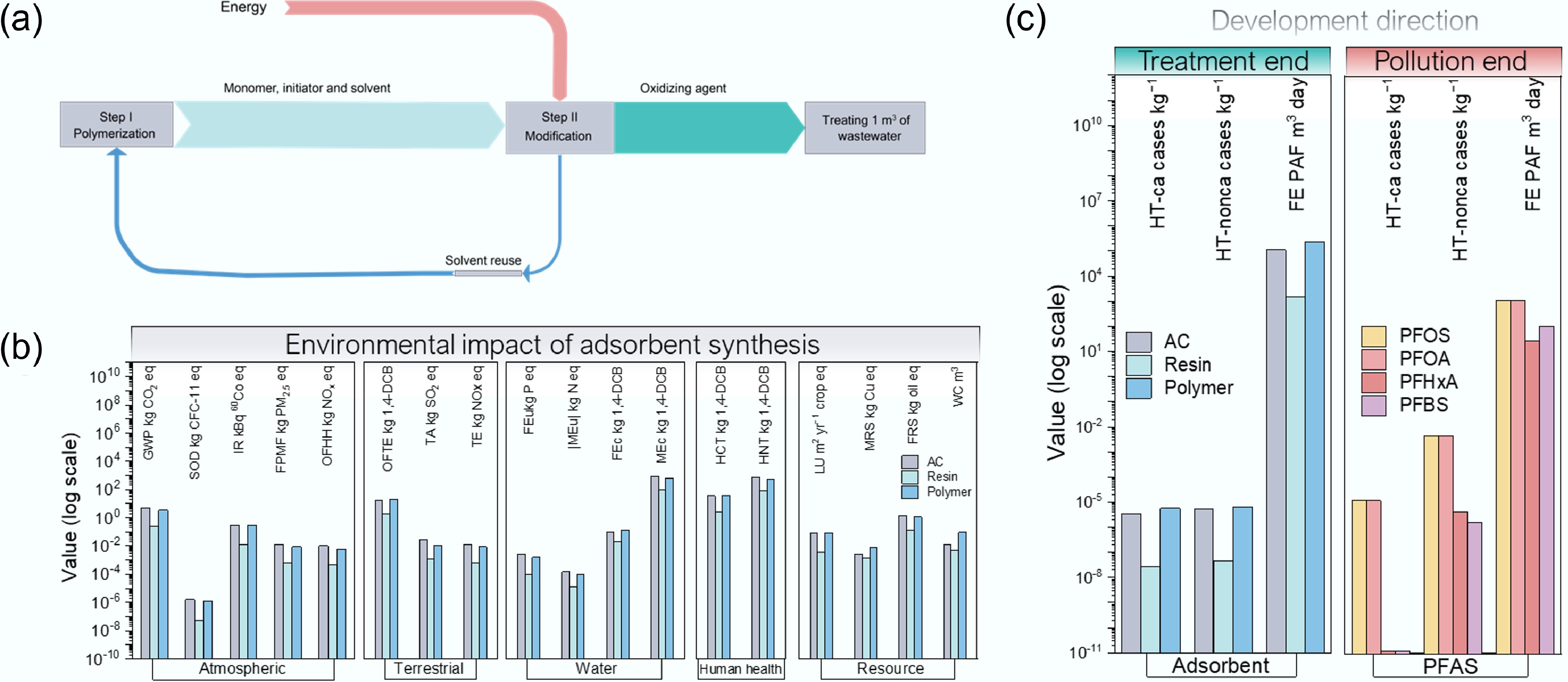
Figure 5.
LCA assessment of the polymer and carbonaceous adsorbents for PFAS remediation. (a) Mass flow of polymer adsorption synthesis, including polymerization of monomer, and modification of the polymer process; (b) Comparison of environmental impacts on MEu, HNT, GWP, TE, and the other 14 categories for polymer and carbonaceous adsorbents (Supplementary Table S7); (c) Environmental damages of polymer and carbonaceous adsorbents and long-chain and short-chain PFAS.
Across the functional unit of 1 m3 treated water, the polymer route exhibits clear advantages over AC in several energy-dominated midpoint categories (Fig. 5b), including decreases in global warming potential (GWP) by 33.6%, fine particulate matter formation (PM2.5) by 43.8%, ozone formation (human health) and ozone formation (terrestrial) by 36.0% and 34.6%, and stratospheric ozone depletion by 20.1%. Conversely, polymer synthesis introduces hotspots in toxicity- and resource-linked categories due to monomer, solvent, and oxidant demand, including increases in freshwater ecotoxicity by 40.5%, terrestrial ecotoxicity by 22.7%, mineral resource scarcity by 167.0%, and water consumption by 600.0%, respectively. Nutrient-related and some ecotoxicity endpoints diverge. Freshwater and marine eutrophication decrease by 42.7% and 35.1%, and marine ecotoxicity decreases by 22.9%, whereas freshwater ecotoxicity remains elevated, implicating solvent/oxidant inventories as dominant contributors.
Using strong-base anion-exchange resin (AER) as a benchmark highlight that most performance gaps are attributable to manufacturing burdens (Fig. 5b). The polymer exhibits higher life-cycle impacts than AER across many categories by ~4.5- to 23-fold (e.g., GWP +1,188%, PM2.5 +1,100%, water consumption +1,941%, mineral resource scarcity +452%). In contrast, AER outperforms AC broadly, with 52%–97% reductions across core categories (e.g., GWP –94.8%, PM2.5 –95.3%, human non-carcinogenic toxicity –89.1%, fossil resource scarcity –90.6%). These trends indicate that realizing the use-phase benefits of polymers requires manufacturing-stage greening, that is, notably solvent recovery, low-impact oxidation, and alternative monomer sourcing.
Treatment-side endpoint factors per kg sorbent were then aligned with pollution-side toxicity factors per kg PFAS to assess environmental reasonableness via a break-even framework (Fig. 5c; PFAS data from Holmquist et al.[26]). For human non-carcinogenic toxicity, the polymer yields a net benefit at 0.0015 kg PFAS kg−1 polymer for PFOA/PFOS. For PFHxA and PFBS, thresholds of 1.4 and 3.5 kg kg−1 are achievable with selective adsorption and routine regeneration; for example, an effective per-cycle capacity of ~0.20 kg kg−1 with moderate matrix derating suggests ~tens of cycles are sufficient for PFBS. For human carcinogenic toxicity, break-even is attainable for PFOA/PFOS (0.462 kg kg−1) but not for PFHxA/PFBS within the tested dataset. Freshwater ecotoxicity is rate-limiting across all species: thresholds of 193 kg kg−1 (PFOA/PFOS), 8032 kg kg−1 (PFHxA), and 2174 kg kg−1 (PFBS) are not practically reachable through performance alone. Thus, net benefit is clearest where polymer dose is minimized, regeneration is routine, and selective uptake extends coverage to short-chain PFAS (PFHxA, PFBS, GenX) without high-energy polishing.
Closing the freshwater ecotoxicity gap relative to AER requires source-level green manufacturing, including: (i) low-impact, high-recovery solvent systems; (ii) replacement of high-impact oxidants with electro-oxidation or H2O2/air in closed reactors with vent abatement; and (iii) monomer resourcing that avoids halogenated/nitrated precursors and heavy-metal catalysis. At the system level, pairing AC/AER up-front for long-chain PFAS and NOM removal with a small polymer polishing stage for short-chain PFAS reduces polymer dose per m3, and increases effective regeneration cycles, improving LCA performance even before synthesis optimization (Fig. 5b, c).
In summary, at the synthesis stage, polymer adsorbents currently carry higher gate-to-grave burdens than AC and AER. Narrowing this gap will require manufacturing-stage greening (solvent recovery, mild oxidation, alternative monomer sourcing) (Fig. 5b). During use, environmental impacts can be reduced through hybrid treatment trains, for example, deploying AC/AER to remove long-chains and NOM, followed by targeted polymer polishing with routine regeneration. These actions thereby minimize dose, increase lifetime capture, and lower per-m3 impacts.
-
Despite rapid advances in polymer-based adsorbents, the primary barriers to effective short-chain PFAS remediation are increasingly system-level rather than purely materials-level. In realistic treatment contexts, competitive adsorption in mixed matrices, management of PFAS-enriched secondary streams, and the environmental burdens associated with sorbent manufacture and regeneration collectively determine whether advanced materials can be deployed at scale. Future progress therefore, requires integrating molecular-level selectivity with process design and sustainability constraints.
Hybrid treatment trains for mixed PFAS matrices
-
In environmental waters, PFAS occur as complex mixtures rather than isolated solutes, coexisting with DOM, background electrolytes, and other organic contaminants that reshape adsorption equilibria and breakthrough behavior. Under these conditions, pursuing universal single-media solutions is often neither technically nor economically optimal. A more practical strategy is to design role-splitting hybrid treatment trains that leverage complementary material strengths.
Carbonaceous media can serve as robust front-end units to preferentially remove long-chain PFAS and strongly adsorb co-contaminants, thereby mitigating site monopolization and competitive fouling effects that disproportionately impair short-chain PFAS uptake[81,82]. Polymer-based adsorbents can then be deployed as downstream polishing units, where programmable microenvironments enable headgroup-oriented recognition and cooperative binding that can remain effective under competitive conditions. Such staged architectures decouple bulk removal from ultra-trace polishing, improving overall resilience while reducing performance demands on any single material class.
Integration of capture–concentrate–destroy pathways
-
Because adsorption inherently concentrates PFAS into smaller volumes (spent media, regenerant brines, or concentrates), the critical question shifts from removal to responsible management of these secondary streams. Without downstream treatment, adsorption risks merely redistributing PFAS burdens rather than eliminating them.
An emerging and promising direction is the deliberate integration of capture–concentrate–destroy treatment loops, in which separation is explicitly coupled to destructive technologies capable of defluorination. Recent studies demonstrate that concentrating PFAS prior to treatment via adsorption, foam fractionation, or regenerable resins substantially improves the energy efficiency and kinetics of destructive processes such as electrochemical oxidation (e.g., boron-doped diamond anodes), and advanced reduction systems (e.g., UV/sulfite-generated hydrated electrons)[83,84]. Within this context, redox-active polymer adsorbents are particularly compelling. These materials enable programmable capture followed by electrically driven regeneration and in situ oxidative or reductive polishing, providing a materials-level bridge between separation and destruction[41]. Such integration aligns molecular design with system-level closure of PFAS mass balances.
Toward green polymer manufacturing
-
While many polymer adsorbents achieve high selectivity through post-functionalization and solvent-intensive synthesis, these same steps often dominate life-cycle environmental burdens and risk undermining net remediation benefits. As performance targets approach regulatory limits, further gains in adsorption affinity alone are unlikely to justify disproportionate increases in synthesis impacts.
A near-term research priority is therefore to shift from performance-only optimization toward constraint-aware design, in which adsorption selectivity, regenerability, and manufacturing footprints are evaluated as a coupled system. Promising directions include minimizing and recycling solvents and reagents through closed-loop processing, replacing multistep wet chemistry with solvent-free or solvent-minimized approaches, and adopting scalable strategies such as mechanochemical synthesis or reactive extrusion[85,86]. Embedding life-cycle considerations early in material development will be essential for translating laboratory advances into environmentally benign technologies.
-
Polymer-based adsorbents provide a versatile platform for remediating highly water-solubility short-chain PFAS by the rational engineering of programmable microenvironments with co-localized, cooperative binding domains. Across the polymer families examined in this review, high-performance short-chain PFAS removal consistently arises from the integration of complementary interaction motifs: headgroup anchoring sites (electrostatic and/or hydrogen-bonding interactions) and tail-accommodation domains (hydrophobic or fluorophilic). Together, these interactions mitigate desolvation penalties, enhance affinity, and preserve selectivity under competitive real-water matrices.
Representative studies report removal efficiencies exceeding 90% with effluent concentrations reaching the ng L−1 range in complex waters, alongside regenerability under relatively mild chemical or electrochemical conditions. However, LCA reveals that the environmental viability of these materials is not governed by adsorption performance alone. Production-stage impacts, which are dominated by solvent use, functionalization reagents, and energy-intensive synthesis, can rival or exceed the benefits associated with PFAS removal if not carefully managed. Under the characterization framework considered here, the per-mass impact potentials of adsorbent-related substances in human toxicity and freshwater ecotoxicity categories may exceed those of PFAS themselves by orders of magnitude.
This review underscores a central conclusion: the net environmental benefit of polymer-based PFAS adsorption is jointly determined by molecular-level selectivity, operational durability, and controllable material- and process-level burdens across realistic deployment scenarios. Future advances will therefore depend more on integrating cooperative binding design with hybrid treatment architectures, capture–concentrate–destroy workflows, and green manufacturing. Framed in this way, polymer adsorbents can evolve from high-performing materials into deployable components of sustainable PFAS remediation systems.
-
It accompanies this paper at: https://doi.org/10.48130/een-0026-0002.
-
The authors confirm their contributions to the paper as follows: Wenjian Yang: study conception and design, analysis and interpretation of results, draft manuscript preparation; Pengfei Chen: data collection; Jianzhen Huang: data collection; Daoyuan Zu: data collection, analysis and interpretation of results; Kui Yang: analysis and interpretation of results; Xiangtong Kong: analysis and interpretation of results; Xiaodong Yang: data collection, funding and supervision; Choonsoo Kim: analysis and interpretation of results; Jinxing Ma: study conception and design, analysis and interpretation of results, funding and supervision, draft manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
We gratefully acknowledge funding support from the National Natural Science Foundation of China (Grant Nos 52388101, 52470028, and 52300029), the Guangdong Natural Science Funds for Distinguished Young Scholars (Grant No. 2022B1515020053), and the National Key Research and Development Program of China (Grant No. 2022YFC3901304).
-
The authors declare that there is no conflict of interest.
-
Polymer adsorbents for short-chain PFAS remediation are critically compared.
Cooperative multi-site binding governs selective short-chain PFAS adsorption.
Regenerable polymer adsorbents enable capture–concentrate strategies.
Life-cycle analysis links adsorption performance with process sustainability.
-
Full list of author information is available at the end of the article.
- The supplementary files can be downloaded from here.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang W, Chen P, Huang J, Zu D, Yang K, et al. 2026. Emerging polymer-based adsorbents for short-chain PFAS: mechanisms, performance, and research outlook. Energy & Environment Nexus 2: e007 doi: 10.48130/een-0026-0002 

Emerging polymer-based adsorbents for short-chain PFAS: mechanisms, performance, and research outlook
- Received: 07 November 2025
- Revised: 12 January 2026
- Accepted: 15 January 2026
- Published online: 23 February 2026
Abstract: As regulation shifts toward highly mobile short-chain PFAS at sub-ng L−1 targets, conventional carbonaceous adsorbents often underperform in competitive real-water matrices. Polymer adsorbents have proliferated to address this gap, yet the rapidly growing body of literature has not been synthesized into mechanism-driven design guidance and a sustainability-oriented life-cycle perspective. Here, a unifying framework is proposed to guide polymer-based sorbent design for short-chain PFAS, by coupling headgroup anchoring, desolvation control, and fluorinated tail accommodation within confined polymer architectures. Representative polymer families (e.g., cyclodextrin polymers, molecularly imprinted polymers, hydrogels, and electroactive polymers) are critically assessed through this framework, linking molecular interactions to adsorption performance under realistic water matrices, and regeneration constraints. A life-cycle assessment is used to identify synthesis-stage environmental hotspots, and define break-even removal thresholds required for net environmental benefit. In summary, this review establishes cooperative microenvironment engineering as a governing principle for deployable short-chain PFAS remediation, and outlines pathways toward sustainable capture–concentrate–destroy systems suitable for practical applications.
-
Key words:
- Adsorption /
- Polymer /
- Short-chain PFAS /
- Molecular interactions /
- Life-cycle assessment